

Abstract
In this review, we discuss transposon-insertion sequencing, variously known in the literature as TraDIS, Tn-seq, INSeq, and HITS. By monitoring a large library of single transposon-insertion mutants with high-throughput sequencing, these methods can rapidly identify genomic regions that contribute to organismal fitness under any condition assayable in the laboratory with exquisite resolution. We discuss the various protocols that have been developed and methods for analysis. We provide an overview of studies that have examined the reproducibility and accuracy of these methods, as well as studies showing the advantages offered by the high resolution and dynamic range of high-throughput sequencing over previous methods. We review a number of applications in the literature, from predicting genes essential for in vitro growth to directly assaying requirements for survival under infective conditions in vivo. We also highlight recent progress in assaying non-coding regions of the genome in addition to known coding sequences, including the combining of RNA-seq with high-throughput transposon mutagenesis.
Keywords: transposon mutagenesis, essential genes, sequencing, systems biology, bacteria, sRNA
Introduction
A common approach to identifying genomic regions involved in survival under a particular set of conditions is to screen large pools of mutants simultaneously. This can be done with defined mutants;1,2 however, the construction of defined mutant libraries is labor intensive and requires accurate genomic annotation, which can be particularly difficult to define for non-coding regions. An alternative to defined libraries is the construction and analysis of random transposon-insertion libraries. The original application of this method used DNA hybridization to track uniquely tagged transposon-insertions in Salmonella enterica serovar Typhimurium over the course of BALB/c mouse infection.3 DNA hybridization was eventually superseded by methods that used microarray detection of the genomic DNA flanking insertion sites, variously known as TraSH, MATT and DeADMAn (reviewed in ref. 4). However, these methods suffered from many of the problems microarrays generally suffer from: difficulty detecting low-abundance transcripts, mis-hybridization, probe saturation and difficulty identifying insertion sites precisely.
The application of high-throughput sequencing to the challenge of determining insertion location and prevalence solves many of these problems. Interestingly, the first application of transposon-insertion sequencing, developed by Hutchison et al., actually predates the development of microarray-based methods.5 However, this was applied to libraries of only approximately 1,000 transposon mutants in highly reduced Mycoplasma genomes, and the difficulty of sequencing at the time prevented wide spread adoption or high resolution. Modern high-throughput sequencing technology allows the methods discussed in this review to routinely monitor as many as one million mutants simultaneously in virtually any genetically tractable microorganism.
Protocols
Several methods were developed concurrently for high-throughput sequencing of transposon-insertion sites: TraDIS,6 INSeq,7 HITS8 and Tn-seq9 followed by Tn-seq Circle10 and refinements to the INSeq protocol.11 All of these protocols follow the same basic workflow with minor variations (see Fig. 1, Table 1): transposon mutagenesis and construction of pools of single insertion mutants; enrichment of transposon-insertion junctions and, finally, in some protocols a purification step either precedes or follows PCR enrichment before sequencing.
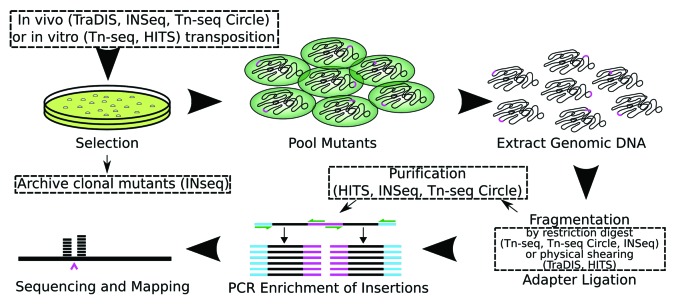
Figure 1. An illustration of the workflow typical of transposon-insertion sequencing protocols. Transposons are represented by pink lines, sequencing adaptors by blue, genomic DNA by black and PCR primers by green. Mutants are generated through either in vivo or in vitro transposition and subsequent selection for antibiotic resistance. These mutants are pooled, and optionally competed in test conditions, then genomic DNA is extracted and fragmented by restriction digest or physical shearing. Sequencing adaptors are ligated, some protocols then perform a step to purify fragments containing transposon insertions, and PCR with transposon- and adaptor-specific primers is used to specifically enrich for transposon-containing fragments. The fragments are then sequenced and mapped back to a reference genome to uniquely identify insertion sites with nucleotide-resolution. Dashed boxes indicate steps which differ between protocols.
Table 1. Applications of transposon-insertion sequencing.
| Study | Publication date | Organism | Total mutants | Unique insertion sites; Density | Application | Tn used | Name coined |
|---|---|---|---|---|---|---|---|
| Hutchison III et al., Science5 |
Oct 1999 |
M. genitalium/M. pneumoniae |
1,291 918 |
685; ~1/850 bp 669; ~1/850 bp |
Required gene sets |
Tn4001 |
Global Transposon Mutagenesis |
| Goodman et al., Cell Host and Microbe7 |
Sep 2009 |
B. thetaiotaomicron |
2 × 35,000 |
35,000;1/182 bp |
Establishment in human gut as a natural habitat |
Mariner |
INSeq |
| Gawronski et al., PNAS8 |
Sep 2009 |
H. influenzae |
75,000 |
55,935; 1/32 bp |
Prolonged survival in lung in vivo |
Mariner |
HITS |
| van Opijnen et al., Nature Methods9 |
Oct 2009 |
S. pneumoniae |
6 × 25,000 |
23,875; 1/91bp |
Transcriptional regulation and carbohydrate transport |
Mariner |
Tn-seq |
| Langridge et al., Genome Research6 |
Dec 2009 |
S.Typhi
1
|
1.1 million |
370,000; 1/13 bp |
Gene requirements, bile tolerance |
Tn5 |
TraDIS |
| Gallagher et al., mBio10 |
Jan 2011 |
P. aeruginosa |
~100,000 |
95,905; 1/65 bp |
Tobramycin resistance |
Mariner |
Tn-seq (circle method) |
| Eckert et al., J Bact.24 |
Jan 2011 |
E. coli |
19 × 95 |
1,645 |
Colonization of bovine intestinal tract; retrospective re-evaluation of a STM study |
Tn5 |
- |
| Christen et al., Mol Syst Biol.16 |
July 2011 |
C. crescentus |
800,000 |
428,735; 1/8 bp |
Gene/ncRNAs/promoter requirements |
Tn5 |
- |
| Griffin et al., PLOS Pathogens27 |
Sept 2011 |
M. tuberculosis |
2 × 100,000 |
36,488;1/120 bp |
Gene requirements and cholesterol utilization |
Mariner |
- |
| Khatiwara et al., AEM35 |
May 2012 |
S. Typhimurium |
16,000 |
~8,000; ~1/610 |
Bile, low nutrient and heat tolerance |
Tn5 |
- |
| Mann et al., PLOS Pathogens46 |
July 2012 |
S. pnuemoniae |
~9,000–24,000 |
~8,000‒22,000 |
Determining roles of sRNAs in pathogenesis |
Mariner |
- |
| van Opijnen and Camilli, Genome Research22 |
July 2012 |
S. pnuemoniae |
~4,000‒30,000 |
Varying |
Stress response and metabolism in vitro and murine in vivo colonization |
Mariner |
- |
| Brutinel and Gralnick, Molecular Microbiology51 |
Aug 2012 |
S. oneidensis |
50,000 |
26,793; ~1/191 bp |
Gene requirements and Metabolism |
Mariner |
- |
| Zhang et al., PLOS Pathogens23 |
Sept 2012 |
M. tuberculosis |
2 × 100,000 |
36,488; ~1/120 bp |
Identifying genes, regulators and ncRNAs required for growth |
Mariner |
- |
| Klein et al., BMC Genomics52 |
Oct. 2012 |
P. gingivalis |
N/A |
54,000; 1/43 bp |
Gene requirements |
Mariner |
- |
| Pickard et al., J Bact36 |
Jan 2013 |
S.Typhi
1
|
1.1 million |
370,000; 1/13 bp |
Bacteriophage infection |
Tn5 |
- |
| Barquist et al., Nuc. Acids Res15 | March 2013 |
S. Typhi1 S. Typhimurium |
1.1 million 930,000 |
370,000; 1/13 bp 550,000; 1/9 bp |
Comparison of coding and non-coding gene requirements between two Salmonella serovars | Tn5 | - |
1 The same mutant library was used in these studies. A collection of studies to date utilizing transposon-insertion sequencing. Columns: (1) study reference, (2) date of publication, (3) organism mutagenized, (4) number of mutants generated, (5) number of unique insertion sites recovered from sequencing, (6) brief description of the application, (7) transposon used, (8) method name coined, if any.
Transposon mutagenesis
Most studies have used either Tn5 or mariner transposon derivatives. Tn5 originated as a bacterial transposon which has been adapted for laboratory use. Large-scale studies have shown that Tn5, while not showing any strong preference for regional GC-content, do have a weak preference for a particular insertion motif.12-14 Transposon-insertion sequencing studies performed with Tn5 transposons in S. enterica serovars have reported a slight bias toward AT-rich sequence regions.6,15 However, this preference does not appear to be a major obstacle to analysis given the extremely high insertion densities obtained with this transposon6,15,16(see Table 1). Additionally, Tn5 has been shown to be active in a wide range of bacterial species, though the number of transformants obtained can vary significantly depending on the transformation efficiency of the host.
Mariner/Himar1 transposons on the other hand originate from eukaryotic hosts and have an absolute requirement for TA bases at their integration site,17,18 with no other known bias besides a possible preference for bent DNA.17 This can be a disadvantage in that it limits the number of potential insertion sites, particularly in GC-rich sequence. However, this specificity can also be used in the prediction of gene essentiality in near-saturated libraries: as every potential integration site is known and the probability of integration at any particular site can be assumed to be roughly equal, it is straight-forward to calculate the probability that any particular region lacks insertions by chance. Himar1 transposition can also be conducted in vitro in the absence of any host factors,19 and inserted transposons can then be transferred to the genomes of naturally transformable bacteria through homologous recombination.20 This can be advantageous when working with naturally transformable bacteria with poor electroporation efficiency.8,9 It is worth noting that Tn5 is also capable of transposition in vitro,21 and could potentially be used to increase insertion density and, hence, the resolution of the assay, particularly in GC-rich genomic regions.
Pool construction
Once mutants have been constructed, they are plated on an appropriate selective media for the transposon chosen, and colonies are counted, picked and pooled. A disadvantage of this is that the mutants must be recreated for follow up or validation studies. Goodman et al. introduced a clever way around this in the INSeq protocol: by individually archiving mutants, then sequencing combinatorial mutant pools, it is possible to uniquely characterize 2n insertion mutants by sequencing only n pools.7 Each mutant is labeled with a unique binary string that indicates which pools it has been added to. These binary strings can then be reconstructed for each insertion observed in these pools by recording their presence or absence in sequencing data, providing a unique pattern relating insertions to archived mutants. The authors control false identifications due to errors in sequencing by requiring that each binary label have a minimum edit distance to every other label, allowing for a robust association of labels with insertions despite sometimes noisy sequencing data. As a proof of concept, the authors were able to identify over 7,000 Bacteroides thetaiotaomicron mutants from only 24 sequenced pools. This effectively uses methods for the generation of random transposon pools to rapidly generate defined mutant arrays, though it is heavily dependent on liquid-handling robotics.
Enrichment of transposon-insertion junctions
Once pools have been constructed, they are grown in either selective or permissive conditions, depending on the experiment, and then genomic DNA is extracted. Fragmentation proceeds either through restriction digestion in the case of transposons modified to contain appropriate sites7,9,10or via physical shearing,6,8 then sequencing adapters are ligated to the resulting fragments. PCR is performed on these fragments using a transposon-specific primer and a sequencing adaptor-specific primer to enrich for fragments spanning the transposon-genomic DNA junction. Some protocols purify fragments containing transposon insertions using biotinylated primers10,11or PAGE7 before and/or after PCR enrichment. The purification step from the Tn-seq Circle protocol is particularly unusual in that restriction-digested fragments containing transposon sequence are circularized before being treated with an exonuclease that digests all fragments without transposon insertions, theoretically completely eliminating background.10 Given the success of protocols that do not include a purification step and the lack of systematic comparisons, it is currently unclear whether including one provides any major advantages.
Reproducibility, Accuracy and Concordance with Previous Methods
A number of studies have looked at the reproducibility of transposon-insertion sequencing. Multiple studies using different protocol variations have repeatedly shown extremely high reproducibility in the number of insertions per gene (correlations of ~90%) in replicates of the same library grown and sequenced independently,7,9,10 and good reproducibility (correlations between 70–90%) in independently constructed non-saturated libraries.9,22 Van Opijnen and Camilli22 compared traditional 1 × 1 competition experiments between wild-type and mutant Streptococcus pneumoniae to results obtained by transposon-insertion sequencing and showed that there was no significant difference in results over a range of tested conditions.
The accuracy of transposon-insertion sequencing in determining library composition has also been assessed. Zhang et al. constructed a library of identified transposon-insertion mutants in known relative quantities, and then were able to recover the relative mutant prevalence with transposon-insertion sequencing.23 Additionally, by estimating the number of PCR templates prior to enrichment, this study showed that there is a high correlation between enrichment input and sequencing output.
Two studies have evaluated concordance between results obtained with transposon-insertion sequencing and microarray monitoring of transposon insertions in order to demonstrate the enhanced accuracy and dynamic range of sequencing over previous methods. In the first, 19 libraries of 95 enterohemorrhagic Escherichia coli (EHEC) transposon mutants that had previously been screened in cattle using signature-tagged mutagenesis (STM), were pooled and re-evaluated using the TraDIS protocol.24 The original STM study had identified 13 insertions in 11 genes attenuating intestinal colonization in a type III secretion system located in the locus of enterocyte effacement (LEE).25 By applying sequencing to the same samples, an additional 41 mutations in the LEE were identified, spanning a total of 21 genes. Additional loci outside the LEE, which have been previously implicated in intestinal colonization but had not been detected by STM, were also reported by TraDIS.
The second study re-evaluated genes required for optimal growth determined by TraSH in Mycobacterium tuberculosis.26,27 The greater dynamic range of sequencing as compared with microarrays allowed easier discrimination between insertions that were nonviable and those that were only significantly underrepresented. The authors estimate that genes called as required by sequencing in their study are at least 100-fold underrepresented in the pool. In comparison, the threshold in the previous microarray experiment reported genes that had log probe ratios at least 5-fold lower than average between transposon-flanking DNA hybridization and whole genomic DNA hybridization. Additionally, the nucleotide-resolution of insertion sequencing allowed the authors to identify genes which had required regions, likely corresponding to required protein domains,23 but which tolerated insertions in other regions. Altogether, the authors increase the set of genes predicted to be required for growth in laboratory conditions in M. tuberculosis by more than 25% (from 614 to 774).
Gene Requirements
The earliest application of transposon-insertion sequencing was to determine the minimal set of genes necessary for the survival of Mycoplasma.5 This essential genome is of great interest to synthetic and systems biology where it is seen as a foundation for engineering cell metabolism, and in infection biology and medicine where it is seen as a promising target for therapies. However, it is important to remember that “essentiality” is always relative to growth conditions: a biosynthetic gene that is non-essential in a growth medium supplying a particular nutrient may become essential in a medium that lacks it. Traditionally, gene essentiality has been determined in clonal populations;1,28,29 since the high-throughput transposon sequencing protocols described here necessarily contain a short period of competitive growth before DNA extraction, many of these studies prefer to refer to the “required” genome for the particular conditions under evaluation.
Because of this short period of competitive growth, and because many otherwise required genes tolerate insertions in their terminus7,27,30or outside essential domains,23 the determination of required genomic regions is not completely straight-forward and a number of approaches have been taken to counter this. These include only calling genes completely lacking insertions as required,9 determining a cut-off based on the empirical or theoretical distribution of gene-wise insertion densities.6,15,27,30 Additionally, windowed methods have been developed which can be used to identify essential regions in the absence of gene annotation,23,31 and have had success in identifying required protein domains, promoter regions and non-coding RNAs (ncRNAs). The organisms that have been evaluated for gene requirements under standard laboratory conditions are summarized in Table 1.
In agreement with previous studies,1,28 many required genes identified by transposon-insertion sequencing are involved in fundamental biological processes such as cell division, DNA replication, transcription and translation,6,7,15,27and many of these requirements appear to be conserved between genera and classes.15,16 However, a recent study defining required gene sets in Salmonella serovars has found that phage repressors, necessary for maintaining the lysogenic state of the prophage, are also required,15 even though mobile genetic elements such as phage are usually considered part of the accessory genome. This study also highlights the need for temperance when interpreting the results of high-throughput assays of gene requirements. For example, many genes in Salmonella Pathogenicity Island 2 (SPI-2) did not exhibit transposon-insertions, despite clear evidence from directed knockouts showing that these genes are non-essential for viability or growth. Under laboratory conditions, SPI-2 is silenced by the nucleoid-forming protein H-NS,32,33 which acts by oligermerizing along silenced regions of DNA blocking RNA polymerase access. A previous study has shown that transposon insertion “cold spots” can be caused by competition between high-density proteins and transposases for DNA.34 This suggests that H-NS may be restricting transposase access to DNA, though this has not previously been observed in transposon-insertion sequencing data, and will require additional work to confirm.
Defining Conditional Gene Requirements
One of the most valuable applications of the transposon-insertion sequencing method is the ability to identify genes important in a condition of interest, by comparing differences in the numbers of sequencing reads from input (control) mutant pools to output (test) pools that have been subject to passaging in a certain growth condition. Insertion counts are compared from cells in the input pool and those after passage, thereby identifying genes that either enhance or detract from survival and/or growth in the given condition, defined by decreased or increased insertion frequency, respectively. A further application of this method involves comparing insertions between biologically linked conditions, such as cellular stresses or different stages of a murine infection, to gain insight into complex systems.22
So far, transposon-insertion sequencing has been used to investigate a number of interesting biological questions: bile tolerance in S. Typhi6 and S. Typhimurium,35 bacteriophage infection of S. Typhi,36 antibiotic resistance in Pseudomonas aeruginosa,10 cholesterol utilization in M. tuberculosis27 and a number of stress and nutrient conditions in S. pneumoniae.22 Transposon-insertion sequencing of populations passed through murine models have been used to assess genes required to establish the gut commensal B. thetaiotaomicron in its niche,7 for Hemophilus influenzae infection,8 as well as S. pneumoniae responses to two in vivo niches, the lung and nasopharynx.22 A further extension of the method examined double mutant libraries, that is transposon mutant libraries generated in a defined deletion background, to tease apart complex networks of regulatory genes.9
Two studies in particular illustrate the power of using transposon-insertion sequencing to identify conditionally required genes. In the first, Goodman et al. set out to determine the genes necessary for the establishment of the commensal B. thetaiotaomicron in a murine model.7 First, the growth requirements of transposon mutant populations in the cecum of germ-free mice was assessed, and genes required for growth in monoassociation with the host were found to be enriched in functions such as energy production and amino acid metabolism. By further comparing monoassociated transposon mutant libraries with those grown in the presence of three defined communities of human gut-associated bacteria, the authors identified a locus upregulated by low levels of vitamin B12 that is only required in the absence of other bacteria capable of synthesizing B12. This showed that the gene requirements of any particular bacterium in the gut are at least partially dependent on the metabolic capabilities of the entire community and emphasizes the importance of testing in vivo conditions to complement in vitro study.
The second study, conducted by van Opijnen and Camilli, aimed to map the genetic networks involved in a range of cellular stress responses in S. pneumoniae.22 Seventeen in vitro conditions were tested, including: pH, nutrient limitation, temperature, antibiotic, heavy metal and hydrogen peroxide stress. Approximately 6% of disrupted genes resulted in increased fitness in some condition, suggesting that some genes are maintained despite being detrimental to the organism under particular conditions. These would be interesting candidates for further functional and evolutionary study, as the maintenance of these genes is presumably highly dependent on the conditions the bacteria faces, and may have implications for our understanding of e.g., gene loss in the process of bacterial host adaptation.37 Two additional in vivo experiments were performed in a murine model, where cells were recovered from the lung and nasopharynx. Combining this data, over 1,800 genotype-phenotype genetic interactions were identified. These interactions were mapped and pathways identified. Between the two in vivo niches, certain stress response pathways were markedly different. For example, temperature stress produced a distinct response in the lung, compared with the nasopharyanx, which is perhaps to be expected as temperature varies greatly between these two sites. By further examining sub-pathways required in the two different niches and comparing them to in vitro requirements, the authors were able to draw conclusions regarding the condition S. pneumoniae faces when establishing an infection. This comprehensive mapping of genotype-phenotype relationships will serve as an important atlas for further studies.
Monitoring ncRNA Contributions to Fitness
To date, four studies have used transposon-insertion sequencing to examine the contribution of non-coding RNAs (ncRNAs) and other non-coding regions to organismal fitness (see Table 1). Two of these examined requirements for non-coding regions in the relatively under-explored bacterial species Caulobacter crescentus16 and M. tuberculosis.23 Both utilized analytical techniques that allowed for the identification of putative required regions in the absence of genome annotation. Twenty-seven small RNAs (sRNAs) had previously been detected in C. crescentus;38 six were found to be depleted in transposon insertions indicating an important role in basic cellular processes. Additionally, the well-characterized ncRNAs tmRNA and RNaseP, as well as 29 non-redundant tRNAs, were found to be required. An additional 90 unannotated non-disruptable regions were identified throughout the genome, implying an abundance of unexplored functional non-coding sequence.
While the non-coding transcripts of M. tuberculosis have been explored more thoroughly than those of C. crescentus, most remain functionally uncharacterized, though there are hints that some of these may be involved in pathogenicity.39 Using a mariner transposon-based assay and a windowed statistical analysis that accounted for the distribution of potential TA integration sites, 35 intergenic regions were identified as putatively required in the M. tuberculosis genome.23 In common with the C. crescentus study, the RNA component of RNase P, required for the maturation of tRNAs and tmRNA, involved in the freeing of stalled ribosomes, were identified as required (Fig. 2A) together with 10 non-redundant tRNAs and potential promoter regions. However, due to the lower overall insertion density and lack of TA sites in some GC-rich regions, there were some regions that could not be assayed and the resolution was limited to 250 bases.

Figure 2. Applications of transposon-insertion sequencing to non-coding RNAs. (A) Plots of genomic regions in Mycobacterium tuberculosis containing the required non-coding RNAs RNase P (top) and tmRNA (bottom). Tracks, from top to bottom, 1. Histogram of insertion counts, 2. Comprehensive heat-map of requirement of 500-bp windows, 3. Position of annotated genes, 4. Position of TA dinucleotide sites, 5. Position of non-coding RNA. Reproduced from reference 23. (B) 1 × 1 competition assays validate attenuating Streptococcus pneumoniae sRNA mutants identified by transposon-insertion sequencing. Mice were infected with defined deletions of sRNAs identified as attenuating by Tn-seq and wild-type S. pneumoniae TIGR4 at the body site indicated and bacterial densities were compared 24 h post-infection. These plots show the derived competitive index in blood (top) and the nasopharnyx (bottom). Each point represents the result of a competition experiment between an sRNA deletion mutant and wild-type TIGR4. A competitive index of 1 indicates equivalent numbers of mutants and wild-type were recovered. Modified from reference 46.
A recent study has examined ncRNA requirements in the S. enterica serovars Typhi and Typhimurium.15 Using the tRNAs as a model set of ncRNAs, this study showed that the high transposon insertion density achieved by the TraDIS protocol is capable of assaying the requirement for genomic regions as small as 70‒80 bases. S. enterica, together with the closely related E. coli, has served as a model organism for the discovery and elucidation of ncRNA function, and extensive annotations of non-coding transcripts are available.40-44 As a result, this study was able to assay approximately 300 non-coding regions with evidence for function or transcription. Among the ncRNAs identified as required were RNase P; the RNA component of the signal recognition particle, involved in targeting proteins to the plasma membrane; and a number of known autoregulatory ribosomal protein leader sequences,45 as well as providing evidence for a novel leader sequence, StyR-8,43 which appears to be involved in the autoregulation of the rpmB gene. In total, this study identified 15 confirmed and putative ncRNAs required for robust competitive growth on rich media in both serovars, including a number of known sRNAs involved in stress response.
A particularly exciting study has been conducted in S. pneumoniae TIGR4 combining RNA-seq with transposon-insertion sequencing.46 To identify sRNA loci, the authors first sequenced size-selected RNA from the wild-type and three two-component system knockouts, identifying 89 putative sRNAs, 56 of which were novel. Fifteen of these candidates, selected on the basis of high expression and low predicted folding free energy, were assayed for their ability to establish invasive disease in a murine model. Of these, eight sRNA deletions showed a significant attenuation of disease. To more broadly establish the roles of sRNAs in infecting particular organs, transposon insertion libraries were administered directly to the nasopharnyx, lungs or blood of mice, and bacteria were harvested following disease progression. Twenty-six, 28 and 18 sRNAs were found to attenuate infection in the nasopharnyx, lung and blood, respectively. These results were then validated with targeted deletions of 11 sRNAs (Fig. 2B). In addition to establishing the role of sRNAs in S. pneumoniae virulence, this study illustrated the power of combining RNA-seq and transposon-insertion sequencing to rapidly assign phenotypes to non-coding sequences.
Limitations
In this review, we have largely focused on the potential of transposon-insertion sequencing. However, this technology does have a number of important limitations, which we collect here and summarize in Table 2. As discussed previously, requirements for particular nucleotides at insertion sites, such as the TA required by the Mariner transposon, or preference for certain sequence composition, such as the AT bias exhibited by Tn5, can limit the density of observed insertions in certain genomic regions. This may impact any downstream analysis, and can potentially bias results, particularly the determination of gene requirements. Even if this bias has been accounted for, transposon-insertion screens will always over-predict gene requirements in comparison to targeted deletion libraries as discussed previously. However, this over-prediction can be controlled either through careful consideration of known insertion biases as in many Mariner-based studies, or by high-insertion densities, such as those achieved in several Tn5-based studies (Table 1). Once the library has been created, only regions that have accumulated insertions in the conditions of library creation will be able to be assayed for fitness effects in further conditions. This means that regions that lead to slow growth phenotypes when disrupted in standard laboratory conditions may be difficult to assay in other conditions. Additionally, the dynamic range of fitness effects detected will depend on the complexity of the input library(s). The absence of insertions may be a particular problem for assaying small genomic elements, such as sRNAs or short ORFs. Finally, the validation of hypotheses derived from transposon-insertion sequencing will require the construction of targeted deletions, as individual mutants cannot be recovered from pools unless specialized protocols have been followed during library construction (as in Goodman, 20097).
Table 2. Advantages and limitations of transposon-insertion sequencing.
| Advantages | Limitations |
|---|---|
| Library construction is extremely rapid in comparison to targeted deletion libraries. |
Requirements for particular nucleotides at transposon-insertion sites or insertion biases can limit resolution. |
| Gene requirements and fitness effects can be quickly assayed in a wide range of conditions. |
Determination of gene essentiality is dependent on insertion density, and is less conclusive than targeted gene deletion in clonal populations. |
| The precise location of transposon insertions can be determined due to the nucleotide resolution of high-throughput sequencing. |
Only genomic regions that tolerate insertions under the conditions of library creation may be assayed for fitness effects in further conditions. |
| Wide dynamic range compared with older microarray-based technologies. |
The dynamic range for fitness effects is dependent on mutant abundance in the initial library and may be limiting for some genes. |
| Requirements and fitness effects of genomic regions can be determined independently of annotation. | Mutants must be reconstructed for follow-up experiments in the absence of specialized protocols and robotics (see e.g., Goodman, 20097). |
The Future of Transposon-Insertion Sequencing
Transposon-insertion sequencing is a robust and powerful technique for the rapid connection of genotype to phenotype in a wide range of bacterial species. Already, a number of studies have demonstrated the effectiveness of this method and the results have been far-reaching: enhancing our understanding of basic gene functions, establishing requirements for colonization and infection, mapping complex metabolic pathways and exploring non-coding genomic “dark matter.” Due to the range of potential applications of transposon-insertion sequencing, along with the decreasing cost and growing accessibility of next-generation sequencing, we believe that this method will become increasingly common in the near future.
A number of bacterial species have already been subjected to transposon-insertion sequencing (Table 1). Microarray-based approaches to monitoring transposon mutant libraries have even been applied to eukaryotic systems,47 and similarly transposon-insertion sequencing can potentially be applied to any system where the creation of large-scale transposon mutant libraries is technologically feasible. Recently, the Genomic Encyclopedia of Bacteria and Archea (GEBA)48 has been expanding our knowledge of bacterial diversity through targeted genomic sequencing of underexplored branches of the tree of life. Applying transposon-insertion sequencing in a comparative manner15 across the bacterial phylogeny will provide an unprecedented view of the determinants for survival in diverse environments. While most transposon-insertion sequencing studies to date have focused on pathogenic bacteria, these techniques could also have applications in energy production, bioremediation and synthetic biology.
The combination of transposon-insertion sequencing with other high-throughput and computational methods is already proving to be fertile ground for enhancing our understanding of bacterial systems. For instance, by using transposon-insertion sequencing in a collection of relatively simple conditions combined with a computational pathway analysis, van Opijnen and Camilli were able to provide a holistic understanding of the genetic subsystems involved in a complex process such as S. pneumoniae pathogenesis.22 In the future, methods to assay phenotype in a high-throughput manner49,50may be combined with transposon-insertion sequencing to provide exhaustive simple genotype-phenotype associations with which to understand complex processes in a systems biology framework. We look forward to the adoption of these data sets by the community as an important tool for rapid hypothesis generation.
Acknowledgments
This work was supported by the Wellcome Trust (grant number WT098051). C.J.B. and A.K.C. were supported by the Medical Research Council (grant number S1710).
Disclosure of Potential Conflicts of Interest
L.B. authored one of the studies reviewed in this article.15 All of the authors are actively involved in studies using the TraDIS transposon-insertion sequencing methodology.
Footnotes
Previously published online: www.landesbioscience.com/journals/rnabiology/article/24765
References
- 1.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2:0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hobbs EC, Astarita JL, Storz G. Small RNAs and small proteins involved in resistance to cell envelope stress and acid shock in Escherichia coli: analysis of a bar-coded mutant collection. J Bacteriol. 2010;192:59–67. doi: 10.1128/JB.00873-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hensel M, Shea JE, Gleeson C, Jones MD, Dalton E, Holden DW. Simultaneous identification of bacterial virulence genes by negative selection. Science. 1995;269:400–3. doi: 10.1126/science.7618105. [DOI] [PubMed] [Google Scholar]
- 4.Mazurkiewicz P, Tang CM, Boone C, Holden DW. Signature-tagged mutagenesis: barcoding mutants for genome-wide screens. Nat Rev Genet. 2006;7:929–39. doi: 10.1038/nrg1984. [DOI] [PubMed] [Google Scholar]
- 5.Hutchison CA, Peterson SN, Gill SR, Cline RT, White O, Fraser CM, et al. Global transposon mutagenesis and a minimal Mycoplasma genome. Science. 1999;286:2165–9. doi: 10.1126/science.286.5447.2165. [DOI] [PubMed] [Google Scholar]
- 6.Langridge GC, Phan MD, Turner DJ, Perkins TT, Parts L, Haase J, et al. Simultaneous assay of every Salmonella Typhi gene using one million transposon mutants. Genome Res. 2009;19:2308–16. doi: 10.1101/gr.097097.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goodman AL, McNulty NP, Zhao Y, Leip D, Mitra RD, Lozupone CA, et al. Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host Microbe. 2009;6:279–89. doi: 10.1016/j.chom.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gawronski JD, Wong SM, Giannoukos G, Ward DV, Akerley BJ. Tracking insertion mutants within libraries by deep sequencing and a genome-wide screen for Haemophilus genes required in the lung. Proc Natl Acad Sci USA. 2009;106:16422–7. doi: 10.1073/pnas.0906627106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Opijnen T, Bodi KL, Camilli A. Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat Methods. 2009;6:767–72. doi: 10.1038/nmeth.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gallagher LA, Shendure J, Manoil C. Genome-scale identification of resistance functions in Pseudomonas aeruginosa using Tn-seq. MBio. 2011;2:e00315–10. doi: 10.1128/mBio.00315-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goodman AL, Wu M, Gordon JI. Identifying microbial fitness determinants by insertion sequencing using genome-wide transposon mutant libraries. Nat Protoc. 2011;6:1969–80. doi: 10.1038/nprot.2011.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shevchenko Y, Bouffard GG, Butterfield YS, Blakesley RW, Hartley JL, Young AC, et al. Systematic sequencing of cDNA clones using the transposon Tn5. Nucleic Acids Res. 2002;30:2469–77. doi: 10.1093/nar/30.11.2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adey A, Morrison HG, Asan, Xun X, Kitzman JO, Turner EH, et al. Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biol. 2010;11:R119. doi: 10.1186/gb-2010-11-12-r119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Green B, Bouchier C, Fairhead C, Craig NL, Cormack BP. Insertion site preference of Mu, Tn5, and Tn7 transposons. Mob DNA. 2012;3:3. doi: 10.1186/1759-8753-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barquist L, Langridge GC, Turner DJ, Phan MD, Turner AK, Bateman A, et al. A comparison of dense transposon insertion libraries in the Salmonella serovars Typhi and Typhimurium. Nucleic Acids Res. 2013 doi: 10.1093/nar/gkt148. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Christen B, Abeliuk E, Collier JM, Kalogeraki VS, Passarelli B, Coller JA, et al. The essential genome of a bacterium. Mol Syst Biol. 2011;7:528. doi: 10.1038/msb.2011.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lampe DJ, Grant TE, Robertson HM. Factors affecting transposition of the Himar1 mariner transposon in vitro. Genetics. 1998;149:179–87. doi: 10.1093/genetics/149.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rubin EJ, Akerley BJ, Novik VN, Lampe DJ, Husson RN, Mekalanos JJ. In vivo transposition of mariner-based elements in enteric bacteria and mycobacteria. Proc Natl Acad Sci USA. 1999;96:1645–50. doi: 10.1073/pnas.96.4.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lampe DJ, Churchill ME, Robertson HM. A purified mariner transposase is sufficient to mediate transposition in vitro. EMBO J. 1996;15:5470–9. [PMC free article] [PubMed] [Google Scholar]
- 20.Johnsborg O, Eldholm V, Håvarstein LS. Natural genetic transformation: prevalence, mechanisms and function. Res Microbiol. 2007;158:767–78. doi: 10.1016/j.resmic.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 21.Goryshin IY, Reznikoff WS. Tn5 in vitro transposition. J Biol Chem. 1998;273:7367–74. doi: 10.1074/jbc.273.13.7367. [DOI] [PubMed] [Google Scholar]
- 22.van Opijnen T, Camilli A. A fine scale phenotype-genotype virulence map of a bacterial pathogen. Genome Res. 2012;22:2541–51. doi: 10.1101/gr.137430.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang YJ, Ioerger TR, Huttenhower C, Long JE, Sassetti CM, Sacchettini JC, et al. Global assessment of genomic regions required for growth in Mycobacterium tuberculosis. PLoS Pathog. 2012;8:e1002946. doi: 10.1371/journal.ppat.1002946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eckert SE, Dziva F, Chaudhuri RR, Langridge GC, Turner DJ, Pickard DJ, et al. Retrospective application of transposon-directed insertion site sequencing to a library of signature-tagged mini-Tn5Km2 mutants of Escherichia coli O157:H7 screened in cattle. J Bacteriol. 2011;193:1771–6. doi: 10.1128/JB.01292-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dziva F, van Diemen PM, Stevens MP, Smith AJ, Wallis TS. Identification of Escherichia coli O157 : H7 genes influencing colonization of the bovine gastrointestinal tract using signature-tagged mutagenesis. Microbiology. 2004;150:3631–45. doi: 10.1099/mic.0.27448-0. [DOI] [PubMed] [Google Scholar]
- 26.Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 27.Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, Sassetti CM. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 2011;7:e1002251. doi: 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jacobs MA, Alwood A, Thaipisuttikul I, Spencer D, Haugen E, Ernst S, et al. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc Natl Acad Sci USA. 2003;100:14339–44. doi: 10.1073/pnas.2036282100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Glass JI, Assad-Garcia N, Alperovich N, Yooseph S, Lewis MR, Maruf M, et al. Essential genes of a minimal bacterium. Proc Natl Acad Sci USA. 2006;103:425–30. doi: 10.1073/pnas.0510013103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zomer A, Burghout P, Bootsma HJ, Hermans PW, van Hijum SA. ESSENTIALS: software for rapid analysis of high throughput transposon insertion sequencing data. PLoS One. 2012;7:e43012. doi: 10.1371/journal.pone.0043012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DeJesus MA, Zhang YJ, Sassetti CM, Rubin EJ, Sacchettini JC, Ioerger TR. Bayesian analysis of gene essentiality based on sequencing of transposon insertion libraries. Bioinformatics. 2013;29:695–703. doi: 10.1093/bioinformatics/btt043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lucchini S, Rowley G, Goldberg MD, Hurd D, Harrison M, Hinton JC. H-NS mediates the silencing of laterally acquired genes in bacteria. PLoS Pathog. 2006;2:e81. doi: 10.1371/journal.ppat.0020081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Navarre WW, Porwollik S, Wang Y, McClelland M, Rosen H, Libby SJ, et al. Selective silencing of foreign DNA with low GC content by the H-NS protein in Salmonella. Science. 2006;313:236–8. doi: 10.1126/science.1128794. [DOI] [PubMed] [Google Scholar]
- 34.Manna D, Porwollik S, McClelland M, Tan R, Higgins NP. Microarray analysis of Mu transposition in Salmonella enterica, serovar Typhimurium: transposon exclusion by high-density DNA binding proteins. Mol Microbiol. 2007;66:315–28. doi: 10.1111/j.1365-2958.2007.05915.x. [DOI] [PubMed] [Google Scholar]
- 35.Khatiwara A, Jiang T, Sung SS, Dawoud T, Kim JN, Bhattacharya D, et al. Genome scanning for conditionally essential genes in Salmonella enterica Serotype Typhimurium. Appl Environ Microbiol. 2012;78:3098–107. doi: 10.1128/AEM.06865-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pickard D, Kingsley RA, Hale C, Turner K, Sivaraman K, Wetter M, et al. A genomewide mutagenesis screen identifies multiple genes contributing to Vi capsular expression in Salmonella enterica serovar Typhi. J Bacteriol. 2013;195:1320–6. doi: 10.1128/JB.01632-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Toft C, Andersson SG. Evolutionary microbial genomics: insights into bacterial host adaptation. Nat Rev Genet. 2010;11:465–75. doi: 10.1038/nrg2798. [DOI] [PubMed] [Google Scholar]
- 38.Landt SG, Abeliuk E, McGrath PT, Lesley JA, McAdams HH, Shapiro L. Small non-coding RNAs in Caulobacter crescentus. Mol Microbiol. 2008;68:600–14. doi: 10.1111/j.1365-2958.2008.06172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arnvig K, Young D. Non-coding RNA and its potential role in Mycobacterium tuberculosis pathogenesis. RNA Biol. 2012;9:427–36. doi: 10.4161/rna.20105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kröger C, Dillon SC, Cameron AD, Papenfort K, Sivasankaran SK, Hokamp K, et al. The transcriptional landscape and small RNAs of Salmonella enterica serovar Typhimurium. Proc Natl Acad Sci USA. 2012;109:E1277–86. doi: 10.1073/pnas.1201061109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burge SW, Daub J, Eberhardt R, Tate J, Barquist L, Nawrocki EP, et al. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res. 2013;41(Database issue):D226–32. doi: 10.1093/nar/gks1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raghavan R, Groisman EA, Ochman H. Genome-wide detection of novel regulatory RNAs in E. coli. Genome Res. 2011;21:1487–97. doi: 10.1101/gr.119370.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chinni SV, Raabe CA, Zakaria R, Randau G, Hoe CH, Zemann A, et al. Experimental identification and characterization of 97 novel npcRNA candidates in Salmonella enterica serovar Typhi. Nucleic Acids Res. 2010;38:5893–908. doi: 10.1093/nar/gkq281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sittka A, Sharma CM, Rolle K, Vogel J. Deep sequencing of Salmonella RNA associated with heterologous Hfq proteins in vivo reveals small RNAs as a major target class and identifies RNA processing phenotypes. RNA Biol. 2009;6:266–75. doi: 10.4161/rna.6.3.8332. [DOI] [PubMed] [Google Scholar]
- 45.Fu Y, Deiorio-Haggar K, Anthony J, Meyer MM. Most RNAs regulating ribosomal protein biosynthesis in Escherichia coli are narrowly distributed to Gammaproteobacteria. Nucleic Acids Res. 2013;41:3491–503. doi: 10.1093/nar/gkt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mann B, van Opijnen T, Wang J, Obert C, Wang YD, Carter R, et al. Control of virulence by small RNAs in Streptococcus pneumoniae. PLoS Pathog. 2012;8:e1002788. doi: 10.1371/journal.ppat.1002788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ross-Macdonald P, Coelho PS, Roemer T, Agarwal S, Kumar A, Jansen R, et al. Large-scale analysis of the yeast genome by transposon tagging and gene disruption. Nature. 1999;402:413–8. doi: 10.1038/46558. [DOI] [PubMed] [Google Scholar]
- 48.Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova NN, et al. A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature. 2009;462:1056–60. doi: 10.1038/nature08656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bochner BR. Global phenotypic characterization of bacteria. FEMS Microbiol Rev. 2009;33:191–205. doi: 10.1111/j.1574-6976.2008.00149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nichols RJ, Sen S, Choo YJ, Beltrao P, Zietek M, Chaba R, et al. Phenotypic landscape of a bacterial cell. Cell. 2011;144:143–56. doi: 10.1016/j.cell.2010.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brutinel ED, Gralnick JA. Anomalies of the anaerobic tricarboxylic acid cycle in Shewanella oneidensis revealed by Tn-seq. Mol Microbiol. 2012;86:273–83. doi: 10.1111/j.1365-2958.2012.08196.x. [DOI] [PubMed] [Google Scholar]
- 52.Klein BA, Tenorio EL, Lazinski DW, Camilli A, Duncan MJ, Hu LT. Identification of essential genes of the periodontal pathogen Porphyromonas gingivalis. BMC Genomics. 2012;13:578. doi: 10.1186/1471-2164-13-578. [DOI] [PMC free article] [PubMed] [Google Scholar]