

Abstract
The Haseman-Elston method is widely used for the mapping of quantitative-trait loci. However, this method does not use all the information in the data, because it only considers the sib-pair trait-value difference. In addition, the Haseman-Elston method was developed for independent sib pairs; its generalization to nonindependent sib pairs is not straightforward. Here we introduce a score test statistic derived from a normal likelihood based on multiplex sibship data, conditional on identical-by-descent sharing statuses. This score test is asymptotically equivalent to the corresponding likelihood-ratio test, but it is much easier to implement. Because the proposed test uses all of the trait values, it makes more efficient use of the data than does the Haseman-Elston method. The proposed test is naturally applicable to sibships of arbitrary size. The finite-sample properties of the proposed score statistic are evaluated via simulations.
Introduction
The Haseman-Elston method (H-E method; Haseman and Elston 1972) has been widely used for the mapping of quantitative-trait loci. This method was originally developed for independent sib pairs. It regresses the squared difference in the trait values of the sib pairs on their estimated proportion of marker alleles that are shared identical by descent (IBD). If the slope of the regression line is significantly less than 0, then there is evidence for linkage between the marker and a trait locus.
However, the squared trait-value difference does not summarize all of the information in the trait data, since the trait data for a sib pair is bivariate in nature (Wright 1997). Recently, regression methods that use other transformations of the trait values as dependent variables have been proposed, to make more efficient use of the trait information (Elston et al. 2000; Xu et al. 2000; Forrest 2001; Sham and Purcell 2001; Wang et al. 2001). Similar to the H-E method, these recent modifications were also developed for independent sib-pair data. The use of these methods for dependent sib pairs that arise from multiplex sibships is not straightforward. Generalization of the H-E method to arbitrary sibship size has attracted much interest in the literature. For instance, one way of dealing with a sibship containing more than two sibs is to treat the distinct sib pairs in the sibship as independent and apply the H-E method (Amos et al. 1989; Collins and Morton 1995). To take into account the dependence among the distinct sib pairs from the same sibship, it has been suggested either to adjust the degrees of freedom of the t test statistic (Wilson and Elston 1993) or to use the generalized linear regression technique (Elston et al. 2000).
To make efficient use of the information contained in the trait data, the maximum-likelihood method has also been used for the mapping of quantitative-trait loci (Kruglyak and Lander 1995; Fulker and Cherny 1996; Wright 1997). Because this approach does not require preliminary data reduction, as the H-E method does, it is more powerful when the normality assumption is satisfied. Another advantage of the maximum-likelihood method is that it handles sibships of arbitrary size in a systematic manner. However, implementing the maximum likelihood method involves intensive computation in maximizing the likelihood function. Actually, for large sibships, even the computation of the likelihood function could be very computation intensive, because calculating the likelihood function requires the joint probabilities of the IBD-sharing statuses among all of the sibs. This is a daunting requirement when one considers that the calculation of the IBD-sharing probability even for sib pairs is not trivial (see reports by Kruglyak and Lander [1995], Almasy and Blangero [1998], and Tiwari and Elston [1997], for algorithms for calculating the IBD-sharing probability for sib pairs.)
Therefore, it is desirable to have a test statistic that has the optimal property of the maximum-likelihood method yet is easy to compute. In the present study, we derive a score statistic based on the likelihood function of nuclear families with arbitrary sibship size. This score statistic is asymptotically equivalent to the likelihood-ratio test statistic that corresponds to the maximum-likelihood method. The limiting distribution of this score statistic is derived. This score statistic is easy to compute and directly applies to sibships of arbitrary size. A simulation study is conducted to investigate the type I error rate and the power of this score statistic, which are also compared with those of H-E method.
Methods
Consider n nuclear families with sibship size ni in the ith family. At any location on the genome, each sib pair can share 0, 1, or 2 alleles IBD. Let πikl be the number of marker alleles shared IBD by the kth sib and the lth sib in the ith family. πikl is a random variable with possible values of 0, 1, or 2. The probability that πikl=0, 1, or 2 between sib pairs can be estimated from the marker data (Fulker et al. 1995; Kruglyak and Lander 1995).
For a sibship of size ni, there are ni(ni-1)/2 distinct sib pairs. Since each sib pair could share 0, 1, or 2 marker alleles IBD, the total number of IBD-sharing configurations for all of these sib pairs is 3ni(ni-1)/2. We denote this number by Ji. Let γij be the probability of the jth configuration for the ith family. This probability can be 0 for some configurations; for instance, in a sibship of size 3, any configuration in which the first sib shares 2 alleles IBD with both the second sib and the third sib, and the second sib and the third sib share 1 allele IBD, has zero probability.
In the same sibship, the IBD-sharing status for one sib pair is independent of that for another sib pair, even when these two sib pairs have one sib in common (Amos et al. 1989). However, the IBD-sharing statuses among all the sib pairs are not jointly independent. This means that γij is not the product of the probabilities of the sib-pair IBD-sharing statuses involved in the jth IBD-sharing configuration. Generally, the following relationship holds:
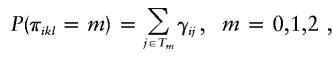 |
where Tm is the set of IBD-sharing configurations in which the kth sib and the lth sib share m alleles IBD.
Let yi=(yi1,…,yini)t be a vector consisting of the trait values of the sibs in the ith family. We assume that, given the jth IBD-sharing configuration, the density function of yi is φ(0,Σij), where
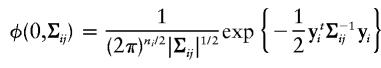 |
is a multivariate normal density function, and Σij=(σkl), with σkl=1 if k=l and σkl=ρm if k≠l, where ρm is the correlation coefficient between the kth sib and the lth sib, given that they share m alleles IBD, where m=0,1,2. To see an example of the matrix Σij, consider a sibship of size 3. In this sibship, the matrix Σij that corresponds to a configuration in which sibs 1 and 2 share 1 allele IBD, sibs 1 and 3 share 1 allele IBD, and sibs 2 and 3 share 2 alleles IBD, is
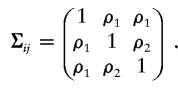 |
We note that we have assumed that the yi vectors have been standardized such that each component of yi has a mean of 0 and a variance of 1. For a discussion about the ways of standardizing yi vectors, see the Data Standardization section.
Let mi denote the marker data in the ith family. Since there are Ji IBD-sharing configuration patterns among the ni sibs in the ith family, the probability of (yi,mi) is
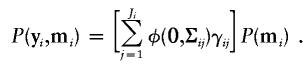 |
The unknown parameters in the above equation are ρ0, ρ1, and ρ2. The log likelihood for n families is, up to an additive constant,
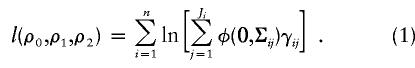 |
For the conditional correlation coefficient of the trait values between members of a sib pair, ρi, i=0,1,2, the following order restriction is generally true (Wright 1997): 0<ρ0⩽ρ1⩽ρ2⩽1. When there is no linkage, the correlation coefficient does not depend on the marker allele–sharing status—that is, 0<ρ0=ρ1=ρ2⩽1.
In the present study, we put the following restriction on ρ0, ρ1, and ρ2:
where δ=ρ2-ρ0 and f is a prespecified known value. That is, ρ1 is a convex combination of ρ0 and ρ2. The situation without such an assumption will be considered in a separate article. For a model without gene-environment interaction, we have (from Kempthorne [1957] and Tang and Siegmund [2001]):
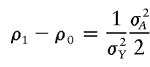 |
and
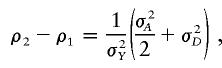 |
where σ2A and σ2D are the variances of the additive and dominance effects of the trait, respectively, and σ2Y is the variance of the trait. It can be seen that f=0.5 (which is equivalent to σD=0) represents an additive effect of the trait gene. It can also be seen that ρ2-ρ1⩾ρ1-ρ0, since σD⩾0, suggesting f<0.5 if the dominance variance is present.
When there is no linkage, δ=0; otherwise, δ>0. Under this parameterization, the unknown parameters in the log-likelihood function in equation (1) becomes (ρ0,δ) instead of (ρ0,ρ1,ρ2). The log-likelihood function in equation (1) now becomes
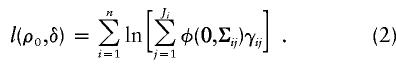 |
The hypotheses of interest are
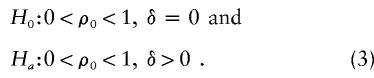 |
Define 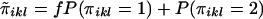 .
. 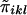 is a measure of the average IBD-sharing extent between the kth sib and the lth sib. When f=0.5, it is the proportion of alleles shared IBD by the kth sib and the lth sib (Haseman and Elston 1972). The distribution of
is a measure of the average IBD-sharing extent between the kth sib and the lth sib. When f=0.5, it is the proportion of alleles shared IBD by the kth sib and the lth sib (Haseman and Elston 1972). The distribution of 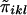 is the same for each pair in each family, regardless of the sibship size. Under the null hypothesis,
is the same for each pair in each family, regardless of the sibship size. Under the null hypothesis, 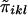 is independent of yi. We denote the mean and variance of
is independent of yi. We denote the mean and variance of 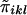 by
by 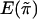 and
and 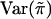 , respectively.
, respectively.
Under the null hypothesis H0, δ=0 and ρ0=ρ1=ρ2. The matrices Σij, j=1,2,…,Ji, are all the same, with all off-diagonal elements equal to ρ0. Let this matrix be denoted by Σi0. For simplicity, we introduce a vector, wi=(wi1,…,wini)t≡Σ-1i0yi. Under the null hypothesis, wi∼N(0,Σ-1i0), since yi∼N(0,Σi0). We note that
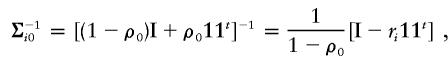 |
where ri=ρ0/[1+(ni-1)ρ0] and 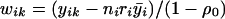 , where
, where 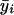 is the average of all the elements of yi.
is the average of all the elements of yi.
In what follows, we present a score statistic for testing the hypotheses in equation (3). The first-order derivatives and the information matrix that are necessary for this purpose are presented in Appendix A.
To better describe the score statistic, we define
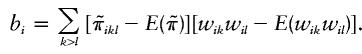 |
That is, bi is the inner product of the vector 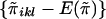 and the vector {wikwil-E(wikwil)}, two vectors each of length 0.5ni(ni-1). Under the null hypothesis, the mean and variance of bi are, respectively, E(bi)=0 and
and the vector {wikwil-E(wikwil)}, two vectors each of length 0.5ni(ni-1). Under the null hypothesis, the mean and variance of bi are, respectively, E(bi)=0 and 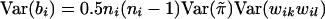 , since
, since 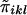 values are independent of wi when there is no linkage.
values are independent of wi when there is no linkage.
It is shown in Appendix B that, to test the null hypothesis H0 against the alternative hypothesis Ha in equation (3), one can use the following score statistic:
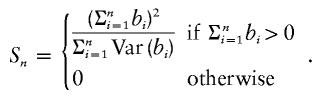 |
In Appendix B, it is derived that Sn is asymptotically distributed as 0.5χ20+0.5χ21 under the null hypothesis in equation (3).
We note that the mean 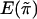 and variance
and variance 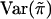 of
of 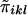 are the same for any sib pair in any family. It can be derived from the study by Amos et al. (1989) that
are the same for any sib pair in any family. It can be derived from the study by Amos et al. (1989) that 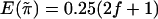 and
and
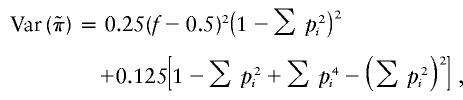 |
where pi is the frequency of the ith marker allele. However, the mean and variance of wikwil depend on the sibship size ni through ri:
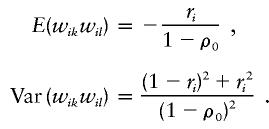 |
These expressions for  , and Var(wikwil) can be used in the calculation of the score statistic Sn; however, we do not endorse such a practice. Instead, we recommend using the sample counterparts of these quantities in the calculation of Sn. The main reason is that doing this tends to make the score statistic Sn more robust in situations in which the normality assumption for the trait values is violated and/or the marker locus is in linkage disequilibrium with the trait locus. In the latter case, it is inappropriate to use the population marker-allele frequencies to calculate
, and Var(wikwil) can be used in the calculation of the score statistic Sn; however, we do not endorse such a practice. Instead, we recommend using the sample counterparts of these quantities in the calculation of Sn. The main reason is that doing this tends to make the score statistic Sn more robust in situations in which the normality assumption for the trait values is violated and/or the marker locus is in linkage disequilibrium with the trait locus. In the latter case, it is inappropriate to use the population marker-allele frequencies to calculate  .
.
To be specific, we recommend replacing 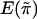 and
and 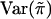 with the sample mean and the sample variance of {πikl} for all sib pairs from all families, respectively, and replacing E(wikwil) and Var(wikwil) with the sample mean and the sample variance of {wikwil} for all sib pairs from all the families that are of the same size, respectively. This is how the score statistic Sn is calculated in our simulation.
with the sample mean and the sample variance of {πikl} for all sib pairs from all families, respectively, and replacing E(wikwil) and Var(wikwil) with the sample mean and the sample variance of {wikwil} for all sib pairs from all the families that are of the same size, respectively. This is how the score statistic Sn is calculated in our simulation.
When transforming yi into wi, we need to know the correlation coefficient, ρ0, between the trait values of sib pairs. Since the true value of ρ0 is unknown, we suggest replacing it with the sample correlation coefficient between independent sib pairs. By standard asymptotic theory, since this sample correlation is a consistent estimator of ρ0, such substitution does not change the asymptotic distribution of Sn.
Data Standardization
In deriving the score statistic Sn, we have assumed that the trait values for a sibship, yi, have a multivariate normal distribution, conditional on the IBD-sharing configuration within the sibship. The conditional marginal mean and variance for each component of yi—say, yik—are 0 and 1, respectively. This implies that the unconditional marginal mean and variance of yik are also 0 and 1, respectively.
When the mean of yik is not 0 and the variance of yik is not 1, we can standardize yi in the following way: Consider the trait values of all sibs in all families, {yik, k=1,…,ni, i=1,…,n}. Let 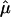 and
and 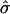 be the sample mean and sample standard deviation, respectively, of these trait values. We standardize yik by
be the sample mean and sample standard deviation, respectively, of these trait values. We standardize yik by
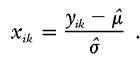 |
Since 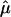 and
and 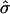 are consistent estimators of their respective population counterparts, xik has a mean of 0 and a variance of 1, asymptotically. Consequently, the score statistic Sn based on {xik} still has the limiting distribution 0.5χ20+0.5χ21, on the basis of the standard asymptotic theory.
are consistent estimators of their respective population counterparts, xik has a mean of 0 and a variance of 1, asymptotically. Consequently, the score statistic Sn based on {xik} still has the limiting distribution 0.5χ20+0.5χ21, on the basis of the standard asymptotic theory.
So far, we have critically assumed that yi has a multivariate normal distribution conditional on the IBD-sharing configuration in the sibship. When this normality assumption is not satisfied, the proposed score statistic Sn may have poor performance. This is also a concern for the H-E method (Allison et al. 1999).
To reduce the impact of nonnormality on the performance of the proposed score statistic, we recommend transforming the trait values first, on the basis of the empirical normal quantile–distribution transformation. The description of this transformation is as follows: Consider the trait values of all sibs in all families, {yik,k=1,…,ni,i=1,…,n}. Let rik be the rank of the yik. The transformation of yik is
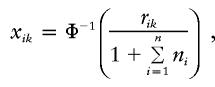 |
where Φ−1 is the inverse of the cumulative function of the standard normal distribution. xik is basically the empirical normal quantile distribution transformation of yik, since 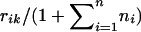 is basically the empirical distribution of all the trait values. We use
is basically the empirical distribution of all the trait values. We use 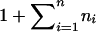 instead of
instead of 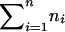 here, because we want to make sure that xik<∞. From standard asymptotic theory, xik follows the standard normal distribution, with a mean of 0 and a variance of 1.
here, because we want to make sure that xik<∞. From standard asymptotic theory, xik follows the standard normal distribution, with a mean of 0 and a variance of 1.
We then assume that the joint distribution of xi=(xi1,xi2,…,xini)t is a multivariate normal distribution conditional on the IBD-sharing configuration: for the kth sib and the lth sib, the correlation coefficients between xik and xil are ρ0, ρ1, or ρ2 if they share 0, 1, or 2 alleles IBD, respectively, at the marker locus. This modeling procedure is often referred to as the “multivariate (empirical) normal copula” model. For more discussions about bivariate copula models, see (for example) reports by Genest and MacKay (1986) and Klaassen and Wellner (1997).
It can be shown that such transformation does not change the asymptotic distribution of the likelihood-ratio statistic for the log-likelihood in equation (2) (authors' unpublished data). Since the asymptotic distribution of the score statistic Sn is the same as that of the likelihood-ratio statistic, it follows that such transformation will not change the asymptotic distribution of Sn either.
Special Cases
In the previous section, we derived a score statistic for sibships of arbitrary size. This statistic is related to some other statistics in the literature and has a simple interpretation when the sibship size is constant across all families.
Independent Sib Pairs
For sib-pair data in which ni=2, i=1,2,…,n, we have
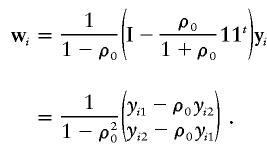 |
Furthermore,
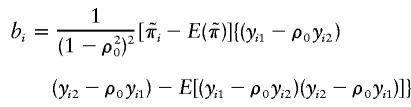 |
and
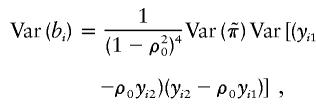 |
where 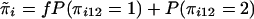 .
.
Since
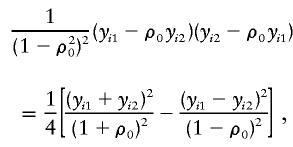 |
it can be seen that, when f=0.5, 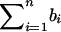 is proportional to the estimated slope in the “new combined HE regression” (HE-COM; Sham and Purcell 2001). In this regression, the dependent variable is
is proportional to the estimated slope in the “new combined HE regression” (HE-COM; Sham and Purcell 2001). In this regression, the dependent variable is
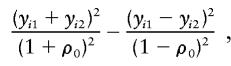 |
which is a weighted sum of the squared sums and the squared differences, and the independent variable is 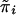 . Therefore, for independent sib-pair data, the proposed score statistic Sn is equivalent to the t test for testing the regression effect in such a regression. The rejection region of such a test is one sided. Sham and Purcell (2001) compared the performance of the H-E method with some other common statistics; HE-COM outperforms the other statistics in all of the situations investigated.
. Therefore, for independent sib-pair data, the proposed score statistic Sn is equivalent to the t test for testing the regression effect in such a regression. The rejection region of such a test is one sided. Sham and Purcell (2001) compared the performance of the H-E method with some other common statistics; HE-COM outperforms the other statistics in all of the situations investigated.
Constant Sibship Size >2
When the sibship size is constant across all families, the proposed statistic Sn takes a simpler form and is related to the usual F-test statistic for testing whether the regression coefficient is 0 in a regression analysis.
Let s be the common sibship size. We have
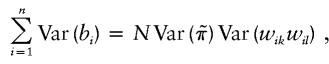 |
where N=0.5ns(s-1) is the total number of sib pairs in all n families. Now E(wikwil) and Var(wikwil) are the same across all families. The nonzero part of Sn becomes
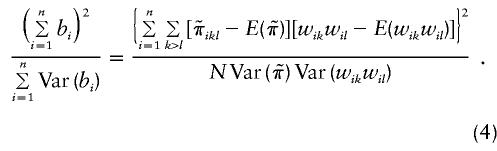 |
On the other hand, if we regress {wikwil} against {πikl}, the usual F statistic for testing whether the regression slope is 0 is equivalent to the quantity on the right hand side in equation (4).
Simulations
In the simulations, we assume that there are three sibs in each family. The trait is assumed to be determined by two equally frequent alleles, D and d, and the marker-allele frequencies are also assumed to be equal. The trait values, (y1,y2,y3), of the three sibs in a family are simulated on the basis of the following model:
where u represents the effect of shared genes other than the one at the locus under consideration or common environmental factors; ε1, ε2, and ε3 are error terms that are independent of each other; and g1, g2, and g3 are the genetic contributions of the linked trait locus, with
 |
The value of μdd, μDd, and μDD are determined from the broad-sense heritability, h, which, following Gillespie (1998), is defined as
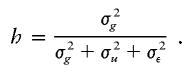 |
In the above expression, σ2g, σ2u, and σ2ε are the variances due to the quantitative-trait locus, shared gene or environmental effect u, and error ε, respectively.
Specifically, we consider the following four trait models in the simulation:
-
Model 1:
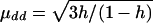 , μDd=0, and μDD= -μdd. εi1 and εi2 are independently distributed as N(0,1).
, μDd=0, and μDD= -μdd. εi1 and εi2 are independently distributed as N(0,1). -
Model 2:
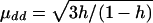 , μDd=0, and μDD= -μdd. εi1 and εi2 are independently distributed as
, μDd=0, and μDD= -μdd. εi1 and εi2 are independently distributed as 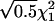 .
. -
Model 3:
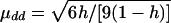 , μDd=0.5μdd, and μDD=-μdd. εi1 and εi2 are independently distributed as N(0,1).
, μDd=0.5μdd, and μDD=-μdd. εi1 and εi2 are independently distributed as N(0,1). -
Model 4:
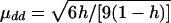 , μDd=0.5μdd, and μDD=-μdd. εi1 and εi2 are independently distributed as
, μDd=0.5μdd, and μDD=-μdd. εi1 and εi2 are independently distributed as 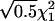 .
.
In all four models, ui is generated from N(0,0.5).
Models 1 and 2 assume that the mean phenotypes are determined additively by the alleles at the disease locus. Models 3 and 4 introduce some dominance. We note that the error distributions in models 1 and 3 are symmetrical, whereas the error distributions in models 2 and 4 are skewed to the right. Under these four models, the type I error and power of the proposed score statistic and the original H-E statistic (Haseman and Elston 1972) are compared. When calculating the score statistic, we use two methods to standardize the trait values. First, we use the sample mean and sample standard deviation in the way described in the Data Standardization section. The corresponding score statistic is denoted by S*n. We also standardize the trait values with the empirical normal quantile distribution transformation, which is also described in that section; the corresponding score statistic is denoted by S**n. In calculating the H-E statistic, we treat the three dependent sib pairs formed from the three sibs in the same family as independent sib pairs. Such practice is valid when number of sib pairs is large, as in the situation of our simulation (Blackwelder and Elston 1985; Wilson and Elston 1993). In all of these analyses, we use an additive model, by setting f=0.5.
The IBD-sharing probabilities at the marker locus are calculated from table II of Haseman and Elston (1972), since it is faster and more convenient than using some existing genetics programs. The simulation program was written in R language (Ihaka and Gentleman 1996). It was run in R (version 1.3.0) on a Compaq XP 1000 workstation running Red Hat Linux 7.1.
In the simulation study of the type I error rate, we set the recombination fraction (θ) between the quantitative-trait locus and the marker at θ=0.5 and set the heritability at h=0. Table 1 reports the observed type I error rates of these three statistics when the nominal significance level is at .1, .05, .01, and .001 and when the number of family members is 100 and 200. For these three statistics, the observed type I error rates are very close to the respective nominal significance levels.
Table 1.
Type I Error Rates Based on 10,000 Replications
|
Nominal Significance Level |
||||
| Model, SampleSize, andScore Statistic | .1 | .05 | .01 | .001 |
| 1: | ||||
| 100: | ||||
| H-E | .0993 | .0523 | .0120 | .0013 |
| S*n | .0996 | .0491 | .0106 | .0014 |
| S**n | .1020 | .0484 | .0106 | .0011 |
| 200: | ||||
| H-E | .1004 | .0521 | .0113 | .0011 |
| S*n | .0999 | .0512 | .0124 | .0018 |
| S**n | .0992 | .0514 | .0133 | .0018 |
| 2: | ||||
| 100: | ||||
| H-E | .1026 | .0520 | .0109 | .0014 |
| S*n | .1043 | .0551 | .0122 | .0013 |
| S**n | .1055 | .0547 | .0122 | .0009 |
| 200: | ||||
| H-E | .1025 | .0510 | .0115 | .0010 |
| S*n | .1047 | .0531 | .0132 | .0018 |
| S**n | .1061 | .0550 | .0115 | .0013 |
| 3: | ||||
| 100: | ||||
| H-E | .1063 | .0526 | .0112 | .0010 |
| S*n | .1023 | .0514 | .0112 | .0012 |
| S**n | .1002 | .0519 | .0109 | .0012 |
| 200: | ||||
| H-E | .0988 | .0500 | .0095 | .0013 |
| S*n | .1015 | .0524 | .0112 | .0018 |
| S**n | .1013 | .0531 | .0109 | .0018 |
| 4: | ||||
| 100: | ||||
| H-E | .1049 | .0534 | .0110 | .0011 |
| S*n | .1049 | .0566 | .0142 | .0014 |
| S**n | .1065 | .0555 | .0127 | .0018 |
| 200: | ||||
| H-E | .1003 | .0505 | .0114 | .0011 |
| S*n | .1011 | .0531 | .0126 | .0012 |
| S**n | .0972 | .0497 | .0114 | .0020 |
To study the power of the four statistics, we fix θ between the trait locus and the marker at 0. Figures 1 and 2 depict the power of the four statistics when the (broad-sense) heritability h is allowed to change from 0.05 to 0.5 with step size 0.05 for sample sizes 100 and 200. In the four models considered, the score statistics (S*n and S**n) perform no worse than does the H-E statistic. The difference in their performance is negligible when the heritability is small (h<0.2 for n=100 and h<0.1 for n=200 in models 1 and 3; h<0.1 for n=100 in models 2 and 4). However, for other heritability values, both score statistics perform better than the H-E statistic. The increase in the performance of the two score statistics is more apparent in models 2 and 4, where the error terms are skewed.
Figure 1.

Power at different levels of heritability at the nominal significance level of .001, for 1,000 replicates, with 100 families included in each.
Figure 2.

Power at different levels of heritability at the nominal significance level of .001, for 1,000 replicates, with 200 families included in each.
We compare the two data-standardization techniques, by comparing the performance of S*n and S**n. For the models where the error terms are normally distributed (i.e., models 1 and 3), the powers of these two score statistics are very close to each other (the powers of these statistics in model 1 are virtually the same). However, for models in which the error terms are skewed (i.e., models 2 and 4), the power of S*n is less than the power when the error terms are normally distributed (compare model 3 vs. model 1, and model 4 vs. model 2). However, the power of S**n almost remains the same (again, compare model 3 vs. model 1, and model 4 vs. model 2). The empirical normal quantile distribution transformation seems to be more robust to nonnormality of the error terms.
Discussion
In the present study, we proposed a score statistic for detecting linkage to quantitative-trait loci. This score statistic inherits the benefit of the likelihood-ratio statistic, in that it makes efficient use of the information from all sibs in sibships of arbitrary size, yet it is much easier to compute. The ease of computation results from two facts: First, as an intrinsic property, the score statistic does not involve maximization of the likelihood function. Because of the explicit formula for the score statistic, its calculation is straightforward. Second, although the calculation of the likelihood function requires the joint probabilities of the IBD-sharing statuses among all sibs in a sibship, it turns out that it suffices to know the pairwise IBD-sharing probabilities in order to calculate the score statistic. Since several genetics programs exist that can export pairwise IBD-sharing probabilities between sib pairs, the calculation of the proposed score statistic can be implemented very easily. This second advantage of the score statistic over the likelihood-ratio statistic is important here, because it makes the calculation of the score statistic immediately feasible in that it does not require the joint sharing probabilities among all sibs in a sibship. Tang and Siegmund (2001) considered a score statistic in a similar context, but it was assumed that the markers were completely informative, in which case the IBD-sharing configuration among a sibship would be known with certainty.
In comparison with the H-E method and its recent modifications, a salient feature of the proposed score statistic is that it handles multiplex sibships in a natural way. Without breaking the sibship into sib pairs, the proposed score statistic is calculated directly from the sibship. For independent sib pairs, this score statistic turns out to be asymptotically equivalent to the HE-COM statistic of Sham and Purcell (2001).
In our simulation study, we considered one additive model and one dominant model. For each model, we considered the cases where the error terms are symmetrical (normally distributed) and skewed (χ2 distributed). Under these simulation models, the proposed score statistic outperforms the H-E method, in terms of power, when the heritability changes from 0.05 to 0.5. The type I error rates of the score statistic and the H-E method are both well under control.
For nonnormal data, we recommend a data-transformation procedure that is based on the standard normal density function and the empirical distribution of the trait values. As pointed out by an anonymous reviewer, such a data-transformation procedure can be done prior to any other quantitative-trait loci mapping method, and this does not guarantee multivariate normality. The latter point is the reason that we used the normal copula model; we assume that the transformed data have a multivariate normal distribution. As the same reviewer pointed out, such a practice could complicate the interpretation of some interesting quantities such as additive variance and heritability; however, as far as the test is concerned, we have shown that, for the model used in our study, such a transformation does not change the asymptotic distribution of the score statistic we derived (authors' unpublished data).
Compared with our likelihood function (2), the variance-component analysis uses a different but related likelihood function. In likelihood function (2), the likelihood for the ith family is a mixture of Ji normal densities. A corresponding variance-component likelihood function would be
where Σi is a symmetric matrix whose diagonal elements are 1 and whose klth off-diagonal element is
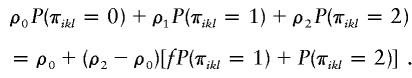 |
We prefer likelihood function (2) to likelihood function (5), because the former contains more information; one can write out likelihood function (5) from likelihood function (2), but not vice versa. If the normality assumption holds, the analysis based on the former should have as much power as that based on the latter. In a simulation study, the likelihood-ratio statistic from likelihood function (2) performs better than that from likelihood function (5) (Dolan et al. 1999). We expect our score statistic to be more robust against violation of the normality assumption, in terms of type I error rate; the asymptotic distribution of a score statistic relies solely on the central limit theorem, although the derivation of the score statistic depends on the model assumption. In contrast, the asymptotic distribution of the likelihood-ratio statistic is directly related to the model assumption. Further studies are needed to assess the power behavior of our approach and of the variance-component method for non-normal data.
We note that likelihood function (2) is based on the joint distribution of marker and trait. Thus, this likelihood is only correct for randomly selected families and is approximately correct for moderately selected sibship data. This likelihood cannot be simply applied to extremely discordant sib-pair data. However, analysis of extremely discordant sib-pair data can be considered to be a missing-data problem—that is, the pairs not selected for genotyping can be viewed as having their marker data missing. For instance, one approach for applying our method to extremely discordant sib-pair data is to include all the untyped pairs and assign the prior IBD-sharing probabilities to these pairs (Kruglyak and Lander 1995; Eaves et al. 1996; Dolan et al. 1999). Other approaches for analyzing extremely discordant sib-pair data include the method based on IBD-sharing scores or weighted IBD-sharing scores (Risch and Zhang 1995; Gu et al. 1996) or the method based on the conditional likelihood of marker data, given trait values (Dudoit and Speed 1999; Sham et al. 2000; Goldstein et al. 2001).
In the present study, we considered only the sib-sib relationship; however, the score statistic can also be applied directly in situations where there is only one relationship involved, no matter what that relationship is. When there is more than one relationship, the situation becomes more complicated. The score statistic that can handle multiple relationships together will be presented in a separate article.
Acknowledgments
This work is supported, in part, by a University of Iowa College of Public Health–College of Medicine New Investigator Research Award (to K.W.), by National Institute of Mental Health grant K01-01541 (to J.H.), and by National Institute of Mental Health grant R01-52841 (Principle Investigator: Dr. Veronica Vieland; Investigators include K.W. and J.H.). We thank two anonymous reviewers for their constructive comments, which helped us to improve our manuscript.
Appendix A: Derivatives and Information Matrix
Let Ωi be a symmetrical matrix whose diagonal elements are all 0 and whose klth off-diagonal element is 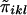 . It is apparent that
. It is apparent that 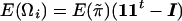 .
.
When evaluated at (ρ0,δ)=(ρ0,0), the derivative of l(ρ0,δ) with respect to ρ0 is
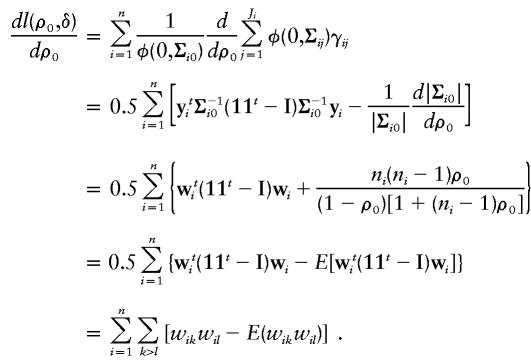 |
Similarly, the derivative of l(ρ0,δ) with respect to δ, when evaluated at (ρ0,δ)=(ρ0,0), is
 |
In deriving these first-order derivatives, we used the following facts:
-
1.
dΣ-1i0/dρ0=-Σ-1i0 dΣi0/dρ0Σ-1i0=-Σ-1i0(11t-I)Σ-1i0,
-
2.
 ,
, -
3.
|Σi0|=(1-ρ0)ni-1[1+(ni-1)ρ0],
-
4.
 . (The proof of this fact is omitted, because of its length.)
. (The proof of this fact is omitted, because of its length.)
The expectations of wti(11t-I)wi and the conditional expectation of wtiΩiwi are taken under the null hypothesis, and are
 |
and
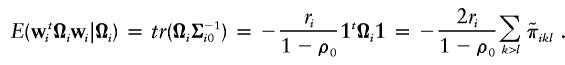 |
The information matrix is
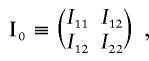 |
where
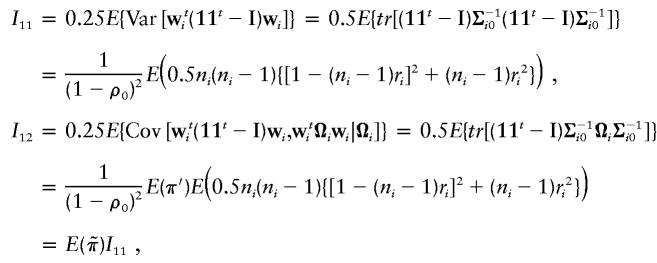 |
and
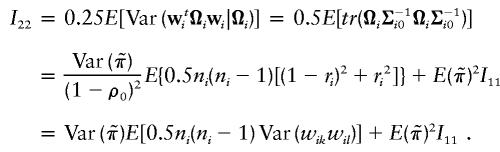 |
In the final expressions for I11, I12, and I22, the expectation is taken with respect to the sibship size ni.
Appendix B : Derivation of the Score Statistic Sn
From asymptotic theory, under the null hypothesis,
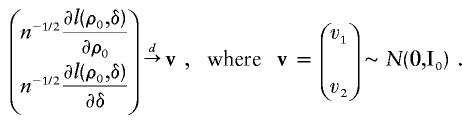 |
Let Θ0={(ρ0,δ):δ=0} and Θ1={(ρ0,δ):δ>0} be the sets of parameters that correspond to the null hypothesis and the alternative hypothesis, respectively. Let 𝔥0={h=(h1,h2):h1∈R,h2=0} and 𝔥1={h=(h1,h2):h1∈R,h2>0}. From theorem 16.7 of van der Vaart (1998), the likelihood ratio statistic is
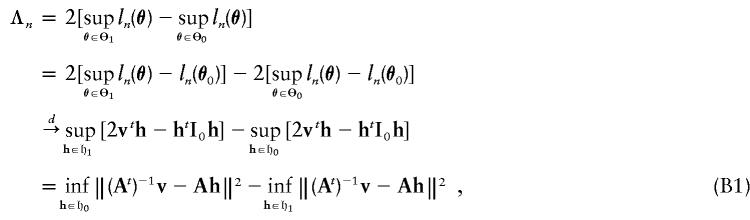 |
where
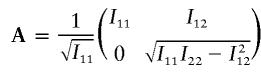 |
Notice that AtA=I0 and (At)-1v∼N(0,I), where I is a 2×2 identity matrix.
The parameter space 𝔥1 is the upper-half space. Since Ah∈𝔥0 if and only if h∈𝔥0, and since Ah∈𝔥1 if and only if h∈𝔥1, {Ah:h∈𝔥1} is also the upper-half space. Since
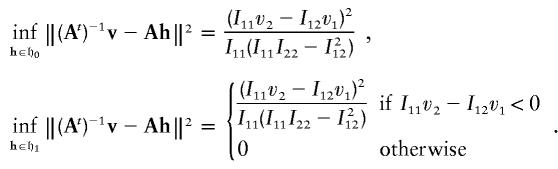 |
Therefore, from equation (B1),
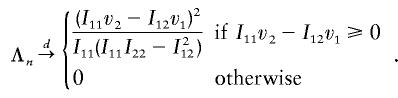 |
whose distribution is 0.5χ20+0.5χ21.
Since
a consistent estimator of I11I22-I212 is
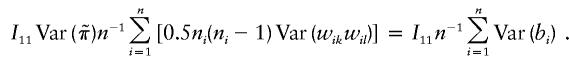 |
A sample version of I11v2-I12v1 is
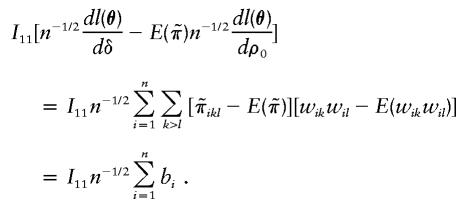 |
Therefore, a sample version of (I11v2-I12v1)2/[I11(I11I22-I212)] is (Σni=1bi)2/Σni=1Var(bi), which is the score statistic Sn reported in the text.
References
- Allison DB, Neale MC, Zannolli R, Schork NJ, Amos CI, Blangero J (1999) Testing the robustness of the likelihood-ratio test in a variance-component quantitative-trait loci–mapping procedure. Am J Hum Genet 65:531–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almasy L, Blangero J (1998) Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet 62:1198–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amos CI, Elston RC, Wilson AF, Bailey-Wilson JE (1989) A more powerful robust sib-pair test of linkage for quantitative traits. Genet Epidemiol 6:435–449 [DOI] [PubMed] [Google Scholar]
- Blackwelder WC, Elston RC (1985) A comparison of sib-pair linkage tests for disease susceptibility loci. Genet Epidemiol 2:85–97 [DOI] [PubMed] [Google Scholar]
- Collins A, Morton NE (1995) Nonparametric tests for linkage with dependent sib pairs. Hum Hered 45:311–318 [DOI] [PubMed] [Google Scholar]
- Dolan CV, Boomsma DI, Neale MC (1999) A simulation study of the effects of assignment or prior identity-by-descent probabilities to unselected sib pairs, in covariance-structure modeling of a quantitative-trait locus. Am J Hum Genet 64:268–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudoit S, Speed TP (1999) A score test for linkage using identity by descent data from sibships. Ann Stat 27:943–986 [Google Scholar]
- Eaves LJ, Neale MC, Maes H (1996) Multivariate multipoint linkage analysis of quantitative trait loci. Behav Genet 26:519–525 [DOI] [PubMed] [Google Scholar]
- Elston RC, Buxbaum S, Jacobs KB, Olson JM (2000) Haseman and Elston revisited. Genet Epidemiol 19:1–17 [DOI] [PubMed] [Google Scholar]
- Forrest WF (2001) Weighting improves the ‘new Haseman-Elston’ method. Hum Hered 52:47–54 [DOI] [PubMed] [Google Scholar]
- Fulker DW, Cherny SS (1996) An improved multipoint sib-pair analysis of quantitative traits. Behav Genet 26:527–532 [DOI] [PubMed] [Google Scholar]
- Fulker DW, Cherny SS, Cardon LR (1995) Multipoint interval mapping of quantitative trait loci, using sib pairs. Am J Hum Genet 56:1224–1233 [PMC free article] [PubMed] [Google Scholar]
- Genest C, MacKay J (1986) The joy of copulas: bivariate distributions with uniform marginals. Am Statistician 40:280–283 [Google Scholar]
- Gillespie JH (1998) Population genetics: a concise guide. Johns Hopkins University Press, Baltimore [Google Scholar]
- Goldstein DR, Dudoit S, Speed TP (2001) Power and robustness of a score test for linkage analysis of quantitative traits using identity by descent data on sib pairs. Genet Epidemiol 20:415–431 [DOI] [PubMed] [Google Scholar]
- Gu C, Todorov A, Rao DC (1996) Combining extremely concordant sib pairs with extremely discordant sib pairs provide a cost effective way to linkage analysis of quantitative trait loci. Genet Epidemiol 13:513–533 [DOI] [PubMed] [Google Scholar]
- Haseman JK, Elston RC (1972) The investigation of linkage between a quantitative trait and a marker locus. Behav Genet 2:3–19 [DOI] [PubMed] [Google Scholar]
- Ihaka R, Gentleman R (1996) R: a language for data analysis and graphics. J Comp Graph Stat 5:299–314 [Google Scholar]
- Kempthorne O (1957) An introduction to genetic statistics. Wiley, New York [Google Scholar]
- Klaassen CAJ, Wellner JA (1997) Efficient estimation in the bivariate normal copula model: normal margins are least favorable. Bernoulli 3:55–77 [Google Scholar]
- Kruglyak L, Lander ES (1995) Complete multipoint sib-pair analysis of qualitative and quantitative traits. Am J Hum Genet 57:439–454 [PMC free article] [PubMed] [Google Scholar]
- Risch N, Zhang H (1995) Extreme discordant sib pairs for mapping quantitative trait loci in humans. Science 268:1584–1589 [DOI] [PubMed] [Google Scholar]
- Sham PC, Purcell S (2001) Equivalence between Haseman-Elston and variance-components linkage analyses for sib pairs. Am J Hum Genet 68:1527–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sham PC, Zhao JH, Cherny SS, Hewitt JK (2000) Variance-components QTL linkage analysis of selected and non-normal samples: conditioning on trait values. Genet Epidemiol 10 Suppl 1:S22–S28 [DOI] [PubMed] [Google Scholar]
- Tang HK, Siegmund D (2001) Mapping quantitative trait loci in oligogenic models. Biostatistics 2:147–162 [DOI] [PubMed] [Google Scholar]
- Tiwari HK, Elston RC (1997) Linkage of multilocus components of variance to polymorphic markers. Ann Hum Genet 61:253–261 [DOI] [PubMed] [Google Scholar]
- van der Vaart AW (1998) Asymptotic statistics. Cambridge University Press, New York [Google Scholar]
- Wang D, Lin S, Cheng R, Gao X, Wright FA (2001) Transformation of sib-pair values for the Haseman-Elston method. Am J Hum Genet 68:1238–1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson AF, Elston RC (1993) Statistical validity of the Haseman-Elston sib-pair test in small samples. Genet Epidemiol 10:593–598 [DOI] [PubMed] [Google Scholar]
- Wright FA (1997) The phenotypic difference discards sib-pair QTL linkage information. Am J Hum Genet 60:740–742 [PMC free article] [PubMed] [Google Scholar]
- Xu X, Weiss S, Xu XP, Wei LJ (2000) A unified Haseman-Elston method for testing linkage with quantitative traits. Am J Hum Genet 67:1025–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]