

Abstract
Iron-mediated free radical damage contributes to secondary damage after intracerebral hemorrhage (ICH). Iron is released from heme after hemoglobin breakdown and accumulates in the parenchyma over days and then persists in the brain for months (e.g., hemosiderin). This non-heme iron has been linked to cerebral edema and cell death. Deferoxamine, a ferric iron chelator, has been shown to mitigate iron-mediated damage, but results vary with less protection in the collagenase model of ICH. This study used rapid-scanning X-ray fluorescence (RS-XRF), a synchrotron-based imaging technique, to spatially map total iron and other elements (zinc, calcium and sulfur) at three survival times after collagenase-induced ICH in rats. Total iron was compared to levels of non-heme iron determined by a Ferrozine-based spectrophotometry assay in separate animals. Finally, using RS-XRF we measured iron levels in ICH rats treated with deferoxamine versus saline. The non-heme iron assay showed elevations in injured striatum at 3 days and 4 weeks post-ICH, but not at 1 day. RS-XRF also detected significantly increased iron levels at comparable times, especially notable in the peri-hematoma zone. Changes in other elements were observed in some animals, but these were inconsistent among animals. Deferoxamine diminished total parenchymal iron levels but did not attenuate neurological deficits or lesion volume at 7 days. In summary, ICH significantly increased non-heme and total iron levels. We evaluated the latter and found it to be significantly lowered by deferoxamine, but its failure to attenuate injury or functional impairment in this model raises concern about successful translation to patients.
Keywords: Stroke, Hemorrhage, Iron, Synchrotron, X-ray fluorescence, Deferoxamine
Introduction
Brain injury after intracerebral hemorrhage (ICH) starts with direct mechanical trauma as blood rapidly enters the parenchyma, and progresses because of numerous deleterious processes causing secondary degeneration. Well-known among these mechanisms are the toxic effects of blood that cause cell death, inflammation, blood brain barrier disruption and cerebral edema. The ever-expanding knowledge of ICH pathophysiology has lead to the development and testing of many neuroprotective treatments in animal models including some drugs that have undergone or are currently in clinical trials (Aronowski and Zhao, 2011; Frantzias et al., 2011; MacLellan et al., 2009).
Among many deleterious processes occurring after ICH, iron is perhaps the most widely studied contributor to secondary degeneration (Aronowski and Zhao, 2011; Xi et al., 2006), and iron is widely thought to have a central role in other neurodegenerative diseases (Kell, 2010). Following ICH, iron is liberated by heme metabolism through heme oxygenase-1 activity, but it is important to note that this does not occur immediately after ICH. Instead, hemolysis only begins about 24 h after ICH occurs and is complete after several days. During this time, activated microglia and macrophages protect the brain by removing intact and damaged erythrocytes from the parenchyma. They also remove released hemoglobin and its component, heme, via uptake of haptoglobin-hemoglobin and hemopexin-heme complexes, respectively (Aronowski and Zhao, 2011). However, these protective measures along with increases in iron binding proteins, ferritin and transferrin, are not sufficient to prevent a substantial increase in oxidative stress, which, for instance, damages DNA after ICH (Aronowski and Zhao, 2011; Nakamura et al., 2005; Wagner et al., 2002; Wu et al., 2012, 2002). Post-ICH oxidative stress originates in part from ‘free’ or loosely bound ferrous iron that promotes the generation of highly reactive free radicals (e.g., OH·) through Fenton chemistry.
Use of animal models shows substantial elevations in iron after ICH. For example, non-heme iron was 2–4× higher in brain tissue samples from 3 to 28 days after a striatal infusion of whole blood (Wu et al., 2003), a common rodent model of ICH. Likewise, iron histochemistry (Perls stain) shows prominent ferric iron concentration within and near the hematoma in rodents (Wu et al., 2003) as does magnetic resonance imaging (MRI) that detects paramagnetic ferritin iron (Wu et al., 2010). Several lines of evidence strongly suggest that excessive iron mediates secondary damage, although not all data concur. First, intracerebral FeCl2 infusion leads to edema and rapid cell death (Huang et al., 2002; Nakamura et al., 2005). We have also observed neuronal death occurring over months after FeCl2 infusion along with marked dendritic atrophy (Caliaperumal, Ma and Colbourne, unpublished data). Thus, iron alone appears to causes injury similar to that seen after ICH, but these data do not prove that this is a key mechanism of secondary degeneration after ICH. Further evidence comes from numerous studies showing that free radical scavengers improve outcome in animal models of ICH. However, these drugs often did not reduce lesion size (Peeling et al., 1998, 2001) and a clinical trial with NXY-059, a free radical scavenger, was also negative (Lyden et al., 2007).
The strongest data to support the hypothesis that iron mediates secondary degeneration comes from the use of iron chelators in animal models. The most widely studied drug is deferoxamine (DFX), a Fe3+ chelator used in patients with iron overload and currently in clinical trials for ICH (Selim et al., 2011). Post-ICH treatment with DFX improves behavioral scores, reduces edema and blood brain barrier dysfunction, and lessens cell death in the whole blood model in rats (Hua et al., 2006; Nakamura et al., 2003; Okauchi et al., 2009, 2010; Song et al., 2008), mice (Wu et al., 2012) and pigs (Gu et al., 2009). However, results in the collagenase model of ICH are less encouraging (Warkentin et al., 2010; Wu et al., 2012). Collagenase is an enzyme that breaks down the basal lamina of vessels resulting in spontaneous hemorrhaging (Rosenberg et al., 1990). It is widely used to model ICH in rodents, but only a few studies have tested whether DFX reduces injury in this model. In one, DFX treatments like those used successfully in whole blood studies failed to lessen edema, behavioral impairments or tissue loss in the collagenase model (Warkentin et al., 2010). Another collagenase study also found that DFX did not lessen edema or lesion volume, but there was a reduction in peri-hematoma cell death and modest functional benefits (Wu et al., 2012). Studies in the whole blood model show that DFX lessens iron levels in cerebrospinal fluid, but interestingly not in the brain (Wan et al., 2006, 2009). Others report that there are fewer iron positive cells, including many macrophages and microglia, as identified with the Perls stain after collagenase-induced ICH (Wu et al., 2012).
There are numerous ways to determine iron levels in biological tissues (McRae et al., 2009). In the ICH field, studies have relied upon the Perls histochemical stain for ferric iron and the non-heme iron assay. The former identifies iron-rich cells, such as macrophages, and hemosiderin, but staining is not easily quantified nor does it reflect total iron. The latter is suitable for quantifying non-heme iron levels (Wu et al., 2008) in homogenized tissue samples but not in histological slides. These two methods cannot quantitatively determine the spread of iron (e.g., relative to the hematoma) nor relate the distribution of neuronal death with iron levels. In contrast, synchrotron rapid scanning X-ray fluorescence (RS-XRF) can both map and quantify elements in tissue slides (Habib et al., 2010; Popescu et al., 2009; Popescu and Nichol, 2011). While RS-XRF has been applied to human stroke (Zheng, Haacke, Webb and Nichol, unpublished data), it has not been used in rodent ICH models. Thus, we used RS-XRF to simultaneously map several elements at moderate resolution (e.g., 50 μm) in coronal sections of rat brain at 3 survival times after collagenase-induced ICH. We also evaluated the effect of DFX treatment on total iron levels as measured with RS-XRF. Tissue loss and behavioral deficits were evaluated too. Finally, for comparative purposes we used a Ferrozine-based non-heme iron assay at 3 times post-ICH.
Methods
Animals
Sixty-nine male Sprague–Dawley rats were obtained from the Biosciences breeding colony at the University of Alberta. They were approximately 11 weeks of age when they were entered into this study. Procedures were in accordance with the Canadian Council on Animal Care and were approved by the Biosciences Animal Care and Use Committee at the University of Alberta. Rats were given free access to food and water throughout the study.
Sixteen rats were used in the first experiment that quantified non-heme iron levels after ICH or in naïve rats. In the second experiment, 36 rats were used to track the time course of iron changes following ICH with RS-XRF. The third experiment used 17 rats to evaluate DFX. In this study, the rats were habituated to their new housing for 3 days and then handled for 4 days (5 min/day) to mitigate handling stress during behavioral testing.
ICH surgery (all experiments)
Surgery was performed aseptically under isoflurane anesthesia (4% induction; ~2% maintenance in 60% N2O with the balance of O2). The head was shaved and disinfected with 70% ethanol and betadine and then 0.2 mL of bupivacaine HCl (Sanofi Canada, Markham, OT, Canada) was injected subcutaneously. At this time rats were injected with 5.0 mL of sterile saline subcutaneously on the back to hydrate the animal during and following the surgery. A midline incision was made and a burr hole was drilled 1.0 mm anterior and 3.5 mm right of Bregma. Note that the lesions appear on the left side in Figures due to the tissue collection method used. After lowering a 26-gauge needle (Hamilton syringe; Hamilton, Reno, NV, USA) 7.0 mm below the surface of the skull, 0.5 μm of sterile saline containing 0.10 U of bacterial collagenase (Type IV-S; Sigma, Oakville, OT, Canada) was injected over 5 min into the striatum (MacLellan et al., 2006; Rosenberg et al., 1990; Warkentin et al., 2010). The needle remained in place for 5 min after the injection and was then slowly removed. A screw (model MN-0080-02P-25; Small Parts Inc., Miami Lakes, FL, USA) was used to seal the hole. Rectal temperature was monitored and maintained near 37.0 °C during the surgery with a heating pad.
Experiment 1: non-heme iron assay
We adapted a method (Rebouche et al., 2004) to measure the amount of non-heme iron in the brains of rats randomly euthanized 1, 3 or 28 days after ICH compared to a naïve control group. Briefly, rats were overdosed with pentobarbital (~100 mg/kg) and perfused with phosphate buffered saline (PBS). The brains were removed and dissected into ipsilateral and contralateral striatum (−2 to +4 mm to Bregma) and dorsal cerebellum (control tissue), each about 0.2 g. Pieces of liver (~0.2 g) were also collected as a control. Teflon coated blades (7280 L, Thermo Scientific, Kalamazoo, MI, USA) and plastic forceps were used to avoid metal contamination. Once the samples were weighed, they were placed in a Dounce homogenizer and distilled water was added to a ratio of 1:10 w/v. The samples were then homogenized, and two aliquots of this solution were transferred to separate microcentrifuge tubes. An equal volume of protein precipitate solution (1 N HCl, 10% trichloroacetic acid in dH2O) was added to each aliquot. The solution was mixed and put in a heating block for 1 h at 95 °C. Samples were then centrifuged at 8200 g for 10 min. Aliquots (100 μL) were taken from each tube and placed in two separate new microcuvettes. Then 600 μL of chromogen solution (0.025 mmol/L Ferrozine, 3 mol/L sodium acetate, 1.5% (v/v) thioglycolic acid in dH2O) was added to each microcuvette and mixed. The samples were left for 30 min at room temperature to allow for color development. The absorbance of each sample, averaged per region of interest for each animal, was then measured at 562 nm using a spectrophotometer and compared to a standard curve made using known quantities of iron (100026-2, High Purity Standards, Charleston, SC, USA) with the above methods. Standards (0, 2, 5, 10 and 15 μg/mL) were made daily, always linear and very similar among days (data not shown).
Experiment 2
Histology
Rats in experiment 2 were randomly euthanized 1, 7 or 21 days following ICH. Rats were injected with sodium pentobarbital (100 mg/kg, i.p.) and then perfused with 0.9% PBS followed by 4% paraformaldehyde in PBS. Ultra pure water was used to make PBS, reducing the chance of metal contamination during perfusion. Brains were extracted and placed in 4% paraformaldehyde over night, and then transferred to 30% sucrose in PBS for at least 2 days. Tissue was then cryostat sectioned at 40 μm using a Teflon coated blade. For lesion volume assessment one section was taken every 200 μm and stained with cresyl violet. Image J 4.0 (Scion Corporation, Fredrick, MD, USA) was used to quantify tissue loss by subtracting the volume of the injured hemisphere from the volume of the normal hemisphere. The volume of each hemisphere was calculated as: (average area of complete coronal section of the hemisphere−area of damage−ventricle)×interval between sections×number of sections (MacLellan et al., 2006; Warkentin et al., 2010). Sections containing the maximum hematoma area were collected on plastic cover slips with very low metal content (Thermanox; Rochester, NY, USA) for RS-XRF analysis.
RS-XRF imaging
The RS-XRF mapping of iron, zinc, sulphur and calcium in coronal sections was carried out at the Stanford Synchrotron Radiation Light-source (SSRL) on beamline 10–2. Sections were imaged at a 50-μm resolution with a dwell time of 50 ms using an incident energy of 13 keV. The slides were mounted vertically at 45° to the incident X-ray beam and 45° to the detector. Fluorescence was normalized against incident X-ray beam intensity. Elements were quantified by comparing the signal strength of the sample against calibration standards designed for XRF (Micromatters, Inc., Sault Ste. Marie, ON, Canada). Quantitative analysis was performed using Sam’s Microanalysis Toolkit (SMAK; Open Source, Samuel Webb, SSRL, Stanford, CA, USA).
Experiment 3: DFX treatment
Drug treatment
In the third experiment, rats were randomly assigned to receive either saline (SAL) or DFX injections (100 mg/kg i.p. at a concentration of 50 mg/mL, Sigma) after ICH. Injections started 6 h after ICH and continued every 12 h until euthanasia for a total of 14 injections. This is a commonly used treatment regimen (Hua et al., 2006; Nakamura et al., 2003; Okauchi et al., 2009; Warkentin et al., 2010).
Neurological deficit scale (NDS) testing
Scores on the NDS were assessed prior to and at 1, 3 and 7 days after surgery. Behavioral testing was done no earlier than 6 h following an injection to reduce the effect of stress on functional performance. Rats were rated on several subtests: spontaneous circling, contralateral hind limb retraction after lateral movement, bilateral forepaw grasp, contralateral forelimb flexion and beam walking ability. Scores on each subtest were combined to give an overall indication of impairment with 0 indicating unimpaired performance and 14 representing maximal deficits (MacLellan et al., 2006; Warkentin et al., 2010). The researcher conducting the testing was blind to group identity.
Histology and RS-XRF
Rats in the experiment 3 were killed at day 7 by overdose with pentobarbital (100 mg/kg, i.p.) and processed for lesion volume and RS-XRF imaging as in Experiment 2.
Statistical analysis (all experiments)
Most data are presented as mean and SEM. The RS-XRF and lesion volume data were assessed using analysis of variance (ANOVA; between subjects design), followed by Tukey post hoc testing when needed. For the ordinal NDS scale, which is presented as medians and inter-quartile range, we used the Kruskal Wallis and Mann Whitney tests to compare groups and the Wilcoxon sign ranks test to compare between days. The Pearson r-value is provided for correlations.
Results
Experiment 1: non-heme iron levels following ICH
There were no exclusions or unexpected mortality in experiment 1.
One-way ANOVAs showed significant main effects for the non-heme iron levels (Fig. 1) within the injured (p<0.001) and non-injured hemispheres (p=0.026), but not the cerebellum (p=0.533) or liver controls (p=0.642). For the injured hemisphere, the day 3 and 28 groups had significantly more iron than naïve or day 1 ICH groups (p≤0.013) and there was more iron at 28 than 3 days post-ICH (p=0.017). For the uninjured side, only day 3 was slightly but significantly higher than naïve rats (p=0.016), but not different from other times post-ICH (p≥0.345). The injured side had considerably more non-heme iron than the contralateral hemisphere on days 3 (p=0.001) and 28 (p=0.016), but not day 1 (p=0.072).
Fig. 1.

Non-heme iron levels (mean±SEM) measured with a Ferrozine-based spectrophotometry assay in naïve rats and at 1, 3 and 28 days after ICH. Samples were taken contra-lateral to ICH (A), on the side of the ICH (B), in the liver (C) and cerebellum (D). Non-heme iron was significantly elevated in the damaged hemisphere at 3 and 28 days compared to the naïve and day 1 ICH group. Day 3 was also significantly higher than naïve rats in the contralateral hemisphere, but not from other times after ICH. No significant differences were observed in either the cerebellum or liver samples. An * denotes significance from naive whereas # denotes a significant difference from day 3 (p<0.05).
Experiment 2: time course and distribution of iron following ICH
There were no exclusions or unexpected mortality in experiment 2.
Significant edema was evident in the histological sections from the ICH group that survived for 24 h, and this was confirmed by image analysis of hemisphere size (Fig. 2a). This prevented accurate lesion size determination. Tissue loss and cavity formation were evident at later survival times with the volume of tissue loss more than doubling from 7 to 21 days (Fig. 2b, p=0.003).
Fig. 2.
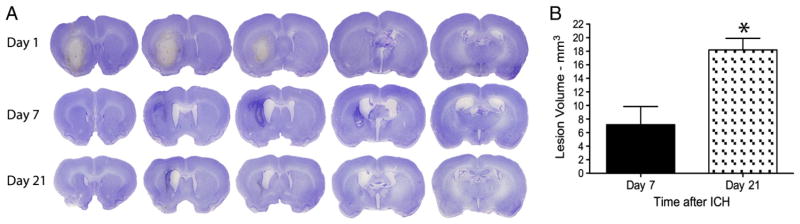
Collagenase-induced ICH primarily damaged the striatum, but other structures were sometimes directly injured, such as the corpus callosum. A) Representative cresyl violet stained lesions illustrate swelling of the damaged hemisphere on day 1, which therefore precluded an accurate determination of tissue loss. B) Tissue loss (mean±SEM) significantly increased between 7 and 21 days post stroke (* denotes p<0.05), which likely stems from continuing cell death, atrophy and more rarely resolution of any residual edema at day 7.
Total iron levels (Fig. 3a) were significantly different among groups in the injured hemisphere (p=0.002, main effect), but not in the contralateral side (p=0.689, main effect). Compared to normal, total iron levels were significantly higher on days 7 (p=0.003) and 21 (p=0.008), but the elevation on day 1 did not attain statistical significance (p=0.368). The injured side had considerably more total iron than the contralateral hemisphere (e.g., all ICH rats included, p<0.001).
Fig. 3.
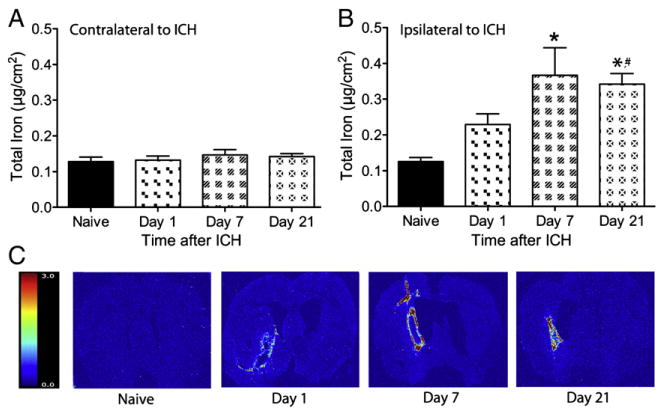
The total concentration (mean±SEM) of iron in contralateral (A) and ipsilateral (B) hemispheres of ICH rats were measured at 1, 7 and 21 following collagenase injection. Contralateral levels of iron did not differ from naïve levels at any time. Iron levels in the ipsilateral hemisphere were elevated from naïve levels at 7 and 21 days following ICH (* p<0.05 vs. naïve rats) and were further elevated at day 21 as compared to day 1 (# p<0.05). Representative images display the distribution and concentration of iron after ICH (C), the color legend represents μg/cm2 of iron.
To determine spread of iron from the hematoma four regions of interest (0.5×0.5 mm) were measured in each hemisphere, starting at the hematoma edge or corresponding location in the non-lesion hemisphere and extending laterally (contralateral: C1-4 and ipsilateral: I1-4; Fig. 4A). Initially at 1 and 7 days iron was contained within the lesion and was not higher in surrounding tissue (p≥0.099, Fig. 4B). However, the quantity of iron in the region directly bordering the hematoma (I1) was slightly higher than naïve levels at 21 days following ICH (p=0.033). This elevation was small, with a mean increase of 0.06 μg/cm2. No other regions had elevated iron (main effects: p≥0.275). Iron levels in the corresponding locations within the contralateral hemisphere were not different among groups (i.e., naïve levels, day 1 after ICH, etc.; main effects: p≥0.631).
Fig. 4.
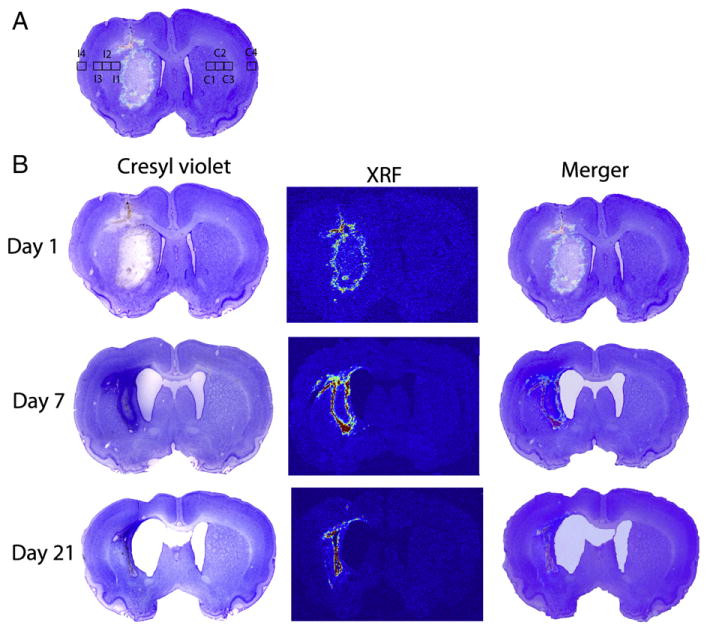
Part A indicates the regions of interest where measurements were taken to determine spread of iron from the hematoma. At 21 days post stroke the level of iron in I1 was significantly higher than a corresponding region in naïve rats. The remaining images (B) demonstrate the injury as identified with cresyl violet staining, the location and concentration of iron mapped with XRF and the merger of these images. High iron regions overlie the border zone of the injury.
Several other elements were simultaneously mapped with RS-XRF. Calcium was visibly elevated in or surrounding the lesion in 4 rats 1 day following ICH, in 7 rats at 7 days and 3 rats at 21 days. Although there were distinct regions of high calcium, the quantity of hemispheric calcium at days 1, 7 and 21 was not different from naïve levels (p≥0.308). Zinc was visibly elevated in 5 brains at 7 days and 2 brains at 21 day following ICH. At 21 days following stroke one brain had sites that had co-localized calcium, zinc and sulfur in a region outside of a dense iron deposit. The distributions and intensities of these changes varied significantly across brains with two examples shown in Fig. 5. Calcium was frequently widespread, located within the core of the injury as well as surrounding tissue, whereas zinc and sulfur changes tended to be more concentrated in small sites surrounding the high iron area. This variability complicated the quantification and interpretation of these findings.
Fig. 5.

Several elements were mapped simultaneously with RS-XRF. The top row of data comes from a single rat brain 7 days after ICH. In this case there was elevated calcium in and around the hematoma. The bi-color map indicates the spatial relationship of these two elements. The second group of images is from a rat at 21 days post-ICH. Elevations of calcium, zinc and sulfur co-localize to a region outside of the iron rich area. All XRF images are scaled independently with the respective legends representing concentrations in μg/cm2.
Experiment 3: effects of DFX treatment
One DFX-treated animal died within minutes of an injection and was excluded from all analysis.
Prior to ICH scores were normal indicating no pre-stroke deficits. There were no group differences at any time (p≥0.328; Fig. 6). However, scores (both groups) were significantly elevated (impairment) following stroke for all days (p<0.001), but with some recovery over days (p≤0.017). Thus, the NDS was sensitive to ICH, but there were no treatment effects.
Fig. 6.

Neurological function (NDS scores) was assessed prior to (baseline — BL) and on days 1, 3 and 7 following ICH. Each symbol represents the NDS score for one animal. Also shown is this median and inter-quartile range. There were no significant group differences at any time indicating that DFX did not improve outcome.
Seven days following ICH the lesion affected the striatal region and in some cases nearby structures, such as the corpus callosum. Lesion volume was not different between groups (p=0.994) with the SAL and DFX groups having a mean lesion volume±SEM of 10.66 mm3±2.84 and 10.63 mm3±1.91, respectively.
Seven days of DFX treatment significantly reduced the amount of iron in both the ipsilateral (p=0.004) and contralateral to lesion hemisphere (p =0.018; Figs. 7A and B). However, iron levels in the injured hemisphere did not correlate with either lesion volume (r=0.361, p=0.169) or behavioral outcome (e.g., day 1 NDS: r=0.213, p=0.428).
Fig. 7.
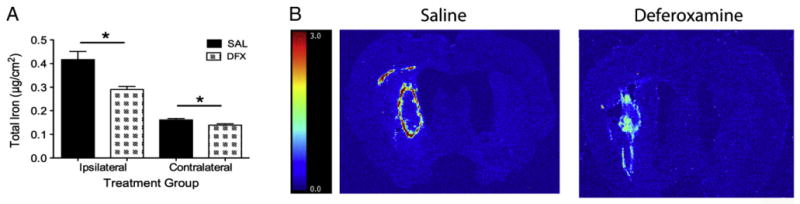
Total iron levels (mean±SEM) were quantified in the ipsilateral and contralateral hemispheres with RS-XRF. Giving DFX significantly lessened iron levels in both hemispheres (A; * p<0.05). Representative iron images are shown in B. The color scale is in μg/cm2.
Discussion
To our knowledge, this is the first experiment using RS-XRF to map and quantify iron, and other elements, after collagenase-induced ICH in rats. Total iron in the damaged hemisphere was significantly and persistently elevated after ICH, but elevations were localized to the hematoma and immediate peri-hematoma regions. Other studies using Perls histochemistry and non-heme spectrophotometry assays (Wu et al., 2003) have drawn similar conclusions. Similarly, we demonstrated a persistent increase in non-heme iron in the collagenase model that was comparable to our RS-XRF data and also to studies using the non-heme iron assay in the whole blood model (Wu et al., 2003). Using the RS-XRF technique we found that post-ICH treatment with DFX lowered total brain iron levels, but there was no improvement on neurological scores nor was there a reduction in brain damage. The quantity of other elements (e.g., Zn++) also varied after ICH with some localized accumulations. These data were more variable, but they have been observed in sections of human brain (Zheng, Haacke, Webb and Nichol, unpublished data). The significance of these findings is not known from these studies. However, calcification commonly occurs after injury, so finding increased levels in damaged areas is not surprising. Localized Zn accumulations are interesting in light of the role Zn plays in neurotoxicity (Shuttleworth and Weiss, 2011) and neuroplasticity (Nakashima and Dyck, 2009), but its role here is unknown. Finally, we speculate that the elevated sulfur levels may reflect alterations in redox state, but additional experiments that map sulfur chemical species are needed to prove this.
As demonstrated, the RS-XRF method spatially quantifies several elements simultaneously in histological sections and is well suited to the study of elemental changes after stroke, including ICH. Neither the Perls stain nor the non-heme assay can achieve this spatial quantification of total iron concentration. The localization of ferric iron with Perls staining to the hematoma and peri-hematoma regions fits with our findings of elevated total iron levels in the hematoma (early after ICH) and immediate peri-hematoma zone whereas normal levels are found further away from the hemorrhage. However, Perls staining is perhaps only suitable for detecting substantial effects and not suitable in all situations such as when a treatment reduces the number of iron-rich cells (all cells have iron) but increases the concentration of iron per cell. Such a situation might occur with anti-inflammatory treatments thereby giving the erroneous indication that iron levels have been attenuated. The RS-XRF technique has several advantages over non-heme iron assays, which in normal situations measure the bulk of iron content, and atomic absorption spectroscopy, which measures total iron. Specifically, the latter techniques destroy the tissue making it impossible to take further measurements (e.g., sections for quantifying lesion volume). As well, spatial quantification is not easily achieved other than to compare relatively large samples from different regions of interest. Both bulk sampling methods and RS-XRF are potentially confounded by cerebral edema that would cause one to underestimate iron concentration (more water with lower cell density per sample), which may partly explain the higher iron levels at later times than in the first few days after ICH. There are potential disadvantages of RS-XRF technique including cost (travel to a synchrotron), additional processing time (e.g., vs. non-heme assay) and limited access to synchrotron time.
Our second experiment showed that the volume of brain damage more than doubled from 7 to 21 days after collagenase infusion, which concurs with previous studies using histology (Nguyen et al., 2008) and MRI endpoints (MacLellan et al., 2008). Protracted neuronal death also occurs in the whole blood model (Felberg et al., 2002), but less so than the collagenase model even when hematoma size is equivalent (MacLellan et al., 2008). We recently observed neuronal death occurring over months and concomitant growth in lesion volume after striatal infusion of FeCl2 (Caliaperumal, Ma and Colbourne, unpublished data). These findings implicate, but not conclusively, early and perhaps protracted iron toxicity in long-term cell death after ICH.
Such findings also strongly suggest that secondary damage, including that sustained in the collagenase model, should be amenable to iron chelation therapy. However, whereas total iron levels were lowered by DFX treatment, we found no evidence to support the claim that the treatment is neuroprotective because both the volume of injury and the behavioral scores were the same as control animals. These findings mirror our previous study where DFX treatment did not lessen cerebral edema, behavioral deficits or tissue loss after collagenase-induced ICH (Warkentin et al., 2010). Notably, that study also used additional behavioral tests and a longer survival time than what we presently used. Thus, it is unlikely that survival time or test sensitivity accounts for our failure to find a neuroprotective effect with DFX. Although, we acknowledge that some behavioral benefit and a small reduction in neuronal death with DFX may have gone undetected. Others have found that DFX does not affect edema or total tissue damage, but they did observe a modest functional benefit in the collagenase model (Wu et al., 2012). Drug dosages and administration regimens have varied among studies, but are comparable in collagenase and whole blood studies. Furthermore, the timing of iron accumulation after collagenase-induced ICH in the present study is comparable to previous work in the whole blood model (Wu et al., 2003) suggesting that both models should be amenable to iron chelation therapy. We previously compared 3 and 7-day DFX treatment regimens and found that neither provided benefit in the collagenase model (Warkentin et al., 2010), but it remains possible that longer treatments might be better or harmful. Thus, the discrepancy between the collagenase and whole blood models is not yet easily reconciled and may not relate to iron per se, but to other differences between models (Kirkman et al., 2011; MacLellan et al., in press). Such factors as the extent and timing of injury (MacLellan et al., 2008) and other mechanisms of secondary degeneration (e.g., extent of inflammation) must be considered. Collectively, animal studies strongly support the use of DFX and other iron chelators (e.g., dipyridyl — (Wu et al., 2011) for hemorrhagic stroke. Should DFX be proven to help ICH patients (Selim et al., 2011), one must then question the predictive validity of the collagenase model. Conversely, if DFX fails to help ICH patients, then the predictive validity of the whole blood model will be challenged.
In summary, our findings show that RS-XRF is an excellent method for evaluating total concentrations of iron and other elements after hemorrhagic stroke. The method is especially useful in situations where the spread or containment of iron may also be affected. In light of the failure of DFX to improve outcome despite lowering iron levels, it seems prudent to question the neuroprotective potential of DFX and the role of iron chelation versus other potential mechanisms of action for this drug.
Acknowledgments
This work is supported by a joint Canadian Institutes of Health Research (CIHR)/Heart and Stroke Foundation of Canada team grant: Synchrotron Medical Imaging (#CIF 99472) awarded to H. Nichol, P. Paterson, F. Colbourne and others. A. Auriat is a CIHR-fellow in Health Research Using Synchrotron Techniques and also supported by a McCormick Fellowship. G. Silasi received funding from a focus on stroke doctoral scholarship. F. Colbourne is a senior medical scholar of Alberta Innovates — Health Solutions. The authors also thank Yonglie Ma, Uzair Ahmed and Nita Plummer for technical assistance as well as Drs. Sam Webb (SSRL) and Weili Zheng for valuable assistance at the synchrotron. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program (P41RR001209).
References
- Aronowski J, Zhao X. Molecular pathophysiology of cerebral hemorrhage: secondary brain injury. Stroke. 2011;42:1781–1786. doi: 10.1161/STROKEAHA.110.596718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felberg RA, Grotta JC, Shirzadi AL, Strong R, Narayana P, Hill-Felberg SJ, Aronowski J. Cell death in experimental intracerebral hemorrhage: the “black hole” model of hemorrhagic damage. Ann Neurol. 2002;51:517–524. doi: 10.1002/ana.10160. [DOI] [PubMed] [Google Scholar]
- Frantzias J, Sena ES, Macleod MR, Al-Shahi Salman R. Treatment of intracerebral hemorrhage in animal models: meta-analysis. Ann Neurol. 2011;69:389–399. doi: 10.1002/ana.22243. [DOI] [PubMed] [Google Scholar]
- Gu Y, Hua Y, Keep RF, Morgenstern LB, Xi G. Deferoxamine reduces intra-cerebral hematoma-induced iron accumulation and neuronal death in piglets. Stroke. 2009;40:2241–2243. doi: 10.1161/STROKEAHA.108.539536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habib AC, Zheng EM, Haacke ME, Webb RC, Nichol H. Visualizing iron deposition in multiple sclerosis cadaver brains. Am Inst Phys. 2010;1266:78–83. [Google Scholar]
- Hua Y, Nakamura T, Keep RF, Wu J, Schallert T, Hoff JT, Xi G. Long-term effects of experimental intracerebral hemorrhage: the role of iron. J Neurosurg. 2006;104:305–312. doi: 10.3171/jns.2006.104.2.305. [DOI] [PubMed] [Google Scholar]
- Huang FP, Xi G, Keep RF, Hua Y, Nemoianu A, Hoff JT. Brain edema after experimental intracerebral hemorrhage: role of hemoglobin degradation products. J Neurosurg. 2002;96:287–293. doi: 10.3171/jns.2002.96.2.0287. [DOI] [PubMed] [Google Scholar]
- Kell DB. Towards a unifying, systems biology understanding of large-scale cellular death and destruction caused by poorly liganded iron: Parkinson’s, Huntington’s, Alzheimer’s, prions, bactericides, chemical toxicology and others as examples. Arch Toxicol. 2010;84:825–889. doi: 10.1007/s00204-010-0577-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkman MA, Allan SM, Parry-Jones AR. Experimental intracerebral hemorrhage: avoiding pitfalls in translational research. J Cereb Blood Flow Metab. 2011;31:2135–2151. doi: 10.1038/jcbfm.2011.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyden PD, Shuaib A, Lees KR, Davalos A, Davis SM, Diener HC, Grotta JC, Ashwood TJ, Hardemark HG, Svensson HH, Rodichok L, Wasiewski WW, Ahlberg G. Safety and tolerability of NXY-059 for acute intracerebral hemorrhage: the CHANT trial. Stroke. 2007;38:2262–2269. doi: 10.1161/STROKEAHA.106.472746. [DOI] [PubMed] [Google Scholar]
- MacLellan CL, Auriat AM, McGie SC, Yan RH, Huynh HD, De Butte MF, Colbourne F. Gauging recovery after hemorrhagic stroke in rats: implications for cytoprotection studies. J Cereb Blood Flow Metab. 2006;26:1031–1042. doi: 10.1038/sj.jcbfm.9600255. [DOI] [PubMed] [Google Scholar]
- MacLellan CL, Silasi G, Poon CC, Edmundson CL, Buist R, Peeling J, Colbourne F. Intracerebral hemorrhage models in rat: comparing collagenase to blood infusion. J Cereb Blood Flow Metab. 2008;28:516–525. doi: 10.1038/sj.jcbfm.9600548. [DOI] [PubMed] [Google Scholar]
- MacLellan CL, Peeling J, Colbourne F. Cytoprotection strategies for experimental intracerebral hemorrhage. In: Carhuapoma JR, Mayer SA, editors. Textbook of Intracerebral Hemorrhage. Cambridge University Press; 2009. [Google Scholar]
- MacLellan CL, Paquette R, Colbourne F. A critical appraisal of experimental intracerebral hemorrhage research. J Cereb Blood Flow Metab. doi: 10.1038/jcbfm.2012.8. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRae R, Bagchi P, Sumalekshmy S, Fahrni CJ. In situ imaging of metals in cells and tissues. Chem Rev. 2009;109:4780–4827. doi: 10.1021/cr900223a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Keep RF, Hua Y, Schallert T, Hoff JT, Xi G. Deferoxamine-induced attenuation of brain edema and neurological deficits in a rat model of intracerebral hemorrhage. Neurosurg Focus. 2003;15:ECP4. doi: 10.3171/foc.2003.15.4.10. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Keep RF, Hua Y, Hoff JT, Xi G. Oxidative DNA injury after experimental intracerebral hemorrhage. Brain Res. 2005;1039:30–36. doi: 10.1016/j.brainres.2005.01.036. [DOI] [PubMed] [Google Scholar]
- Nakashima AS, Dyck RH. Zinc and cortical plasticity. Brain Res Rev. 2009;59:347–373. doi: 10.1016/j.brainresrev.2008.10.003. [DOI] [PubMed] [Google Scholar]
- Nguyen AP, Huynh HD, Sjovold SB, Colbourne F. Progressive brain damage and alterations in dendritic arborization after collagenase-induced intracerebral hemorrhage in rats. Curr Neurovasc Res. 2008;5:171–177. doi: 10.2174/156720208785425710. [DOI] [PubMed] [Google Scholar]
- Okauchi M, Hua Y, Keep RF, Morgenstern LB, Xi G. Effects of deferoxamine on intracerebral hemorrhage-induced brain injury in aged rats. Stroke. 2009;40:1858–1863. doi: 10.1161/STROKEAHA.108.535765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okauchi M, Hua Y, Keep RF, Morgenstern LB, Schallert T, Xi G. Deferoxamine treatment for intracerebral hemorrhage in aged rats: therapeutic time window and optimal duration. Stroke. 2010;41:375–382. doi: 10.1161/STROKEAHA.109.569830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeling J, Yan HJ, Chen SG, Campbell M, Del Bigio MR. Protective effects of free radical inhibitors in intracerebral hemorrhage in rat. Brain Res. 1998;795:63–70. doi: 10.1016/s0006-8993(98)00253-4. [DOI] [PubMed] [Google Scholar]
- Peeling J, Del Bigio MR, Corbett D, Green AR, Jackson DM. Efficacy of disodium 4-[(tert-butylimino)methyl]benzene-1,3-disulfonate N-oxide (NXY-059), a free radical trapping agent, in a rat model of hemorrhagic stroke. Neuropharmacology. 2001;40:433–439. doi: 10.1016/s0028-3908(00)00170-2. [DOI] [PubMed] [Google Scholar]
- Popescu BF, Nichol H. Mapping brain metals to evaluate therapies for neurodegenerative disease. CNS Neurosci Ther. 2011;17:256–268. doi: 10.1111/j.1755-5949.2010.00149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popescu BF, George MJ, Bergmann U, Garachtchenko AV, Kelly ME, McCrea RP, Luning K, Devon RM, George GN, Hanson AD, Harder SM, Chapman LD, Pickering IJ, Nichol H. Mapping metals in Parkinson’s and normal brain using rapid-scanning x-ray fluorescence. Phys Med Biol. 2009;54:651–663. doi: 10.1088/0031-9155/54/3/012. [DOI] [PubMed] [Google Scholar]
- Rebouche CJ, Wilcox CL, Widness JA. Microanalysis of non-heme iron in animal tissues. J Biochem Biophys Methods. 2004;58:239–251. doi: 10.1016/j.jbbm.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, Mun-Bryce S, Wesley M, Kornfeld M. Collagenase-induced intracerebral hemorrhage in rats. Stroke. 1990;21:801–807. doi: 10.1161/01.str.21.5.801. [DOI] [PubMed] [Google Scholar]
- Selim M, Yeatts S, Goldstein JN, Gomes J, Greenberg S, Morgenstern LB, Schlaug G, Torbey M, Waldman B, Xi G, Palesch Y. Safety and tolerability of deferoxamine mesylate in patients with acute intracerebral hemorrhage. Stroke. 2011;42:3067–3074. doi: 10.1161/STROKEAHA.111.617589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuttleworth CW, Weiss JH. Zinc: new clues to diverse roles in brain ischemia. Trends Pharmacol Sci. 2011;32:480–486. doi: 10.1016/j.tips.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S, Hua Y, Keep RF, He Y, Wang J, Wu J, Xi G. Deferoxamine reduces brain swelling in a rat model of hippocampal intracerebral hemorrhage. Acta Neurochir Suppl. 2008;105:13–18. doi: 10.1007/978-3-211-09469-3_3. [DOI] [PubMed] [Google Scholar]
- Wagner KR, Packard BA, Hall CL, Smulian AG, Linke MJ, De Courten-Myers GM, Packard LM, Hall NC. Protein oxidation and heme oxygenase-1 induction in porcine white matter following intracerebral infusions of whole blood or plasma. Dev Neurosci. 2002;24:154–160. doi: 10.1159/000065703. [DOI] [PubMed] [Google Scholar]
- Wan S, Hua Y, Keep RF, Hoff JT, Xi G. Deferoxamine reduces CSF free iron levels following intracerebral hemorrhage. Acta Neurochir Suppl. 2006;96:199–202. doi: 10.1007/3-211-30714-1_43. [DOI] [PubMed] [Google Scholar]
- Wan S, Zhan R, Zheng S, Hua Y, Xi G. Activation of c-Jun-N-terminal kinase in a rat model of intracerebral hemorrhage: the role of iron. Neurosci Res. 2009;63:100–105. doi: 10.1016/j.neures.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warkentin LM, Auriat AM, Wowk S, Colbourne F. Failure of deferoxamine, an iron chelator, to improve outcome after collagenase-induced intracerebral hemorrhage in rats. Brain Res. 2010;1309:95–103. doi: 10.1016/j.brainres.2009.10.058. [DOI] [PubMed] [Google Scholar]
- Wu J, Hua Y, Keep RF, Schallert T, Hoff JT, Xi G. Oxidative brain injury from extravasated erythrocytes after intracerebral hemorrhage. Brain Res. 2002;953:45–52. doi: 10.1016/s0006-8993(02)03268-7. [DOI] [PubMed] [Google Scholar]
- Wu J, Hua Y, Keep RF, Nakamura T, Hoff JT, Xi G. Iron and iron-handling proteins in the brain after intracerebral hemorrhage. Stroke. 2003;34:2964–2969. doi: 10.1161/01.STR.0000103140.52838.45. [DOI] [PubMed] [Google Scholar]
- Wu J, Yang S, Xi G, Song S, Fu G, Keep RF, Hua Y. Microglial activation and brain injury after intracerebral hemorrhage. Acta Neurochir Suppl. 2008;105:59–65. doi: 10.1007/978-3-211-09469-3_13. [DOI] [PubMed] [Google Scholar]
- Wu G, Xi G, Hua Y, Sagher O. T2* magnetic resonance imaging sequences reflect brain tissue iron deposition following intracerebral hemorrhage. Transl Stroke Res. 2010;1:31–34. doi: 10.1007/s12975-009-0008-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Wu T, Xu X, Wang J. Iron toxicity in mice with collagenase-induced intracerebral hemorrhage. J Cereb Blood Flow Metab. 2011;31:1243–1250. doi: 10.1038/jcbfm.2010.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Wu T, Li M, Wang J. Efficacy of the lipid-soluble iron chelator 2,2′-dipyridyl against hemorrhagic brain injury. Neurobiol Dis. 2012;45:388–394. doi: 10.1016/j.nbd.2011.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi G, Keep RF, Hoff JT. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 2006;5:53–63. doi: 10.1016/S1474-4422(05)70283-0. [DOI] [PubMed] [Google Scholar]