

Abstract
C→U RNA editing of neurofibromatosis 1 (NF1) mRNA changes an arginine (CGA) to a UGA translational stop codon, predicted to result in translational termination of the edited mRNA. Previous studies demonstrated varying degrees of C→U RNA editing in peripheral nerve–sheath tumor samples (PNSTs) from patients with NF1, but the basis for this heterogeneity was unexplained. In addition, the role, if any, of apobec-1, the catalytic deaminase that mediates C→U editing of mammalian apolipoprotein B (apoB) RNA, was unresolved. We have examined these questions in PNSTs from patients with NF1 and demonstrate that a subset (8/34) manifest C→U editing of RNA. Two distinguishing characteristics were found in the PNSTs that demonstrated editing of NF1 RNA. First, these tumors express apobec-1 mRNA, the first demonstration, in humans, of its expression beyond the luminal gastrointestinal tract. Second, PNSTs with C→U editing of RNA manifest increased proportions of an alternatively spliced exon, 23A, downstream of the edited base. C→U editing of RNA in these PNSTs was observed preferentially in transcripts containing exon 23A. These findings were complemented by in vitro studies using synthetic RNA templates incubated in the presence of recombinant apobec-1, which again confirmed preferential editing of transcripts containing exon 23A. Finally, adenovirus-mediated transfection of HepG2 cells revealed induction of editing of apoB RNA, along with preferential editing of NF1 transcripts containing exon 23A. Taken together, the data support the hypothesis that C→U RNA editing of the NF1 transcript occurs both in a subset of PNSTs and in an alternatively spliced form containing a downstream exon, presumably an optimal configuration for enzymatic deamination by apobec-1.
Introduction
Neurofibromatosis type 1 (NF1 [MIM 162200]) is a dominantly inherited disease affecting ∼1/3,500 individuals worldwide. The NF1 gene has been mapped and characterized extensively, and several features of interest have emerged in relation to its altered function in disease. The NF1 gene spans ∼350 kb on chromosome 17q11.2 and encodes 60 exons, which are ubiquitously transcribed (Li et al. 1995). The nuclear NF1 transcript is ∼11 kb and encodes neurofibromin, a tumor-suppressor–gene product of 2,818 residues, which contains a region of ∼366 amino acids that functions as a GTPase-activating protein (GAP) (reviewed in Cichowski and Jacks 2001). GAP activity has been demonstrated for neurofibromin—more specifically, in the context of Ras activation, both in vitro and in vivo—and is postulated to be an important feature of the loss of function accompanying mutations in the NF1 gene (Basu et al. 1992; DeClue et al. 1992; Nakafuku et al. 1993; Bollag et al. 1996; Guha et al. 1996; Lau et al. 2000; Sherman et al. 2000).
Although the disorder becomes fully penetrant at a relatively early age, the clinical features of NF1 are quite variable even between affected siblings. In addition, as many as half of the newly diagnosed cases of NF1 represent new mutations, a feature likely accounted for by the high spontaneous-mutation rate at this locus (Andersen et al. 1993; Ars et al. 2000). Taken together, the range of mutations and the phenotypic variability suggest that modifier genes may play a significant role in the natural history of this disease (Skuse and Ludlow 1995). An important consideration in understanding the potential mechanisms for exerting phenotypic variability in this disease is the role of RNA processing of transcripts arising from somatic and germline mutations in the NF1 gene; for example, alterations in mRNA splicing have been demonstrated to occur in ∼50% of patients with NF1 (Park and Pivnick 1998), suggesting an important mechanism for amplifying the phenotypic variability alluded to above.
Another posttranscriptional mechanism, C→U editing of RNA, was postulated for inactivation of the GAP function of NF1 some years ago, by Skuse and colleagues (Skuse et al. 1996; Skuse and Cappione 1997). These investigators identified a C→U change, occurring at nt 3916 in the NF1 transcript, that changes a genomically templated arginine (CGA) to a UGA stop codon in the edited mRNA (Skuse et al. 1996; Skuse and Cappione 1997). The edited transcript encodes a truncated protein product that terminates in the amino-terminal portion of the GAP-related domain and would thus result in functional inactivation of the tumor-suppressor function of neurofibromin (Skuse et al. 1996; Ashkenas 1997; Cappione et al. 1997; Skuse and Cappione 1997). Nevertheless, certain features of these original findings were unexplained. The level of C→U editing of NF1 RNA, for example, was quite variable (4%–17%) among the tumors examined, with a trend suggesting that the more malignant tumors demonstrated higher levels of editing (Skuse et al. 1996; Skuse and Cappione 1997). In addition, the enzymatic factors mediating this C→U change were not identified, and the relationship, if any, to editing of intestinal apolipoprotein B (apoB) (APOB [MIM 107730]) mRNA, the prototype example of C→U deamination of nuclear RNA, was not resolved. In particular, C→U editing of mammalian intestinal apoB RNA demonstrates absolute dependence on the expression of the catalytic deaminase, apobec-1 (APOBEC-1 [MIM 600130]) (Hirano et al. 1996; Morrison et al. 1996), whereas preliminary studies by Skuse et al. (1996) revealed no clear evidence that apobec-1 plays a role in the editing of NF1 RNA.
We have reexamined these questions through analysis of peripheral nerve–sheath tumor samples (PNSTs) from 34 patients with NF1. The majority, 26 of 34, of tumors demonstrated low levels of C→U RNA editing, in the range of 0%–2.5%, with 10 of these 26 demonstrating <1% editing, the limits of reliable detection of the assay. However, the remaining 8 of the 34 tumors demonstrated 3%–12% C→U editing of NF1 RNA, which was reproducible and validated by sequencing of the cDNA products. These tumors demonstrated two distinguishing characteristics. First, these PNSTs express apobec-1 mRNA, the catalytic deaminase of the holoenzyme that edits apoB RNA (Teng et al. 1993; Lau et al. 1994; Yamanaka et al. 1994; Davidson and Shelness 2000), suggesting an important role for apobec-1 in the context of a target beyond apoB RNA. Second, NF1 RNA from these PNSTs contains increased proportions of an alternatively spliced exon, 23A, downstream of the edited base in which editing occurs preferentially. These findings, together with results of both in vivo and in vitro experiments with apobec-1, strongly suggest an important mechanistic linkage between NF1 RNA splicing and C→U editing and provide a basis for understanding the heterogeneity of posttranscriptional regulation of NF1 expression.
Material and Methods
Tissue Procurement
The use of human tumor and normal adjacent tissue in this investigation was reviewed and approved by the institutional review boards of the University of Chicago Hospitals, the University of Utah Hospital, Massachusetts General Hospital, Harvard University Hospital, and Washington University School of Medicine. Samples of PNSTs were collected from a total of 34 patients with NF1, whose clinical diagnosis was based on National Institutes of Health consensus-conference criteria (National Institutes of Health Consensus Development Conference 1988). No attempt was made to identify familial versus sporadic cases. In addition, paired normal and tumor samples were obtained from 11 patients (without NF1) undergoing surgical resection for colon adenocarcinoma. The majority of these samples were moderately differentiated tumors located in the proximal, mid, or descending colon. Each was snap-frozen in liquid nitrogen and was stored at −80°C until processed. Frozen tissue sections were stained with hematoxylin and eosin to confirm the histological composition of these specimens.
Endogenous Editing of NF1 mRNA
Total RNA and genomic DNA from the NF1 and colon-cancer tissues was isolated by TRIzol reagent (LifeTechnologies). First-strand cDNA was synthesizedby Superscript II reverse transcriptase (RT) (LifeTechnologies) primed with random hexamers. PCR was performed with Pyrococcus woesei DNA polymerase (Roche Molecular Biochemicals), to amplify NF1 cDNA flanking the edited base (fig. 1), by primers SA61 (5′- CAAGGAGAACTCCCTATAGCGATG-3′; 5′ at nt 3650) and SA64 (5′-CTGTTCTGAGGGAAACGCTGG-3′; 5′ located at nt 4201). An aliquot of each PCR was analyzed by agarose-gel electrophoresis to monitor yield and the integrity of the product. Typical yield was 0.4–0.5 μg/50 μl PCR. To selectively amplify NF1 mRNA containing exon 23A, primer RML33 (5′-TGTAGCTTTATTCAGTAGGGAGTGGCAAG-3′; 5′ located at nt 4142) was substituted for SA64, in the PCR (fig. 1). To determine the proportions of NF1 mRNA containing exon 23 A, 10 μCi α[32P]- dCTP (3,000 Ci/mmol) was included in the reaction, and the products were analyzed by 8% native PAGE and were quantitated by phosphorimaging (Molecular Dynamics). Genomic DNA corresponding to exon 22 was amplified by primers 23-1-5 (5′-AAAAACACGGTTCTATGTGAAAAG-3′, located in intron 22) and 23-1-3 (5′-TTTGTATCATTCATTTTGTGTGTA-3′, located in intron 23-1), located in the adjacent introns (fig. 1). Primer-extension analysis was employed to determine the extent of editing of NF1 RNA at nt 3916, by primer SK1 (5′- CATGTTGCCAATCAGAGGATGTG-3′, located at nt 3949) (Skuse et al. 1996), which is complementary to a 23-nt region 12 nt downstream of the edited base. The primer SK1 is that used by Skuse et al. (1996). [32P]-labeled SK1 primer was annealed to 10 ng NF1 cDNA flanking the edited base. Primer extension was conducted in the presence of 0.05 μM each of dATP, dCTP, and TTP and 1.6 μM ddGTP, in the presence of T7 DNA polymerase. In this assay, the radiolabeled primer extends to the first upstream C (at nt 3916) and undergoes chain termination, giving an extension product of 34 nt. If the C at nt 3916 is edited to T, the products extend to the next upstream C, at nt 3908, giving an extension product of 42 nt. The primer-extension products were resolved by PAGE with 10% acrylamide containing 8 M urea, followed by phosphorimaging. We have validated and characterized this assay extensively in the context of editing of apoB RNA (Giannoni et al. 1994; Anant et al. 1998; Madsen et al. 1999). Standardization of the assay was undertaken by use of mixtures of NF1 cDNA containing known quantities of C3916 and T3916 products from sequence-validated templates constructed from previously amplified NF1 cDNA (550 bp), in which a T at nt 3916 was introduced by mutagenesis. Known quantities of PCR product were used to establish the optimal conditions for primer extension. This established that the reliable limits of detection with 10 ng template correspond to 1% U (data not shown). Levels of apparent editing <1% were considered background “primer read through.” All assays included a C standard and a T standard, for reference.
Figure 1.
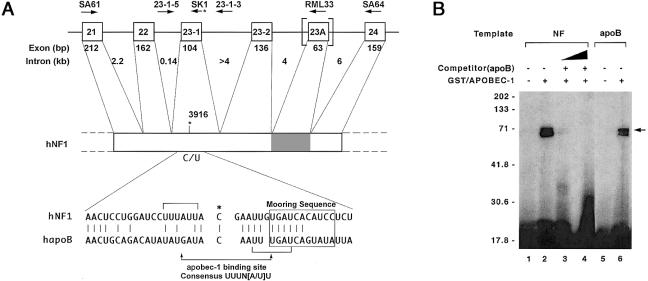
A, Organization of NF1. Top, Exons 21–24 of NF1, along with exon and intron sizes. The alternatively spliced exon 23A is within the vertical brackets, and the corresponding region in NF1 mRNA is shaded. Identities, orientations, and locations of the various primers are indicated above the exon and intron designations. Bottom, Alignment of 41-nt region (nt 3896–3936) of NF1 mRNA surrounding the edited base, compared to human apoB mRNA (hapoB). The edited C in both sequences is indicated by an asterisk (*), and sequence identity is indicated by vertical lines. The apobec-1 binding site and mooring sequence are indicated by horizontal brackets and a box, respectively. B, apobec-1, a NF1 RNA–binding protein. UV cross-linking was performed by incubating 250 ng GST/APOBEC-1 with either a 550-nt [32P]-labeled NF1 RNA (NF [lanes 1–4]) containing exon 23A (nt 3650–4201) or a 105-nt [32P]-labeled apoB RNA (apoB [lanes 5 and 6]), flanking the edited base. After treatment with RNaseT1 and UV irradiation, the cross-linked products were analyzed by 10% SDS-PAGE. For competition analysis, 50- (lane 3) and 100-fold (lane 4) excess of cold apoB RNA was added to the reaction. The results shown are representative of three such experiments. Molecular-weight markers are indicated to the left of the gel, and the location of the cross-linked GST/APOBEC-1 band is indicated by the arrow to the right of the gel.
In Vitro RNA-Editing Assay
Synthetic templates were prepared to yield transcripts that either included (550 nt) or did not include (487 nt) exon 23A and that contained a C at nt 3916. A 20-fmol sample of each RNA was individually incubated in a buffer containing 20 mM HEPES. HCl pH 7.8, and 100 mM KCl, at 30°C for 4 h, in the presence of 250 ng glutathione-S-transferase (GST)/apobec-1 and 50 μg of a 30% ammonium sulfate precipitate of bovine liver S-100 extracts (MacGinnitie et al. 1995). The RNA was extracted, subjected to primer extension with [32P]-labeled SK1 primer (see above), and subsequently analyzed, under denaturing conditions, by PAGE with 8% polyacrylamide gel containing 8 M urea. Editing of RNA was quantitated by phosphorimager analysis (Molecular Dynamics).
Protein-Truncation Assay
NF1 cDNA encoding exons 21–24 was amplified by RT-PCR, as described above, with primers NF-PPT-5′ (5′-GGGTGTAATACGACTCACTATAGGCCACCATGGAGCAGAAACTCATCTCTGAACAAGGAGAACTCCCTATAGCGATGGCTC-3′; T7 promoter is underlined, and myc-tag is boldface italic) and SA64. NF-PTT-5′ was designed to contain an upstream T7 promoter and a myc epitope tag, in frame with the NF1 cDNA. These primers were designed so that the region amplified contained the same numbers of methionine residues in the translation products from transcripts either with or without exon 23A. The full-length product (∼22 kD) contains six methionine residues, whereas the truncated protein (∼11 kD) contains five. The RT-PCR products were subjected to coupled transcription-translation (Promega) with rabbit reticulocyte lysates in the presence of translation-grade [35S]-methionine (Amersham-Pharmacia), according to the manufacturer’s recommendations. The translation reaction was subsequently immunoprecipitated with anti-myc monoclonal antibody 9E-10 (Santa Cruz Biotechnology) and was analyzed by gradient 10%–20% SDS-PAGE and autoradiography.
Detection of apobec-1 and apobec-1 Complementation Factor (ACF) mRNA
RT-PCR was conducted by use of primers, listed below, for apobec-1 and ACF. First-strand cDNA synthesis was undertaken as described above, using random-hexamer priming; cDNA amplification used gene-specific primers located in the 5′ and 3′ regions of the coding sequence. PCR cycling conditions for apobec-1 and ACF were 95°C for 3 min, 1 cycle; 95°C for 30 s, 55°C for 1 min, and 72°C for 1 min, 30 cycles; and 72°C for 10 min. As a control, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA was simultaneously amplified by the indicated primers. The amplified products were sequenced on both strands to confirm their identity. Primers used are as follows: for GAPDH, primers 5′-TCGGAGTCAACGGATTTGGTCG-3′ and GAP 5′-AGGCAGGGATGATGTTCTGGAGAG-3′; for apobec-1, primers SA146 5′-GACGACGACAAGGGATCCATGAGTTCCGAGACAGGC-3′ and SA147 5′-GGAACAAGACCCGTCGACTCATTTCAACCCTGTGGCCCACAG-3′; and, for ACF, primers ACF2 5′-CTCGAGTCAGAAGGTGCCATATCCATC-3′ and ACF6 5′-CAGATATTAGAAGAGATTTGTC-3′. The primer pairs for apobec-1 generate a full-length cDNA, whereas the primers for ACF generate an amplicon of 447 bp. Where indicated in the text, nested PCR was undertaken for apobec-1, by use of the primers listed below, which yield a product of 273 bp (nt 116–388). Primers used for nested PCR were as follows: JM-8 5′-ACCCCAGAGAACTTCGTAAAGAGGCC-3′ and JM-9 5′-CCGAGCTACGTAGATCACTAGAGTCA-3′.
UV Cross-Linking of NF1 and apoB RNAs to apobec-1
NF1 cDNA flanking the edited base (fig. 1) was amplified by primers SA61 and SA64, subcloned into plasmid pGEM-3zf(+) (Promega) and was sequenced to confirm the presence of wild-type sequence. The plasmid was linearized with restriction endonuclease KpnI and was subjected to in vitro transcription with SP6 RNA polymerase, in the presence of α[32P]-CTP (3,000 Ci/mmol). The [32P]-labeled NF1 RNA (50,000 cpm, at 4 × 108 cpm/μg) was incubated with 25 ng recombinant GST/APOBEC-1 (MacGinnitie et al. 1995) for 15 min at room temperature, in a buffer containing 10 mM HEPES pH 8.0, 100 mM KCl, 1 mM EDTA, 0.25 mM DTT, and 2.5% glycerol. The reaction was then sequentially treated with RNase T1 (final concentration, 1 unit/μl) and heparin (final concentration, 5 mg/ml), followed by UV irradiation (250 mJ/cm2) in a Stratalinker (Stratagene). As control, RNA binding was performed with a 105-nt rat apoB RNA that previously had been demonstrated to bind apobec-1 (Anant et al. 1995; Nakamuta et al. 1995; Anant and Davidson 2000). Competition analysis was performed with radiolabeled NF1 RNA and 5- and 10-fold excess of cold apoB RNA, as described in detail elsewhere (Anant et al. 1995).
Adenovirus-Mediated Infection of HepG2 Cells
Recombinant adenovirus expressing either rat apobec-1 or a control, lacZ cDNA, were cloned as described elsewhere (Kozarsky et al. 1996) and were titered to a multiplicity of infection (MOI) of 109–1010 pfu/ml. HepG2 cells were infected with 3 × 103–MOI recombinant adenovirus and cell lysates, and total RNA was prepared for analysis after 48 h. Primer-extension analysis was performed to quantitate C→U editing of endogenous NF1 and apoB RNA, as outlined above and as described in detail elsewhere (Giannoni et al. 1994). Cell lysates were analyzed by denaturing SDS-PAGE and western blotting, to demonstrate, as described elsewhere (Anant and Davidson 2000), the presence of apobec-1, by rabbit anti–apobec-1 IgG.
Results
NF1 RNA Sequence Alignment Flanking the Edited Base at nt 3916
The cis-acting elements regulating C→U editing of apoB RNA have been extensively characterized and include an AU-rich bulk-RNA context, a mooring sequence 3′ of the edited base, and efficiency elements both upstream and downstream (Shah et al. 1991; Backus and Smith 1992; Driscoll et al. 1993; Backus et al. 1994; Anant et al. 1995; Hersberger and Innerarity 1998; Anant and Davidson 2000). Alignment of an ∼40-nt stretch of NF1 RNA sequence flanking nt 3916 reveals ∼50% identity with apoB RNA, as has been noted elsewhere (Skuse et al. 1996), including 6 of 11 matches in the region corresponding to the mooring sequence (fig. 1). More recently, a consensus apobec-1 binding motif, UUUN[A/U]U, has been identified in both apoB RNA and other validated targets of high-affinity binding by apobec-1 (Anant and Davidson 2000). Further inspection of the NF1 RNA sequence flanking the edited base reveals this consensus apobec-1 binding site immediately upstream of the targeted cytidine (fig. 1). Therefore, we examined directly the ability of recombinant apobec-1 to bind synthetic NF1 RNA template, using UV cross-linking. The data demonstrate that apobec-1 binds NF1 RNA (fig. 1B) and that this binding is competed by apoB RNA (fig. 1B, lanes 3 and 4).
C→U Editing of RNA of NF1 Samples
RNA from 34 PNSTs was subjected to primer-extension assay, to detect C→U editing of NF1 RNA, and the reaction products were analyzed by urea PAGE and phosphorimaging. A representative series of samples is shown in figure 2A. Several of the samples (24, 25, and 28; see fig. 2A) demonstrated 12%–16% C→U editing of RNA, similar to the values and range noted by Skuse and colleagues (Skuse et al. 1996; Cappione et al. 1997). However, having established, by using synthetic RNA templates (data not shown), the limits of detection, we were able to exclude bona fide editing in samples in which there was less than ∼1% U (samples 20–22; see fig. 2A). This technical point is also emphasized in the results of primer-extension analysis of genomic DNA from the most extensively edited tumors, in which there is no detectable T in either of the sample lanes or in the control C template (fig. 2B). Direct sequencing of the cDNA products confirmed the presence of a T in the edited cDNA, corresponding to nt 3916 (fig. 2C). The cumulative results for the 34 samples assayed are listed in table 1. The findings demonstrate that a subset (8/34) of tumors manifest C→U editing of RNA (defined as >3% U) and that the remainder (26/34) demonstrate either very low levels (1%–2.5% U [in the case of 16/34]) or undetectable (<1% U [in the case of 10/34]) editing activity. These findings suggest that C→U editing of NF1 RNA is not a universal finding in tumors but, rather, appears to be confined to a subset of samples.
Figure 2.

NF1 mRNA undergoing C→U editing in a subset of neurofibromatosis-related PNSTs. A, Total RNA extracted from neurofibromatosis tumor tissue—and extent of editing of NF1 mRNA, as determined by primer-extension assay as described in the “Material and Methods” section. The primer-extension products were separated on a gel of 10% polyacrylamide containing urea and were autoradiographed. A representation of experiments performed in triplicate for 34 neurofibromatosis tumor samples is shown. The locations of the primer (P) and of unedited (C) and edited (T) primer-extension products are indicated to the right of the gel. B, C→U editing, which is shown not to be a somatic mutation. Genomic DNA flanking exon 23-1 was amplified by PCR using intron-specific primers and subsequently was subjected to primer-extension analysis. As controls, PCR products containing either C or T at nt 3916 of NF1 cDNA were also analyzed. The locations of the primer (P) and of unedited (C) and edited (T) primer-extension products are indicated to the right of the gel. C, Editing at nt 3916 of NF1 RNA, confirmed by sequencing of individual cDNA clones. The locations of the unedited (Unedited C; top) and edited (Edited T; bottom) nucleotide in a representative clone are indicated by arrows above the gels.
Table 1.
C→U Editing of NF1 RNA in PNSTs
|
Editing(%) |
Expression |
|||||
| Category and Sample | Histology | Mean | SD | Range (n=3) | apobec-1 | ACF |
| No editing (<1% U): | ||||||
| NF20 | Plexiform | .64 | .34 | .3–1.0 | − | + |
| NF22 | Triton tumor | .65 | .35 | .3–.9 | − | + |
| NF6 | Neurofibroma | .66 | .43 | .2–1.1 | − | + |
| NF35 | Glioblastoma | .68 | .22 | .45–.9 | − | + |
| NF21 | Malignant schwannoma | .69 | .65 | .2–1.3 | − | + |
| NF3 | Neurofibroma | .72 | .65 | .1–1.4 | − | + |
| NF4 | Neurofibroma | .72 | .68 | .1–1.5 | − | + |
| NF2 | Neurofibroma | .86 | .65 | .75–1.1 | − | + |
| NF18 | Malignant schwannoma | .95 | .65 | .9–1.3 | − | + |
| NF5 | Neurofibroma | .99 | .56 | .95–1.7 | − | + |
| NF7 | Malignant schwannoma | 1.01 | .46 | .9–1.5 | − | + |
| NF17 | Neurofibroma | 1.08 | .24 | .85–1.5 | − | + |
| NF13 | Neurofibroma | 1.14 | .65 | .75–1.8 | − | + |
| NF8 | Malignant schwannoma | 1.15 | .75 | .65–1.9 | − | + |
| NF16 | Neurofibroma | 1.15 | .49 | .55–1.7 | − | + |
| NF9 | Neurofibroma | 1.24 | .43 | .8–1.7 | − | + |
| NF27 | Malignant schwannoma | 1.35 | .36 | .9–1.7 | − | + |
| NF23 | Neurofibroma | 1.37 | .54 | .75–1.9 | − | + |
| Low editing (<2.5% U): | ||||||
| NF14 | White matter | 1.48 | .39 | 1.0–1.9 | − | + |
| NF34 | Glioblastoma | 1.52 | .43 | 1.1–1.95 | − | + |
| NF11 | Neurofibroma | 1.57 | .54 | 1.0–2.1 | − | + |
| NF1 | Neurofibroma | 1.58 | .58 | 1.0–2.2 | − | + |
| NF30 | Ganglioneuroma | 1.83 | .16 | 1.65–2.1 | − | + |
| NF15 | Peripheral nerve | 1.85 | .86 | 1.1–2.5 | − | + |
| NF31 | Schwannoma | 1.99 | .38 | 1.5–2.5 | − | + |
| NF19 | Neurofibroma | 2.36 | 1.03 | 1.33–3.4 | − | + |
| NF10 | Neurofibroma | 3.16 | .98 | 2.2–4.1 | + | + |
| NF33 | Glioblastoma | 3.18 | .68 | 2.6–3.9 | + | + |
| NF26 | Nonmalignant schwannoma | 3.48 | .62 | 2.8–4.2 | + | + |
| NF32 | Neurofibroma | 5.06 | .89 | 4.1–5.9 | + | + |
| Bona-fide editing (>3% U): | ||||||
| NF29 | Granulomatous cell | 5.52 | 1.08 | 4.3–6.7 | + | + |
| NF25 | Malignant schwannoma | 10.35 | 2.53 | 8.1–12.5 | + | + |
| NF24 | Malignant schwannoma | 12.02 | 3.12 | 8.9–15.3 | + | + |
| NF28 | Ganglioneuroblastoma | 12.65 | 1.65 | 10.5–14.8 | + | + |
C→U Editing of NF1 RNA: Detection by Protein-Truncation Assay
The products of the initial round of RT-PCR were amplified in-frame with a myc-tag and T7 promoter and were used to program a coupled in vitro transcription/translation reaction mixture. Both full-length and truncated products were recovered (fig. 3). The molar incorporation of [35S]-methionine into immunoprecipitable protein products revealed the observed proportion of full-length to truncated protein products to be ∼10:1, close to the value expected on the basis of C→U editing (i.e., ∼10%–15%) of NF1 RNA in the respective sample of origin.
Figure 3.

Protein-truncation assay. NF1 cDNA flanking the edited base was amplified by primers NF-PTT-5′ and SA64, as described in the “Material and Methods” section. The RT-PCR products were subjected to in vitro coupled transcription/translation in the presence of [35S] methionine. The translation products were immunoprecipitated with α-myc-tag antibody and were resolved by 10%–20% SDS-PAGE. The locations of the full-length (FL) and truncated (Tr) translation products, which correspond to the unedited and edited NF1 mRNA, respectively, are indicated to the left of the gel. Control reactions were undertaken by use of a synthetic, sequence-validated template containing either C or T at nt 3916. The results shown are representative of three such experiments.
Tumors Demonstrating C→U Editing: Expression of apobec-1 mRNA
The expression of gene products associated with the machinery for editing of apoB RNA was investigated as a potential variable contributing to the heterogeneity observed in C→U editing of NF1 RNA. Two gene products in particular were examined. These correspond to apobec-1, the catalytic deaminase responsible for C→U editing of apoB, and ACF, the competence factor that supports site-specific deamination of apoB RNA by apobec-1 (Teng et al. 1993; Lellek et al. 2000; Mehta et al. 2000). apobec-1 mRNA was detectable in all eight tumors that manifest >3% C→U editing (fig. 4 and table 1). In all cases, the amplification products were sequenced, to confirm the identity of apobec-1 (data not shown). Tumor samples that demonstrated either no C→U editing (samples 21 and 22; see fig. 2A) or very low (i.e., ∼1%–2%) levels did not yield a detectable product for apobec-1 mRNA (fig. 4 and table 1), even when nested PCR was used. Moreover, normal tissue from sites in which apobec-1 is not detectable (e.g., liver, kidney, and brain) demonstrated <1% C→U editing of NF1 RNA (fig. 5). The observation that remains unexplained is the apparent lack of apobec-1 expression in tumors demonstrating very low (i.e., ∼1%–2%) levels C→U editing (fig. 4 and table 1). We speculate that this finding may be due to the mixed cellular composition typically found in the PNSTs, in which editing of NF1 RNA occurs in a small subset of cells and in which apobec-1 mRNA is below detection limits. ACF mRNA, by contrast, was detected in all samples (fig. 4 and table 1), a finding consistent with previous reports that its expression is ubiquitous in humans (Lellek et al. 2000; Mehta et al. 2000). As an additional control, all RNA samples supported amplification of GAPDH mRNA (fig. 4).
Figure 4.
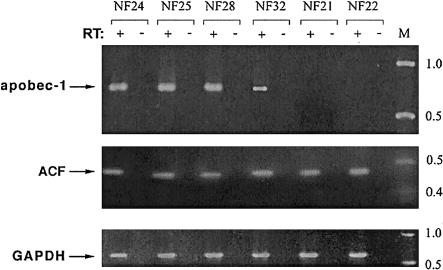
apobec-1 mRNA, expressed in a subset of neurofibromatosis tumors. RT-PCR by gene-specific primers (“+” lanes) was performed to amplify apobec-1, ACF, and GAPDH (top, middle, and bottom, respectively). As a control, PCR without prior RT (“−” lanes) was performed. The locations of molecular-weight standards are indicated to the right of the gel.
Figure 5.
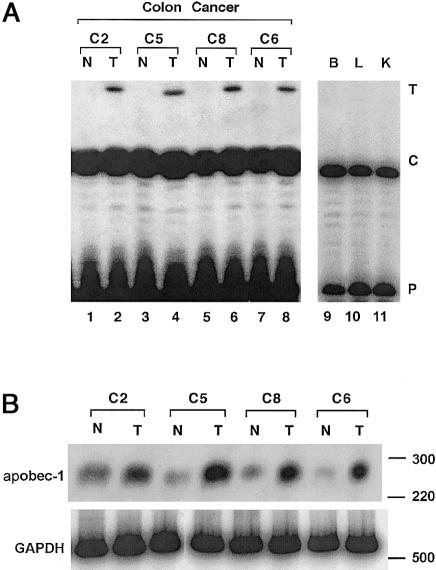
Editing of NF1 mRNA in colon cancers. A, Endogenous editing of NF1 RNA, as determined by primer-extension analysis and described in figure 2A. The data demonstrate increased editing in colon tumor tissue (lanes T), compared to matched normal control tissue (lanes N). In addition, no editing (<1% U) was observed in NF1 mRNA from normal brain (lane B), liver (lane L), or kidney (lane K). B, apobec-1 mRNA expression induced in colon cancers. RT-PCR was performed in the presence of α[32P]-dCTP, by use of primers specific for apobec-1 and GAPDH, to amplify a 274- and a 510-bp fragment, respectively. Increased expression of apobec-1 is observed in colon tumor tissue (lanes T), compared to paired normal tissue (lanes N). The locations of the molecular-weight (in bp) markers are indicated to the right of the gel.
Non-Neuronal Tumors Expressing apobec-1 and Demonstrating Editing of NF1 RNA
In humans, apobec-1 is normally confined to the luminal gastrointestinal tract (Teng et al. 1993; Yamanaka et al. 1994; Funahashi et al. 1995). Human colorectal cancers, however, have been demonstrated to express increased levels of apobec-1 mRNA, compared to normal adjacent mucosa (Lee et al. 1998). NF1 RNA was amplified from these tumors as well as from the adjacent normal tissue and was subjected to primer-extension assay. The results demonstrate higher levels of C→U editing of RNA in the tumor samples (4%–8% U), compared to paired normal tissue, and confirm the increase in apobec-1 mRNA abundance (1%–3% U) (figs. 5A and B). These findings suggest that C→U editing of NF1 RNA is not confined to tumors arising from Schwann-like cells but may be a feature of tumors that support apobec-1 expression; however, a more extensive survey of tumors of the gastrointestinal tract will be required, to allow us to formally examine the implications of editing of NF1 RNA, in relation to apobec-1–gene expression.
Editing of NF1 RNA: Occurrence in Transcripts Containing Exon 23A
To further pursue the mechanisms underlying the heterogeneity of C→U editing of NF1, transcripts were selectively amplified (by a strategy reviewed both in the “Material and Methods” section and in fig. 1), to examine alternative splicing of exon 23A, downstream of the edited base. As illustrated in figure 6A, these samples reveal a range of proportions of this alternatively spliced exon. These amplicons were individually isolated and were subjected to primer-extension analysis, and the extent of C→U editing was determined. As indicated in figure 6B, C→U editing was found only in transcripts containing exon 23A. This approach was applied to the range of tumors studied, and the results demonstrated a strong positive correlation between the relative proportions of exon 23A–containing transcripts and the extent of C→U editing of RNA (fig. 6C). These findings strongly suggest a functional relationship between the presence of exon 23A and the editing of NF1 RNA.
Figure 6.
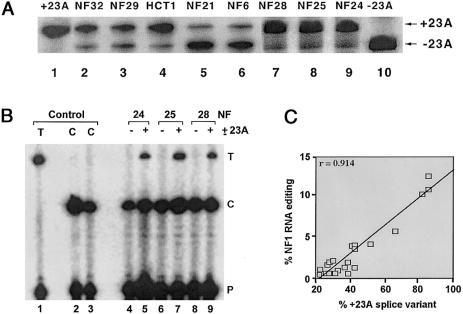
Preferential editing of NF1 mRNA containing exon 23A. A, Levels of alternative splicing of exon 23A in various tumor samples, as determined by RT-PCR and described in the “Material and Methods” section. As controls, PCRs were performed with synthetic RNA templates either containing (+23A) or lacking (−23A) exon 23A. The results shown are representative of experiments performed in triplicate, with all the tumor and normal samples. B, RT-PCR products of NF1 RNA containing (+23A) and lacking (−23A) the alternatively spliced exon, which were purified by electrophoresis and which subsequently were subjected to primer-extension analysis. As a control, primer-extension analysis was performed with cDNA templates containing only either a C or a T at nt 3916. The results shown are representative of three such experiments. C, Correlation between editing of NF1 RNA and inclusion of exon 23A (data are percent of total amount). This correlation was significant (P<.001).
apobec-1 as the Mediator of C→U Editing of NF1 RNA In Vivo and In Vitro
We employed two complementary approaches to confirm the suspected role of apobec-1 in C→U editing of NF1. In the first approach, we incubated synthetic NF1 transcripts that contained the unedited cytidine at nt 3916 and that either included (+23A) or lacked (−23A) the alternatively spliced downstream exon, with apobec-1 and S100 extracts. The results of these assays (fig. 7A) reveal that C→U editing of NF1 RNA can be reproduced in vitro and that transcripts containing exon 23A are more extensively edited (12% ± 1.7% U; n=3), compared to transcripts lacking this alternatively spliced exon (3.9% ± 0.8% U; n=3; P<.001). In the second approach, we used adenovirus-mediated transduction of apobec-1 to demonstrate a gain of function in cell culture. For this purpose, we turned to HepG2 cells, a human liver–derived cell line that expresses apoB RNA and NF1 mRNA but that does not express endogenous apobec-1 (Giannoni et al. 1994). HepG2 cells were infected with either adenovirus apobec-1 or lacZ, and cell lysates were prepared for protein and RNA extraction 48 h later. The results confirm the efficient induction of editing of apoB RNA after introduction of apobec-1 (see the western blot in fig. 7B) and reveal C→U editing at the canonical site (nt 6666), as well as hyperediting of upstream cytidines (C6661), as noted elsewhere (Sowden et al. 1996; Yamanaka et al. 1996) (fig. 7C, left); RNA from these same cells demonstrated C→U editing of NF1 RNA (fig. 7C, middle), with no such change, in either transcript, after lacZ transfection. Further analysis of these NF1 RNA species reveals that C→U editing was detectable only in transcripts containing exon 23A (fig. 7C, right), confirming and extending findings from the tumors analyzed above.
Figure 7.

apobec-1 mediation of C→U editing of NF1 RNA. A, Assays of in vitro editing of RNA, performed with 20 fmol synthetic NF1 RNA either containing (+23A) or lacking (−23A) exon 23 A, in the presence of 250 ng recombinant GST/APOBEC-1 and 50 μg bovine liver extract (Liv S100). After in vitro incubation, the RNA was extracted, and cDNA was prepared for analysis by primer extension (see the “Material and Methods” section). The mobilities of the primer (P) and of the unedited (C) and edited (T) cDNA are indicated to the right of the gel. B, Adenovirus-mediated expression of apobec-1. HepG2 cells, from a human hepatoma cell line, were infected with recombinant adenovirus encoding either apobec-1 (Ad/apobec-1) or the bacterial β-galactosidase (Ad/lacZ). Forty-eight hours after infection, cell lysates were prepared and were subjected to western blot analysis with rabbit α-apobec-1 IgG. Migration of the 27-kD apobec-1 immunoreactive band is indicated by an arrow to the right of the gel, and molecular-weight standards are indicated to the left of the gel. C, Total RNA extracted from HepG2 cells infected with either Ad/apobec-1 or Ad/lacZ and subjected to RT-PCR for amplification of apoB (left panel) and NF1 (middle panel) mRNAs flanking the edited base. Primer-extension analysis was performed and was analyzed by PAGE with 8% acrylamide containing urea. To differentiate the editing of RNA in the two alternatively spliced NF1 transcripts (lanes ±23A), the PCR products were fractionated through agarose gels, and the two products were isolated and were individually subjected to primer-extension analysis (right panel). The locations of the primer and of unedited (C) and edited (U) primer-extension products, for both apoB and NF1, are indicated to the left or right of the gels. This is a representative of three independent assays.
Discussion
Several mechanisms exist for amplification of the repertoire of genes encoded in chromosomal DNA. Posttranscriptional regulation encompasses one such level of control and includes editing of RNA, which has recently emerged as an important restriction point in the species- and tissue-specific modulation of gene expression (Maas and Rich 2000). The central conclusion of the present study demonstrates that C→U editing of NF1 RNA occurs in a subset of tumor samples that have at least two distinguishing characteristics that distinguish them from those tumors in which editing of RNA does not occur; these characteristics are (1) the presence of apobec-1 mRNA and (2) the preferential inclusion of a downstream exon, 23A, in the edited transcript. The findings (a) add support to the hypothesis that, in the regulation of mRNA metabolism, apobec-1 may play a role beyond that of its originally identified target, apoB, and (b) provide evidence that alternative splicing of NF1 RNA may be an important component of C→U editing of this transcript. Each of these observations merits additional discussion.
apobec-1 is an RNA-specific cytidine deaminase whose expression, in humans, is confined to the luminal gastrointestinal tract (Hadjiagapiou et al. 1994; Lau et al. 1994). apobec-1 functions as a dimeric subunit component of a multicomponent holoenzyme and mediates site-specific deamination of a single cytidine in the nuclear apoB transcript. This C→U RNA-editing reaction results in the production of a truncated protein, apoB48, required for intestinal lipid transport (Chen et al. 1987; Powell et al. 1987; Teng et al. 1993). Accordingly, the finding that apobec-1 mRNA is expressed in neuronal tumors from patients with NF1 represents the first demonstration, in humans, of apobec-1 expression in cells other than epithelial cells lining the gastrointestinal tract (Hadjiagapiou et al. 1994; Lau et al. 1994). apobec-1 mRNA expression is widespread in the mouse and rat, and C→U RNA editing activity can be demonstrated in tissue extracts from numerous organs (Greeve et al. 1993; Nakamuta et al. 1995; Hirano et al. 1997; Qian et al. 1997). Nevertheless, the mechanism by which apobec-1 expression is confined to the gastrointestinal tract in humans has not been clarified. Analyses undertaken by several groups have revealed few informative features of the proximal promoter region of the human APOBEC-1 gene, and no additional insight has been gained through comparative analyses of the murine, rat, and human loci (Greeve et al. 1993; Nakamuta et al. 1995; Hirano et al. 1997; Qian et al. 1997). apobec-1 mRNA expression has been examined in a number of human carcinomas (Greeve et al. 1999), in light of the phenotype associated with forced overexpression of apobec-1 in transgenic animals, in which promiscuous C→U RNA editing of other target transcripts has been associated with hepatocellular carcinoma (Yamanaka et al. 1996, 1997). These studies, in human cancer tissues, concluded that apobec-1 mRNA is detectable only in cancers arising in the luminal gastrointestinal tract (Greeve et al. 1999), a finding that previously had been noted in normal tissue (Greeve et al. 1993; Hadjiagapiou et al. 1994; Lau et al. 1994). Accordingly, overexpression of apobec-1 does not appear to be a general feature of malignancy; this said, the factors that permit expression of apobec-1 mRNA in a subset of PNSTs from patients with NF1 will require additional investigation.
In considering the possibility that apobec-1 was involved in C→U editing of NF1, we were intrigued by the earlier observations, by Skuse and colleagues, that the NF1 RNA region flanking the edited base contains only ∼50% identity to the canonical apoB RNA sequence, particularly within an 11-nt motif referred to as the “mooring sequence” (Backus and Smith 1992; Skuse et al. 1996). We suspected, as did Skuse and colleagues, that this degree of mismatch would severely impair the efficiency of C→U RNA editing (Skuse et al. 1996). This suspicion is consistent with the finding that C→U editing of NF1 RNA produces ⩽20% UGA (edited RNA)—in contrast to the situation with intestinal apoB mRNA, in which, typically, >90% exists in the UAA (edited) form (Chen et al. 1987; Powell et al. 1987). The mechanisms accounting for these quantitative differences remain to be elucidated but include, in addition to structural features of the transcript that are associated with the optimal nucleotide sequence, distinctive requirements for trans-acting factors.
The functional significance, if any, of C→U editing of NF1 RNA remains unresolved; in particular, it is widely recognized that second-hit somatic mutations occur in the wild-type NF1 allele in many PNSTs in patients with neurofibromatosis (reviewed in Cichowski and Jacks 2001). Furthermore, it should be emphasized that, with the exception of C→T changes at nt 3916, we did not systematically exclude somatic mutations in the PNSTs investigated in this study; nevertheless, at least from a theoretical standpoint, the finding that a subset of tumors from patients with NF1 demonstrate C→U RNA editing supports the hypothesis that modifier genes (in this case, apobec-1) may play a role in the heterogeneity of molecular defects. The corollary hypothesis is that C→U editing of NF1 RNA creates a translational stop codon, potentially leading to premature truncation of neurofibromin. We attempted to demonstrate the presence of a truncated protein corresponding to the edited mRNA in cell lysates from clonal intestinal-cancer cell lines supporting >15% C→U editing, but we were successful only in identifying the full-length form of the protein (data not shown). Whether this reflects technical limitations of the reagents or other explanations is currently unknown. Thus, the question of whether the edited NF1 RNA encodes a truncated protein in vivo is still unresolved.
Studies of NF1 mRNA in affected patients have revealed an ∼50% incidence of splicing abnormalities (Park and Pivnick 1998). The majority of these splicing defects are predicted to result in protein truncations, and the current findings are certainly consistent with this general expectation; however, the results from this recent survey of splicing abnormalities in NF1 failed to reveal any specific feature associated with exon 23A, nor was there any particular clustering of alternative splice defects, which would favor inclusion of this exon (Costa et al. 2001). Exon 23A itself plays an important role in the function of neurofibromin, as inferred from targeted deletion of this region in mice, which results in a learning deficit (Costa et al. 2001). These findings suggest that there may be important parallels between the murine and human genes for NF1, parallels that could be investigated with surrogate models. Indeed, an important series of questions emerging from current studies concerns the possibility that C→U editing of NF1 RNA may be experimentally approached by murine models. This is an attractive possibility, since our lab and others have generated mutant strains in which apobec-1 has been deleted through homologous recombination. Additionally, the auxiliary subunit of the enzyme that edits apoB RNA recently has been cloned (Lellek et al. 2000; Mehta et al. 2000), and its role in alternative splicing and C→U editing of NF1 RNA will be of interest. In this regard, studies have demonstrated that an apobec-1–related RNA-specific deaminase, ADAR2, which mediates A→I editing of double-stranded RNA, also plays a role in alternative splicing (Rueter et al. 1999); this function will need to be examined in the context of apobec-1 and NF1 RNA. These and other issues related to the molecular mechanisms of posttranscriptional regulation will be the focus of future reports.
Acknowledgments
This work was supported by National Institutes of Health (NIH) grants HL-38180 and DK-56260 and NIH Digestive Disease Research Core Center grant DK-52574 (all to N.O.D.). The authors acknowledge the generous assistance of Drs. David N. Louis (Massachusetts General Hospital, Boston) and Priscilla Short (University of Chicago Hospitals) for their provision of samples for these analyses. In addition, the authors acknowledge their colleagues, Valerie Blanc, Libby Newberry, and Jeffrey Henderson, for their valuable insights and discussion.
Electronic-Database Information
Accession numbers and the URL for data in this article are as follows:
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for NF1 [MIM 162200], APOB [MIM 107730], and APOBEC-1 [MIM 600130])
References
- Anant S, Davidson NO (2000) An AU-rich sequence element (UUUN[A/U]U) downstream of the edited C in apolipoprotein B mRNA is a high-affinity binding site for Apobec-1: binding of Apobec-1 to this motif in the 3′ untranslated region of c-myc increases mRNA stability. Mol Cell Biol 20:1982–1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anant S, MacGinnitie AJ, Davidson NO (1995) apobec-1, the catalytic subunit of the mammalian apolipoprotein B mRNA editing enzyme, is a novel RNA-binding protein. J Biol Chem 270:14762–14767 [PubMed] [Google Scholar]
- Anant S, Yu H, Davidson NO (1998) Evolutionary origins of the mammalian apolipoproteinB RNA editing enzyme, apobec-1: structural homology inferred from analysis of a cloned chicken small intestinal cytidine deaminase. Biol Chem 379:1075–1081 [DOI] [PubMed] [Google Scholar]
- Andersen LB, Fountain JW, Gutmann DH, Tarle SA, Glover TW, Dracopoli NC, Housman DE, Collins FS (1993) Mutations in the neurofibromatosis 1 gene in sporadic malignant melanoma cell lines. Nat Genet 3:118–121 [DOI] [PubMed] [Google Scholar]
- Ars E, Serra E, Garcia J, Kruyer H, Gaona A, Lazaro C, Estivill X (2000) Mutations affecting mRNA splicing are the most common molecular defects in patients with neurofibromatosis type 1. Hum Mol Genet 9:237–247 [DOI] [PubMed] [Google Scholar]
- Ashkenas J (1997) Gene regulation by mRNA editing. Am J Hum Genet 60:278–283 [PMC free article] [PubMed] [Google Scholar]
- Backus JW, Schock D, Smith HC (1994) Only cytidines 5′ of the apolipoprotein B mRNA mooring sequence are edited. Biochim Biophys Acta 1219:1–14 [DOI] [PubMed] [Google Scholar]
- Backus JW, Smith HC (1992) Three distinct RNA sequence elements are required for efficient apolipoprotein B (apoB) RNA editing in vitro. Nucleic Acids Res 20:6007–6014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu TN, Gutmann DH, Fletcher JA, Glover TW, Collins FS, Downward J (1992) Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature 356:713–715 [DOI] [PubMed] [Google Scholar]
- Bollag G, Clapp DW, Shih S, Adler F, Zhang YY, Thompson P, Lange BJ, Freedman MH, McCormick F, Jacks T, Shannon K (1996) Loss of NF1 results in activation of the Ras signaling pathway and leads to aberrant growth in haematopoietic cells. Nat Genet 12:144–148 [DOI] [PubMed] [Google Scholar]
- Cappione AJ, French BL, Skuse GR (1997) A potential role for NF1 mRNA editing in the pathogenesis of NF1 tumors. Am J Hum Genet 60:305–312 [PMC free article] [PubMed] [Google Scholar]
- Chen SH, Habib G, Yang CY, Gu ZW, Lee BR, Weng SA, Silberman SR, Cai SJ, Deslypere JP, Rosseneu M, Gotto AM, Li WH, Chan L (1987) Apolipoprotein B-48 is the product of a messenger RNA with an organ-specific in-frame stop codon. Science 238:363–366 [DOI] [PubMed] [Google Scholar]
- Cichowski K, Jacks T (2001) NF1 tumor suppressor gene function: narrowing the GAP. Cell 104:593–604 [DOI] [PubMed] [Google Scholar]
- Costa RM, Yang T, Huynh DP, Pulst SM, Viskochil DH, Silva AJ, Brannan CI (2001) Learning deficits, but normal development and tumor predisposition, in mice lacking exon 23a of Nf1. Nat Genet 27:399–405 [DOI] [PubMed] [Google Scholar]
- Davidson NO, Shelness GS (2000) Apolipoprotein B: mRNA editing, lipoprotein assembly, and presecretory degradation. Annu Rev Nutr 20:169–193 [DOI] [PubMed] [Google Scholar]
- DeClue JE, Papageorge AG, Fletcher JA, Diehl SR, Ratner N, Vass WC, Lowy DR (1992) Abnormal regulation of mammalian p21ras contributes to malignant tumor growth in von Recklinghausen (type 1) neurofibromatosis. Cell 69:265–273 [DOI] [PubMed] [Google Scholar]
- Driscoll DM, Lakhe-Reddy S, Oleksa LM, Martinez D (1993) Induction of RNA editing at heterologous sites by sequences in apolipoprotein B mRNA. Mol Cell Biol 13:7288–7294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funahashi T, Giannoni F, DePaoli AM, Skarosi SF, Davidson NO (1995) Tissue-specific, developmental and nutritional regulation of the gene encoding the catalytic subunit of the rat apolipoprotein B mRNA editing enzyme: functional role in the modulation of apoB mRNA editing. J Lipid Res 36:414–428 [PubMed] [Google Scholar]
- Giannoni F, Bonen DK, Funahashi T, Hadjiagapiou C, Burant CF, Davidson NO (1994) Complementation of apolipoprotein B mRNA editing by human liver accompanied by secretion of apolipoprotein B48. J Biol Chem 269:5932–5936 [PubMed] [Google Scholar]
- Greeve J, Altkemper I, Dieterich JH, Greten H, Windler E (1993) Apolipoprotein B mRNA editing in 12 different mammalian species: hepatic expression is reflected in low concentrations of apoB-containing plasma lipoproteins. J Lipid Res 34:1367–1383 [PubMed] [Google Scholar]
- Greeve J, Lellek H, Apostel F, Hundoegger K, Barialai A, Kirsten R, Welker S, Greten H (1999) Absence of APOBEC-1 mediated mRNA editing in human carcinomas. Oncogene 18:6357–6366 [DOI] [PubMed] [Google Scholar]
- Guha A, Lau N, Huvar I, Gutmann D, Provias J, Pawson T, Boss G (1996) Ras-GTP levels are elevated in human NF1 peripheral nerve tumors. Oncogene 12:507–513 [PubMed] [Google Scholar]
- Hadjiagapiou C, Giannoni F, Funahashi T, Skarosi SF, Davidson NO (1994) Molecular cloning of a human small intestinal apolipoprotein B mRNA editing protein. Nucleic Acids Res 22:1874–1879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hersberger M, Innerarity TL (1998) Two efficiency elements flanking the editing site of cytidine 6666 in the apolipoprotein B mRNA support mooring-dependent editing. J Biol Chem 273:9435–9442 [DOI] [PubMed] [Google Scholar]
- Hirano K, Min J, Funahashi T, Davidson NO (1997) Cloning and characterization of the rat apobec-1 gene: a comparative analysis of gene structure and promoter usage in rat and mouse. J Lipid Res 38:1103–1119 [PubMed] [Google Scholar]
- Hirano K, Young SG, Farese RV Jr, Ng J, Sande E, Warburton C, Powell-Braxton LM, Davidson NO (1996) Targeted disruption of the mouse apobec-1 gene abolishes apolipoprotein B mRNA editing and eliminates apolipoprotein B48. J Biol Chem 271:9887–9890 [DOI] [PubMed] [Google Scholar]
- Kozarsky KF, Bonen DK, Giannoni F, Funahashi T, Wilson JM, Davidson NO (1996) Hepatic expression of the catalytic subunit of the apolipoprotein B mRNA editing enzyme (apobec-1) ameliorates hypercholesterolemia in LDL receptor-deficient rabbits. Hum Gene Ther 7:943–957 [DOI] [PubMed] [Google Scholar]
- Lau N, Feldkamp MM, Roncari L, Loehr AH, Shannon P, Gutmann DH, Guha A (2000) Loss of neurofibromin is associated with activation of RAS/MAPK and PI3-K/AKT signaling in a neurofibromatosis 1 astrocytoma. J Neuropathol Exp Neurol 59:759–767 [DOI] [PubMed] [Google Scholar]
- Lau PP, Zhu HJ, Baldini A, Charnsangavej C, Chan L (1994) Dimeric structure of a human apolipoprotein B mRNA editing protein and cloning and chromosomal localization of its gene. Proc Natl Acad Sci USA 91:8522–8526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RM, Hirano K, Anant S, Baunoch D, Davidson NO (1998) An alternatively spliced form of apobec-1 messenger RNA is overexpressed in human colon cancer. Gastroenterology 115:1096–1103 [DOI] [PubMed] [Google Scholar]
- Lellek H, Kirsten R, Diehl I, Apostel F, Buck F, Greeve J (2000) Purification and molecular cloning of a novel essential component of the apolipoprotein B mRNA editing enzyme-complex. J Biol Chem 275:19848–19856 [DOI] [PubMed] [Google Scholar]
- Li Y, O'Connell P, Breidenbach HH, Cawthon R, Stevens J, Xu G, Neil S, Robertson M, White R, Viskochil D (1995) Genomic organization of the neurofibromatosis 1 gene (NF1). Genomics 25:9–18 [DOI] [PubMed] [Google Scholar]
- Maas S, Rich A (2000) Changing genetic information through RNA editing. Bioessays 22:790–802 [DOI] [PubMed] [Google Scholar]
- MacGinnitie AJ, Anant S, Davidson NO (1995) Mutagenesis of apobec-1, the catalytic subunit of the mammalian apolipoprotein B mRNA editing enzyme, reveals distinct domains that mediate cytosine nucleoside deaminase, RNA binding, and RNA editing activity. J Biol Chem 270:14768–14775 [PubMed] [Google Scholar]
- Madsen P, Anant S, Rasmussen HH, Gromov P, Vorum H, Dumanski JP, Tommerup N, Collins JE, Wright CL, Dunham I, MacGinnitie AJ, Davidson NO, Celis JE (1999) Psoriasis upregulated phorbolin-1 shares structural but not functional similarity to the mRNA-editing protein apobec-1. J Invest Dermatol 113:162–169 [DOI] [PubMed] [Google Scholar]
- Mehta A, Kinter MT, Sherman NE, Driscoll DM (2000) Molecular cloning of apobec-1 complementation factor, a novel RNA-binding protein involved in the editing of apolipoprotein B mRNA. Mol Cell Biol 20:1846–1854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JR, Paszty C, Stevens ME, Hughes SD, Forte T, Scott J, Rubin EM (1996) Apolipoprotein B RNA editing enzyme-deficient mice are viable despite alterations in lipoprotein metabolism. Proc Natl Acad Sci USA 93:7154–7159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakafuku M, Nagamine M, Ohtoshi A, Tanaka K, Toh-e A, Kaziro Y (1993) Suppression of oncogenic Ras by mutant neurofibromatosis type 1 genes with single amino acid substitutions. Proc Natl Acad Sci USA 90:6706–6710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamuta M, Oka K, Krushkal J, Kobayashi K, Yamamoto M, Li WH, Chan L (1995) Alternative mRNA splicing and differential promoter utilization determine tissue-specific expression of the apolipoprotein B mRNA-editing protein (Apobec1) gene in mice: structure and evolution of Apobec1 and related nucleoside/nucleotide deaminases. J Biol Chem 270:13042–13056 [DOI] [PubMed] [Google Scholar]
- National Institutes of Health Consensus Development Conference (1988) Neurofibromatosis: conference statement. Arch Neurol 45:575–578 [PubMed] [Google Scholar]
- Park VM, Pivnick EK (1998) Neurofibromatosis type 1 (NF1): a protein truncation assay yielding identification of mutations in 73% of patients. J Med Genet 35:813–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell LM, Wallis SC, Pease RJ, Edwards YH, Knott TJ, Scott J (1987) A novel form of tissue-specific RNA processing produces apolipoprotein-B48 in intestine. Cell 50:831–840 [DOI] [PubMed] [Google Scholar]
- Qian X, Balestra ME, Innerarity TL (1997) Two distinct TATA-less promoters direct tissue-specific expression of the rat apo-B editing catalytic polypeptide 1 gene. J Biol Chem 272:18060–18070 [DOI] [PubMed] [Google Scholar]
- Rueter SM, Dawson TR, Emeson RB (1999) Regulation of alternative splicing by RNA editing. Nature 399:75–80 [DOI] [PubMed] [Google Scholar]
- Shah RR, Knott TJ, Legros JE, Navaratnam N, Greeve JC, Scott J (1991) Sequence requirements for the editing of apolipoprotein B mRNA. J Biol Chem 266:16301–16304 [PubMed] [Google Scholar]
- Sherman LS, Atit R, Rosenbaum T, Cox AD, Ratner N (2000) Single cell Ras-GTP analysis reveals altered Ras activity in a subpopulation of neurofibroma Schwann cells but not fibroblasts. J Biol Chem 275:30740–30745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skuse GR, Cappione AJ (1997) RNA processing and clinical variability in neurofibromatosis type I (NF1). Hum Mol Genet 6:1707–1712 [DOI] [PubMed] [Google Scholar]
- Skuse GR, Cappione AJ, Sowden M, Metheny LJ, Smith HC (1996) The neurofibromatosis type I messenger RNA undergoes base-modification RNA editing. Nucleic Acids Res 24:478–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skuse GR, Ludlow JW (1995) Tumour suppressor genes in disease and therapy. Lancet 345:902–906 [DOI] [PubMed] [Google Scholar]
- Sowden M, Hamm JK, Smith HC (1996) Overexpression of APOBEC-1 results in mooring sequence-dependent promiscuous RNA editing. J Biol Chem 271:3011–3017 [DOI] [PubMed] [Google Scholar]
- Teng B, Burant CF, Davidson NO (1993) Molecular cloning of an apolipoprotein B messenger RNA editing protein. Science 260:1816–1819 [DOI] [PubMed] [Google Scholar]
- Yamanaka S, Poksay KS, Arnold KS, Innerarity TL (1997) A novel translational repressor mRNA is edited extensively in livers containing tumors caused by the transgene expression of the apoB mRNA-editing enzyme. Genes Dev 11:321–333 [DOI] [PubMed] [Google Scholar]
- Yamanaka S, Poksay KS, Balestra ME, Zeng GQ, Innerarity TL (1994) Cloning and mutagenesis of the rabbit ApoB mRNA editing protein: a zinc motif is essential for catalytic activity, and noncatalytic auxiliary factor(s) of the editing complex are widely distributed. J Biol Chem 269:21725–21734 [PubMed] [Google Scholar]
- Yamanaka S, Poksay KS, Driscoll DM, Innerarity TL (1996) Hyperediting of multiple cytidines of apolipoprotein B mRNA by APOBEC-1 requires auxiliary protein(s) but not a mooring sequence motif. J Biol Chem 271:11506–11510 [DOI] [PubMed] [Google Scholar]