

Abstract
A key step toward the discovery of a gene related to a trait is the finding of an association between the trait and one or more haplotypes. Haplotype analyses can also provide critical information regarding the function of a gene; however, when unrelated subjects are sampled, haplotypes are often ambiguous because of unknown linkage phase of the measured sites along a chromosome. A popular method of accounting for this ambiguity in case-control studies uses a likelihood that depends on haplotype frequencies, so that the haplotype frequencies can be compared between the cases and controls; however, this traditional method is limited to a binary trait (case vs. control), and it does not provide a method of testing the statistical significance of specific haplotypes. To address these limitations, we developed new methods of testing the statistical association between haplotypes and a wide variety of traits, including binary, ordinal, and quantitative traits. Our methods allow adjustment for nongenetic covariates, which may be critical when analyzing genetically complex traits. Furthermore, our methods provide several different global tests for association, as well as haplotype-specific tests, which give a meaningful advantage in attempts to understand the roles of many different haplotypes. The statistics can be computed rapidly, making it feasible to evaluate the associations between many haplotypes and a trait. To illustrate the use of our new methods, they are applied to a study of the association of haplotypes (composed of genes from the human-leukocyte-antigen complex) with humoral immune response to measles vaccination. Limited simulations are also presented to demonstrate the validity of our methods, as well as to provide guidelines on how our methods could be used.
Introduction
The recent sequencing of the human genome (International Human Genome Sequencing Consortium 2001; Venter et al. 2001) provides an immense amount of data that has strong potential to change the way we understand common human diseases and ultimately may improve their diagnosis, prognosis, and treatment (Collins 1999). Nonetheless, the deciphering of complex genetic mechanisms poses a significant challenge. It is anticipated that, by use of improved genetic-marker maps, particularly those with single-nucleotide polymorphisms (SNPs) (The International SNP Map Working Group 2001), association studies will improve our ability to detect susceptibility alleles for common complex diseases (Risch 2000; Cardon and Bell 2001; Schork et al. 2001). Furthermore, it is likely that haplotypes, which are specific combinations of nucleotides on the same chromosome, will provide more information on the complex relationship between DNA variation and phenotypes than any single SNP can provide (Stephens et al. 2001a).
Haplotype analyses tend to focus either on fine mapping, to localize a susceptibility gene via linkage disequilibrium (LD) with adjacent genetic markers, or on the influence that the entire haplotype has on the trait. For simple diseases, numerous statistical methods have been proposed for Mendelian LD fine mapping (Hastbacka et al. 1992; Kaplan et al. 1995; Terwilliger 1995; Devlin et al. 1996; Guo 1997; Xiong and Guo 1997; Lazzeroni 1998; Rannala and Slatkin 1998), most of which require observed disease and normal chromosomes (i.e., haplotypes composed of marker and disease-locus alleles) and are based on patterns of pairwise LD measures between the genetic markers and the disease gene. However, it is difficult to speculate about how well the current LD-mapping strategies will work for complex diseases.
In contrast to fine mapping, classical genetics has demonstrated that the phenotypic effect of several mutations at different sites within a gene can depend on whether the mutations occur on the same chromosome (in cis position, as a haplotype) or on opposite homologous chromosomes (in trans position). There is strong evidence that several mutations in cis position within a single gene can interact to create a “super allele” that has a large effect on the observed phenotype. Some examples in humans include a gene that influences intestinal lactase activity (Hollox et al. 2001); a gene responsible for human lipoprotein lipase (Clark et al. 1998); the HPC2/ELAC2 gene, which increases the risk for prostate cancer (Tavtigian et al. 2001); and a gene that influences actions of catecholamines, which influence bronchodilation and, hence, asthma (Drysdale 2000). The biologic explanation for these haplotype effects is that several mutations in a gene cause several amino acid changes in the ultimate protein product, and the joint effect of these amino acid changes can have a much larger influence on the function of the protein product than any single amino acid change. This emphasizes the importance of examining candidate genes by SNP haplotyping.
For unrelated subjects, haplotypes can be directly observed whenever there is no more than one heterozygous site. If there are H heterozygous sites, then the number of pairs of haplotypes that are consistent with the observed marker phenotypes is 2H-1. Although the observations on codominant genetic markers are often referred to as “genotypes,” we shall refer to them as “marker phenotypes,” reserving the term “genotype” for when linkage phase is known. The traditional method to determine haplotypes is either pedigree analysis or molecular haplotyping (limited to short DNA sequences) (Michalatos-Beloin et al. 1996). Both of these methods require enormous work either to collect a sufficient number of pedigree members or to perform the necessary laboratory work. Although new genetic technology (e.g., conversion technology [Yan et al. 2000]) may improve molecular haplotyping, the current methods are not adequate for large-scale epidemiological studies of human traits. To account for ambiguous haplotypes among unrelated subjects, several algorithms—including a parsimony algorithm (Clark 1990), a Bayesian population genetic model that uses coalescent theory (Stephens et al. 2001b; Zhang et al. 2001), and maximum likelihood (Terwilliger and Ott 1994; Excoffier and Slatkin 1995; Hawley and Kidd 1995; Long et al. 1995)—have been proposed. An advantage of the likelihood approach is that, in addition to the estimated haplotype frequencies, the posterior probabilities of the pairs of haplotypes that are consistent with the observed marker phenotypes can be computed for each subject. Our methods depend on the likelihood approach and use the posterior probabilities to account for haplotype ambiguities.
For case-control studies with unrelated subjects, haplotype frequencies are often compared between diseased cases and unaffected controls by use of a likelihood-ratio statistic. The expectation-maximization (EM) algorithm is used to maximize the log-likelihood for the pool of all subjects, 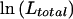 , and then, separately, that for cases,
, and then, separately, that for cases, 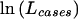 , and that for controls,
, and that for controls, 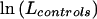 . The likelihood-ratio statistic is then
. The likelihood-ratio statistic is then 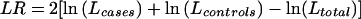 , which has an approximate χ2 distribution. For sparse data, empirical P values may be more reliable. However, maximization of the ln(L) is computer intensive, which can limit computation of empirical P values. Furthermore, this method does not provide statistical evaluations of individual haplotypes, nor does it readily generalize to other types of traits, such as quantitative traits.
, which has an approximate χ2 distribution. For sparse data, empirical P values may be more reliable. However, maximization of the ln(L) is computer intensive, which can limit computation of empirical P values. Furthermore, this method does not provide statistical evaluations of individual haplotypes, nor does it readily generalize to other types of traits, such as quantitative traits.
Our alternative approach is based on efficient score statistics, which provide both global tests and haplotype-specific tests. By use of generalized linear models (GLMs), we generalize our methods to a wide variety of traits and discuss ways to compute empirical P values and to score haplotypes and multilocus genotypes. Our new methods are illustrated by application to a study of the association of HLA haplotypes with measles vaccination response, and limited simulations are provided to illustrate some of the statistical properties of our methods.
Statistical Methods
GLMs
To clarify our approach, we first derive score statistics for situations in which the underlying genotype is not ambiguous (e.g., when a single codominant marker locus is evaluated or when haplotypes can be directly measured). Furthermore, we take a general approach so that measured environmental covariates can be included to develop score statistics for the genetic markers, adjusted for environmental factors. Let y denote a measured trait, let Xe denote a vector of measured environmental factors, including the intercept as the first element, and let Xg denote a vector of numerical codes for the marker genotype, g. We assume that the covariates influence the mean of the trait and not the scale, so that their effects can be summarized by a function of the linear predictor η=X′eα+X′gβ, where α denotes the regression parameters for the intercept and environmental factors and where β represents the effect of the genotype on the trait. We derive score statistics to test the null hypothesis of no association of the trait with the genotype, Ho:β=0.
To present some background on GLMs, we concatenate the covariates and their regression terms into single vectors, so that Z=(Xe|Xg) and γ=(α|β). The likelihood of a subject's trait y given the vector Z can be expressed as a GLM for exponential family data (McCullagh and Nelder 1983) according to
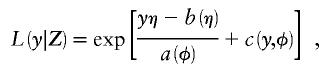 |
where a, b, and c are known functions (special cases of which are discussed later), φ is the dispersion parameter, and η=h(Z′γ) for an arbitrary function 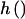 ; for our exposition, we assume canonical link functions such that η=Z′γ. The mean trait value is given by
; for our exposition, we assume canonical link functions such that η=Z′γ. The mean trait value is given by 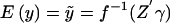 , so that the link function f is given by
, so that the link function f is given by 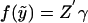 .
.
Score Tests in Absence of Ambiguity: Single-Locus or Measured Haplotypes
When the underlying genotype is not ambiguous, the score statistic for the vector Z is
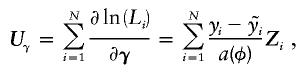 |
where 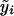 is the fitted value for the ith subject according to its covariate vector Zi, γ is the regression parameter, and N is the number of subjects. After regressing the trait only on the environmental covariates, to determine
is the fitted value for the ith subject according to its covariate vector Zi, γ is the regression parameter, and N is the number of subjects. After regressing the trait only on the environmental covariates, to determine 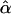 , we set β=0 and use
, we set β=0 and use 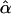 to determine
to determine 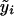 , so that
, so that 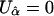 . Then, the score statistic for the genetic markers, adjusted for environmental covariates, is
. Then, the score statistic for the genetic markers, adjusted for environmental covariates, is
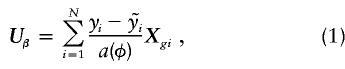 |
which is a measure of the covariation of the residuals (from the regression on environmental factors) with the genotype code.
The variance of Uβ under the null hypothesis, which accounts for the adjustment for the environmental covariates, is determined by Vβ=Vββ-VβαV-1ααVαβ, where Vij are the appropriate submatrices of the matrix V(Uγ). Based on GLMs, it can be shown that
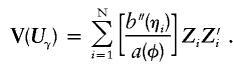 |
Without environmental covariates, there is only an intercept to correct for, so that the variance simplifies to
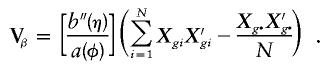 |
A global score statistic can then be computed as S=U′βV-1βUβ. It is worth noting that, for binary data and a single Xg covariate representing the allele dosage (e.g., Xg=0, 1, 2), the score statistic times (N-1)/N is the same as Armitage's test for trend in proportions, which is a popular statistic used to compare genotype frequencies between cases and controls (Sasieni 1997; Devlin and Roeder 1999).
Score Tests for Ambiguous Haplotypes
When linkage phase is unknown, the marker phenotypes, m, may be consistent with a set of underlying multilocus genotypes, G. We shall consider the joint likelihood of the trait and markers, conditional on the environmental covariates, L(y,m|Xe), which is the standard way to account for “missing data” by the EM algorithm. In our case, the underlying unobserved genotypes are considered the missing data. The likelihood for a given subject is
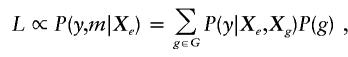 |
where the sum is over the set G of genotypes consistent with marker phenotypes, m. The score statistic for the effects of haplotypes, adjusted for environmental covariates, can be shown to be
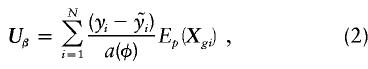 |
where 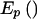 denotes the expectation over the posterior distribution of genotypes under the null hypothesis, given the observed marker data. That is,
denotes the expectation over the posterior distribution of genotypes under the null hypothesis, given the observed marker data. That is, 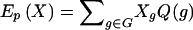 , where the posterior probability of a genotype for a subject is
, where the posterior probability of a genotype for a subject is 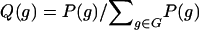 . To compute the genotype probabilities, P(g), we first estimate the haplotype probabilities by application of the EM algorithm (Excoffier and Slatkin 1995) to the pool of all subjects, which is appropriate under the null hypothesis, and then multiply the relevant haplotype probabilities (i.e., assuming Hardy-Weinberg equilibrium). Note the similarities of equations (1) and (2), where Xg is replaced by its posterior expectation when linkage phase is ambiguous.
. To compute the genotype probabilities, P(g), we first estimate the haplotype probabilities by application of the EM algorithm (Excoffier and Slatkin 1995) to the pool of all subjects, which is appropriate under the null hypothesis, and then multiply the relevant haplotype probabilities (i.e., assuming Hardy-Weinberg equilibrium). Note the similarities of equations (1) and (2), where Xg is replaced by its posterior expectation when linkage phase is ambiguous.
Similar to the case when genotypes are not ambiguous, the variance matrix of the vector Uβ can be determined by Vβ=Vββ-VβαV-1ααVαβ, but the ambiguity of the underlying genotypes must now be considered. The variance matrix V(Uγ) can be determined by the matrix of negative second partial derivatives of ln(L), evaluated under the null hypothesis. In our situation, this is equivalent to computation of the variance of the score vector when the EM algorithm is used to account for missing data. As shown by Louis (1982), 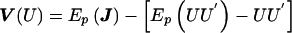 , where J is the negative of the second derivative matrix for the complete data and where
, where J is the negative of the second derivative matrix for the complete data and where 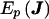 is the expected complete data information over the posterior distribution (i.e., conditional on the set G). The first term of V(U) is the variance if there were no “missing data” due to unknown linkage phase, and the second term is the penalty for incomplete linkage phase. Applying this variance computation to our score vector results in
is the expected complete data information over the posterior distribution (i.e., conditional on the set G). The first term of V(U) is the variance if there were no “missing data” due to unknown linkage phase, and the second term is the penalty for incomplete linkage phase. Applying this variance computation to our score vector results in
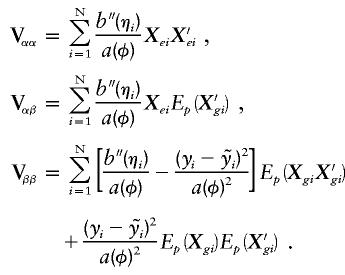 |
The term Vαα is the usual covariance matrix for complete data, and Vαβ is of similar form but with Xgi replaced by its posterior expectation. In contrast, the term Vββ is equivalent to the variance of the score vector proposed by Louis (1982) for incomplete data, thereby accounting for ambiguous haplotypes.
To implement the score statistics for different types of traits, we need merely to assume a distribution for the trait and to make the appropriate substitutions for the expected value of the trait, 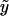 ; the dispersion parameter, a(φ); and the ratio b′′(η)/a(φ). These functions are defined in table 1 for a few common distributions. Without environmental covariates,
; the dispersion parameter, a(φ); and the ratio b′′(η)/a(φ). These functions are defined in table 1 for a few common distributions. Without environmental covariates, 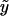 is the same for all subjects and can be estimated as the sample mean for a quantitative trait, as the sample fraction of diseased subjects for a binary trait, or as the sample event rate for a trait with a Poisson distribution. It is straightforward to extend our methods to other types of traits and distributions by defining the appropriate functions; for example, we have extended our methods to analyze ordinal traits (e.g., unaffected, moderately affected, and severely affected) by use of score statistics for the proportional odds model, a multivariate GLM (details are available on request).
is the same for all subjects and can be estimated as the sample mean for a quantitative trait, as the sample fraction of diseased subjects for a binary trait, or as the sample event rate for a trait with a Poisson distribution. It is straightforward to extend our methods to other types of traits and distributions by defining the appropriate functions; for example, we have extended our methods to analyze ordinal traits (e.g., unaffected, moderately affected, and severely affected) by use of score statistics for the proportional odds model, a multivariate GLM (details are available on request).
Table 1.
GLM Functions for Different Distributions[Note]
| Distribution |  |
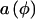 |
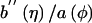 |
| Normal | η | σ2mse | 1/σ2mse |
| Binomial | 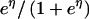 |
1 | 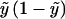 |
| Poisson | eη | 1 | 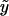 |
Note.— η=X′eα+X′gβ.
Score Statistics
With the above results, we can compute a global score statistic according to
This score statistic has a large sample χ2 distribution with degrees of freedom equal to the rank of Vβ, although the generalized inverse of Vβ may be required when it is not of full rank. This score statistic is asymptotically equivalent to the likelihood-ratio test statistic but avoids the need to compute maximum-likelihood estimates of β. Although maximum-likelihood estimates of the haplotype probabilities under the null hypothesis are required, to compute subject-specific posterior probabilities, the score statistic is not penalized for having estimated the haplotype probabilities, because the score vector for β and the score vector for the haplotype probabilities are independent under the null hypothesis (see Appendix A).
An advantage of our approach is that in addition to a global statistic, we can readily compute score statistics for the components of the Xg vectors, such as individual haplotypes, according to the following expression for the kth component, 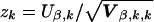 , which has a standard normal distribution for large samples (although we use a χ21 distribution for z2k to compute a two-sided P value). Furthermore, if the effect of a single haplotype is much larger than that for all other haplotypes, then the maximum value of z2k, maximized over all evaluated haplotypes indexed by k, is likely to have greater power than the quadratic statistic of equation (3). However, when several haplotypes are associated with the trait, perhaps because of incomplete LD, which may be the more common situation, the quadratic equation (3) may be the better alternative. Although the distribution of max(z2k) is not well defined and although P values could be approximated by the Bonferonni correction, it may be more reliable to compute the empirical P value by simulations, as discussed in the next subsection.
, which has a standard normal distribution for large samples (although we use a χ21 distribution for z2k to compute a two-sided P value). Furthermore, if the effect of a single haplotype is much larger than that for all other haplotypes, then the maximum value of z2k, maximized over all evaluated haplotypes indexed by k, is likely to have greater power than the quadratic statistic of equation (3). However, when several haplotypes are associated with the trait, perhaps because of incomplete LD, which may be the more common situation, the quadratic equation (3) may be the better alternative. Although the distribution of max(z2k) is not well defined and although P values could be approximated by the Bonferonni correction, it may be more reliable to compute the empirical P value by simulations, as discussed in the next subsection.
Empirical P Values
When the haplotype data is sparse, we may need to compute empirical P values by simulations. A substantial advantage of the use of score statistics, over likelihood-ratio tests that require maximization of the likelihood, is the ability to rapidly compute the score statistics. Empirical P values can be computed by repeatedly first permuting the marker phenotypes among the subjects and then computing the score statistics. Under the null hypothesis that none of the haplotypes are associated with the trait, it is appropriate to pool all subjects and then use the EM algorithm to compute the haplotype probabilities and the posterior genotype probabilities for each subject. These posterior probabilities need to be computed only once, because they are not altered by permutation of the marker phenotypes. It is computationally efficient to compute the values of 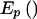 for each subject only once and to store them for reuse. However, the storage issues are not as simple for Vβ, because one would need to store the matrix
for each subject only once and to store them for reuse. However, the storage issues are not as simple for Vβ, because one would need to store the matrix 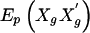 for each subject, which could consume significant computer memory. Instead, we store the values of Xg for each subject, along with their posterior probabilities, to rapidly compute
for each subject, which could consume significant computer memory. Instead, we store the values of Xg for each subject, along with their posterior probabilities, to rapidly compute 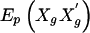 for each permutation of the marker phenotypes.
for each permutation of the marker phenotypes.
Haplotype and Genotype Scoring
The multilocus genotypes, composed of two haplotypes, can be scored a number of ways. A simple scheme is to count the number of haplotypes that a subject possesses. This is accomplished by creation of a vector X with a value of 0, 1, or 2 for the count of haplotypes, which has length H-1, where H is the number of distinguishable haplotypes (one haplotype is ignored, analogous to treating it as a baseline in regression). When there are a large number of haplotypes, this global score statistic will be weakly powered. One way to avoid diminished power, as well as to improve the approximation of the score statistic by the χ2 distribution, is to pool rare haplotypes into a single baseline group.
Another approach is to scan a large chromosomal region for subsegments that may be associated with the trait. Begin with single-locus associations, followed by “sliding” scores for two-locus haplotypes (i.e., sliding a score for haplotypes defined by adjacent pairs of loci across the entire measured chromosomal region), followed by sliding scores for three-locus haplotypes, and so forth (Clayton and Jones 1999). Alternatively, when particular haplotypes are found to be associated with the trait, it may be of interest to determine whether the alleles of a haplotype have a stronger effect in cis position than in trans position. For example, suppose that there are two loci, with alleles A and a at the first locus and B and b at the second locus. To evaluate whether the genotype AB/ab has a stronger effect on the trait than the genotype Ab/aB has, we could create a covariate that has values of 1, for genotype AB/ab; of −1, for genotype Ab/aB; and of 0, for all other genotypes.
The above examples provide some guidelines on how one could consider scoring multilocus genotypes. More-complicated scoring schemes, such as ways to score for particular interactions among alleles, could be developed. An advantage of our general approach is that, once a scoring scheme is defined and appropriate X covariates are created, computation of the score statistics, as well as of the empirical P values, is straightforward.
EM Algorithm for Haplotypes
Although the EM algorithm, which is used to estimate haplotype frequencies for unrelated subjects, has been implemented in a number of software packages (Terwilliger and Ott 1994; Excoffier and Slatkin 1995; Hawley and Kidd 1995; Long et al. 1995; Slatkin and Excoffier 1996; Schneider et al. 2000), we briefly review some of the key features of our algorithm that decrease computation time. The input data are arranged as a matrix, with N rows representing N subjects and 2K columns representing pairs of alleles for K loci whose phase is unknown. Two aspects achieve the speed of our algorithm: (1) the use of functions to quickly index multilocus-marker phenotypes (as well as similar methods for haplotypes); and (2) the storage, in memory, of intermediate results to avoid recomputations. The input-data matrix can be reduced to sufficient statistics, which are the counts of the distinguishable configurations of multilocus-marker phenotypes. These counts are stored, and, then, for each distinguishable configuration, all possible pairs of haplotypes are enumerated. Each enumerated haplotype is given an integer value that represents its grouping according to the unique haplotypes, which is then used as an index to rapidly determine its corresponding probability during the iterations of the EM algorithm.
Availability of Software
The software that implements the computation of score statistics for the association of ambiguous haplotypes with a variety of traits was developed in the programming language S-PLUS (Insightful). Our software is available from our Web site (Statistical and Genetic Epidemiology, Health Sciences Research–Mayo Clinic), although interested users must also have S-PLUS on their computer system.
Application of Score Statistics
Because failure of measles vaccination to stimulate antibody formation is a major individual and public health risk, a study was conducted at the Mayo Clinic to determine the association between HLA alleles and immune response following measles vaccination. The HLA genes produce proteins that bind with antigenic peptides and display them to T cells, which ultimately stimulate an immune response. For this reason, the HLA genes are ideal candidates to evaluate for their association with serologic response to vaccination. A total of 220 unrelated subjects were evaluated for their antibody levels, measured by the enzyme-linked immunosorbent assay test and described elsewhere (Poland et al. 2002). The study was approved by the Mayo Clinic institutional review board, and informed consent was obtained from participants. The level of antibody response is a quantitative measurement, although values <0.8 are considered to be a clinically negative response. Hence, we can evaluate the association between HLA alleles and either a binary trait (having values of 1, if negative response, and of 0, if positive response) or a quantitative trait. Eleven HLA loci were measured, and the association of each locus with both the binary and quantitative traits were evaluated by use of our score tests. On the basis of individual locus evaluations, three loci—DQB, DRB, and HLA-B—demonstrated statistically significant associations with binary response; the number of alleles at each of these loci in our sample were 12, 11, and 24, respectively, thereby demonstrating the highly polymorphic information for which the HLA region is well known. Both binary and quantitative traits were then evaluated for their associations with haplotypes composed of these three loci. Among the 220 subjects, there were a total of 678 enumerated haplotypes that were consistent with the observed alleles at the three loci. However, only 178 haplotypes had nonzero probability estimates. After we pooled the rare haplotypes (with estimated frequencies <0.005) into a single group, there remained 40 haplotypes to evaluate. For the binary trait, the global score statistic was 65.874, and, with 40 df, the P value from the χ2 distribution was .006; this P value was identical to the empirical P value based on 1,000 simulation repetitions. The empirical P value for the max(z2k) statistic was .004. For the quantitative trait, the results were not as striking. The global score statistic was 46.605, with a χ2 P value of .219, which is in close agreement with the empirical P value of .224. The empirical P value for the max(z2k) statistic was .318.
The haplotypes most strongly associated with both binary and quantitative traits, as judged by the haplotype-specific scores, are given in table 2. In terms of relative significance, the haplotype-specific scores were consistent between the binary and quantitative traits, both of which yielded the same extreme haplotypes. The scores for the quantitative trait are negative, because the four extreme haplotypes are associated with a lower-than-average response, and the scores for the binary trait are positive, because y=1 if the response is lower than a minimum cutoff. An advantage of our method is demonstrated by the haplotype-specific scores, which allow the evaluation of which haplotypes have the strongest association with a trait. For the results given in table 2, the P values between the asymptotic χ21 distribution and simulations are very similar. A comparison of all 40 haplotype-specific P values for both binary and quantitative traits is provided in figure 1. For the quantitative trait, which is assumed to have a normal distribution, the χ21 and empirical P values were remarkably close. However, for the binary trait, the empirical P values tended to be greater than the χ21 P values, suggesting that the χ21 approximation may not be adequate. Although the purpose of our analyses is to illustrate the utility of our new methods, as well as the adequacy of the χ2 distribution relative to simulation P values, we should note that first exploring individual locus associations, as we had done to select the three most interesting loci, and then evaluating haplotypes on the basis of the most significant ones can bias the haplotype P values. This approach does not invalidate our comparisons of asymptotic versus simulation P values, conditional on which loci were selected, but it is an approach that requires caution when attempting to build significant haplotypes from prior significance testing. To further evaluate the adequacy of the χ2 distribution, simulations were performed, as discussed in the next section.
Table 2.
Haplotypes Most Strongly Associated with Antibody Response to Measles Vaccination
|
Binary Trait |
Quantitative Trait |
||||||||
|
Haplotype |
P Value |
P Value |
|||||||
| DQB | DRB | HLA-B | Haplotype Frequency | Score | χ21 | Simulation | Score | χ21 | Simulation |
| 63 | 13 | 60 | .006 | 2.144 | .032 | .033 | −1.651 | .099 | .096 |
| 31 | 4 | 44 | .029 | 2.534 | .011 | .014 | −2.267 | .023 | .020 |
| 21 | 7 | 13 | .011 | 3.693 | .000 | .001 | −2.315 | .020 | .022 |
| 21 | 3 | 8 | .105 | 3.823 | .000 | .000 | −2.450 | .014 | .012 |
Figure 1.

Comparison of P values for haplotype-specific score statistics. P values were computed by the χ2 distribution (i.e., χ21) versus simulations, to test the association of HLA haplotypes with failure to demonstrate an immune response (binary trait [A]) or with a quantitative antibody level (quantitative trait [B]).
Simulation Methods to Evaluate Type I Error Rates
To evaluate the type I error rates of the χ2 approximations for both the global statistic and the haplotype-specific statistics, simulations were performed by using our observed marker phenotypes for the three HLA loci and randomly assigning traits to the 220 subjects. For binary traits, the percentage of diseased subjects was simulated at 20% and 50% (e.g., 50% typical in case-control studies). Quantitative traits were simulated by three different distributions: (1) a standard normal distribution; (2) an exponential distribution, to evaluate the influence of skewed traits (both the simulated trait and a log transformation of the trait were analyzed); and (3) a t distribution with 5 df, to evaluate the influence of kurtosis (this t distribution has a kurtosis of 9, which is much greater than the kurtosis of 3 for a standard normal distribution, and, hence, has much heavier tails than a normal distribution).
Simulation Results
The type I error rates for the score statistics with a binary trait are presented in table 3. When the fraction of diseased subjects was 50%, which is typical for case-control study designs, the global score statistic tended to be somewhat conservative. In contrast, the global score statistic tended to be anticonservative when the fraction of diseased subjects was only 20% and, particularly, when a large number of rare haplotypes were evaluated. This suggests that the χ2 approximation is inadequate for such sparse data and that P values based on simulations would be preferred. However, when considering the average P values for the haplotype-specific score statistics, averaged over all evaluated haplotypes, it appears that the χ2 distribution with 1 df yields a good approximation for the haplotype-specific score statistics.
Table 3.
Type I Error Rates for Global and Haplotype-Specific Score Statistics for a Binary Trait
|
Simulation P Values for |
||||
| Haplotype-SpecificScore Statisticsb |
||||
| Frequency of Skipped Haplotypesa (No. Evaluated), for Percentage Diseased | Nominal Type I Error Rate | Global Score Statistic | Median | Mean |
| 50: | ||||
| <.005 (n = 40) | .05 | .027 | .036 | .032 |
| .01 | .002 | .000 | .003 | |
| <.01 (n = 21) | .05 | .025 | .049 | .048 |
| .01 | .002 | .006 | .005 | |
| 20: | ||||
| <.005 (n = 40) | .05 | .120 | .049 | .051 |
| .01 | .073 | .014 | .015 | |
| <.01 (n = 21) | .05 | .070 | .043 | .043 |
| .01 | .022 | .014 | .014 | |
“Skipped haplotypes” are not evaluated but are pooled into a single baseline group when their frequencies are less than the threshold in column 2.
Median and mean P values are over the number of evaluated haplotype-specific scores.
The type I error rates for quantitative traits are presented in table 4. When the trait had a normal distribution, the χ2 approximation for the global statistic, as well as that for the haplotype-specific statistics, appeared to be adequate. When the trait was skewed (i.e., had an exponential distribution) or when the trait distribution had heavy tails (i.e., was the t distribution), the χ2 P values for the global statistics were somewhat anticonservative, although this problem was diminished by eliminating the rare haplotypes and, for the skewed trait, by taking the log transformation of the trait. It is of interest that the χ2 distribution with 1 df yields a good approximation for the haplotype-specific score statistics, thereby suggesting that the haplotype-specific score statistics tend to be fairly robust to departures from a normal distribution.
Table 4.
Type I Error Rates for Global and Haplotype-Specific Score Statistics for Quantitative Traits
|
Simulation P Values for |
||||
| Haplotype-SpecificScore Statisticsb |
||||
| Frequency of Skipped Haplotypesa (No. Evaluated), for Trait Distribution | Nominal Type I Error Rate | Global Score Statistic | Median | Mean |
| Normal: | ||||
| <.005 (n = 40) | .05 | .063 | .051 | .052 |
| .01 | .022 | .011 | .011 | |
| <.01 (n = 21) | .05 | .048 | .052 | .053 |
| .01 | .014 | .011 | .011 | |
| Exponential: | ||||
| <.005 (n = 40) | .05 | .084 | .047 | .047 |
| .01 | .037 | .018 | .019 | |
| <.01 (n = 21) | .05 | .078 | .047 | .048 |
| .01 | .024 | .017 | .017 | |
| Exponential, log transformed: | ||||
| <.005 (n = 40) | .05 | .069 | .051 | .051 |
| .01 | .023 | .013 | .015 | |
| <.01 (n = 21) | .05 | .060 | .050 | .050 |
| .01 | .018 | .012 | .012 | |
| t Distribution (5 df): | ||||
| <.005 (n = 40) | .05 | .077 | .047 | .047 |
| .01 | .032 | .019 | .019 | |
| <.01 (n = 21) | .05 | .064 | .043 | .044 |
| .01 | .028 | .015 | .016 | |
“Skipped haplotypes” are not evaluated but are pooled into a single baseline group when their frequencies are less than the threshold in column 1.
Median and mean P values are over the number of evaluated haplotype-specific scores.
Discussion
Our proposed methods, which are based on score equations for GLMs, provide several significant advantages over other current methods of analysis for associations of ambiguous haplotypes with traits. The most obvious advantage is that a wide variety of traits can be evaluated, in contrast to the likelihood-ratio statistic that is widely used to compare haplotype frequencies between cases and controls. We have currently developed score statistics that can be used for binary, Poisson-count, ordinal, and quantitative traits. Further developments are planned for other types of study designs and traits, such as matched case-control designs and censored survival data. Other advantages of our general approach are (1) that haplotype-specific scores can be easily computed, allowing evaluation of individual haplotypes when the global score statistic indicates statistical significance; (2) that, in addition to the global quadratic statistic, the max(z2k) statistic, as well as its simulation P value, can be easily computed to evaluate whether only a few haplotypes are strongly associated with a trait; (3) that simulation P values can be computed in an efficient manner; and (4) that adjustment for nongenetic covariates can be readily implemented.
Although simulation P values may be most reliable when the data is sparse, the computational burden to compute them increases as the number of loci in the haplotypes increases. Our simulations suggest that the P values approximated by the χ2 distribution for the global score statistic are adequate when a case-control study is balanced between cases and controls or when a trait is not highly skewed and when rare haplotypes are eliminated. It may be reasonable to screen for haplotype associations by use of the χ2 approximation and then to confirm suggestive findings with simulation P values. To provide some guidance on the computational efficiency of the score statistic versus the likelihood-based method (for case-control data), we computed the relative central-processing-unit time for 100 simulations. For our example data, which included 220 subjects and three loci, 19 subjects had one possible haplotype pair (i.e., no ambiguity), 55 subjects had two possible haplotype pairs, and 146 subjects had four possible haplotype pairs. The likelihood method took 13.4 times longer (in the central processing unit) than the score statistic. However, efficiency depends on the amount of haplotype ambiguity and, hence, on the number of loci in the haplotype. To illustrate this, we simply repeated our three loci, to create six loci per subject. With a greater amount of haplotype ambiguity (i.e., 4 subjects with 1 possible haplotype pair; 15 subjects with 2 possible haplotype pairs; 66 subjects with 8 possible haplotype pairs; and 146 subjects with 32 possible haplotype pairs), the likelihood method took 25.0 times longer than the score statistic, so the benefit of the score statistics increases as the haplotype ambiguity becomes greater.
Some limitations of the likelihood approach to the estimation of haplotype frequencies are that it requires large amounts of computer memory and processing time, both of which grow exponentially with the number of heterozygous loci, and that the final solution can depend on the initial starting values used for the haplotype frequencies, so that it may be necessary to use several different starting values to find the solution that yields the maximum likelihood. Some recent studies on the utility of the likelihood method (Fallin and Schork 2000; Tishkoff et al. 2000; Fallin et al. 2001) have reported the following properties: (1) it provides accurate estimates of the most frequent haplotypes but tends to be inaccurate for rare haplotypes (Tishkoff et al. 2000); (2) the inaccuracies for small frequencies can be caused by their large sampling variation (Fallin and Schork 2000), as well as by the allele frequencies and amount of LD (McKeigue 2000); and (3) the accuracy of the method increases as the LD among the marker loci increases (Fallin and Schork 2000). Finally, when the assumption of Hardy-Weinberg proportions for genotypes is violated with an excess of heterozygotes, the error of the EM procedure is increased, although the error is not increased with an excess of homozygotes (Fallin and Schork 2000). Further simulations are required to evaluate how robust our score statistics are to departures from the Hardy-Weinberg assumption, as well as to evaluate the power of our methods. We are also developing methods that account for departures from Hardy-Weinberg genotype proportions and plan to evaluate whether they provide a robust method when incorporated into our haplotype-scoring algorithms.
Acknowledgment
This research was supported by U.S. Public Health Services, National Institutes of Health; contract grant numbers R01 DE13276, N01 AI45240, and R01 2AI33144.
Appendix A
A brief outline of why the score vector for β and the score vector for the haplotype probabilities are independent under the null hypothesis is provided, although many of the detailed intermediate steps are skipped for the sake of brevity. If h denotes the vector of haplotype probabilities, which determine the genotype probabilities, then we wish to show that 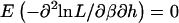 . To see this, note that
. To see this, note that
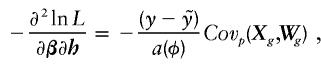 |
where Wg = 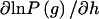 , P[g = (i/j)] = [2−I(i = j)]hihj, i and j are particular haplotypes, and Covp(X,W) is the covariance of X and W over the posterior distribution of genotypes. The expected value of this expression is zero, because
, P[g = (i/j)] = [2−I(i = j)]hihj, i and j are particular haplotypes, and Covp(X,W) is the covariance of X and W over the posterior distribution of genotypes. The expected value of this expression is zero, because 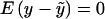 .
.
Electronic-Database Information
URLs for data in this article are as follows:
- Insightful, http://www.insightful.com/default_class5.asp (for the programming language S-PLUS)
- Statistical and Genetic Epidemiology, Health Sciences Research–Mayo Clinic, http://www.mayo.edu/statgen/ (for software that implements computation of score statistics)
References
- Cardon L, Bell J (2001) Association study designs for complex diseases. Nat Rev Genet 2:91–99 [DOI] [PubMed] [Google Scholar]
- Clark AG (1990) Inference of haplotypes from PCR-amplified samples of diploid populations. Mol Biol Evol 7:111–122 [DOI] [PubMed] [Google Scholar]
- Clark AG, Weiss KM, Nickerson DA, Taylor SL, Buchanan A, Stengård J, Salomaa V, Vartiainen E, Perola M, Boerwinkle E, Sing CF (1998) Haplotype structure and population genetic inferences from nucleotide-sequence variation in human lipoprotein lipase. Am J Hum Genet 63:595–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton D, Jones H (1999) Transmission/disequilibrium tests for extended marker haplotypes. Am J Hum Genet 65:1161–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins F (1999) Shattuck lecture: medical and societal consequences of the Human Genome Project. New Engl J Med 341:28–37 [DOI] [PubMed] [Google Scholar]
- Devlin B, Risch N, Roeder K (1996) Disequilibrium mapping: composite likelihood for pairwise disequilibrium. Genomics 36:1–16 [DOI] [PubMed] [Google Scholar]
- Devlin B, Roeder K (1999) Genomic control for association studies. Biometrics 55:997–1004 [DOI] [PubMed] [Google Scholar]
- Drysdale CM (2000) Complex promoter and coding region β2-adrenergic receptor haplotypes alter receptor expression and predict in vivo responsiveness. Proc Natl Acad Sci USA 97:10483–10488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier L, Slatkin M (1995) Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population. Mol Biol Evol 12:921–927 [DOI] [PubMed] [Google Scholar]
- Fallin D, Cohen A, Essioux L, Chumakov I, Blumenfeld M, Cohen D, Schork N (2001) Genetic analysis of case/control data using estimated haplotype frequencies: application to APOE locus variation and Alzheimer's disease. Genome Res 11:143–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallin D, Schork N (2000) Accuracy of haplotype frequency estimation for biallelic loci, via the expectation-maximization algorithm for unphased diploid genotype data. Am J Hum Genet 67:947–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo SW (1997) Linkage disequilibrium measures for fine-scale mapping: a comparison. Hum Hered 47:301–314 [DOI] [PubMed] [Google Scholar]
- Hastbacka J, de la Chapelle A, Kaitila I, Sistonen P, Weaver A, Lander E (1992) Linkage disequilibrium mapping in isolated founder populations: diastrophic dysplasia in Finland. Nat Genet 2:204–211 [DOI] [PubMed] [Google Scholar]
- Hawley ME, Kidd KK (1995) HAPLO: a program using the EM algorithm to estimate the frequencies of multi-site haplotypes. J Hered 86:409–411 [DOI] [PubMed] [Google Scholar]
- Hollox EJ, Poulter M, Zvarik M, Ferak V, Krause A, Jenkins T, Saha N, Kozlov AI, Swallow DM (2001) Lactase haplotype diversity in the old world. Am J Hum Genet 68:160–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium (2001) Initial sequencing and analysis of the human genome. Nature 409:860–921 [DOI] [PubMed] [Google Scholar]
- International SNP Map Working Group, The (2001) A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409:928–933 [DOI] [PubMed] [Google Scholar]
- Kaplan NL, Hill WG, Weir BS (1995) Likelihood methods for locating disease genes in nonequilibrium populations. Am J Hum Genet 56:18–32 [PMC free article] [PubMed] [Google Scholar]
- Lazzeroni LC (1998) Linkage disequilibrium and gene mapping: an empirical least-squares approach. Am J Hum Genet 62:159–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JC, Williams RC, Urbanek M (1995) An E-M algorithm and testing strategy for multiple-locus haplotypes. Am J Hum Genet 56:799–810 [PMC free article] [PubMed] [Google Scholar]
- Louis T (1982) Finding the observed information matrix when using the EM algorithm. J R Stat Soc B 44:226–233 [Google Scholar]
- McCullagh P, Nelder JA (1983) Generalized linear models. Chapman and Hall, London [Google Scholar]
- McKeigue PM (2000) Efficiency of estimation of haplotype frequencies: use of marker phenotypes of unrelated individuals versus counting of phase-known gametes. Am J Hum Genet 67:1626–1627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalatos-Beloin S, Tishkoff S, Bentley K, Kidd K, Ruano G (1996) Molecular haplotyping of genetic markers 10 kb apart by allele-specific long-range PCR. Nucleic Acids Res 24:4841–4843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poland G, Ovsyannikova I, Jacobson R, Vierkant R, Jacobsen S, Pankratz V, Schaid D (2002) Identification of an association between HLA class II alleles and low antibody levels after measles immunization. Vaccine 20:430–438 [DOI] [PubMed] [Google Scholar]
- Rannala B, Slatkin M (1998) Likelihood analysis of disequilibrium mapping, and related problems. Am J Hum Genet 62:459–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risch N (2000) Searching for genetic determinants in the new millennium. Nature 405:847–856 [DOI] [PubMed] [Google Scholar]
- Sasieni PD (1997) From genotypes to genes: doubling the sample size. Biometrics 53:1253–1261 [PubMed] [Google Scholar]
- Schneider S, Roessli D, Excoffier L (2000) Arlequin. Genetics and Biometry Laboratory, Department of Anthropology and Genetics. University of Geneva, Geneva [Google Scholar]
- Schork N, Fallin D, Thiel B, Xu X, Broeckel U, Jacob H, Cohen D (2001) The future of genetic case-control studies. In: Rao DC (ed) Advances in genetics. Vol 42. Academic Press, pp 191–212 [DOI] [PubMed] [Google Scholar]
- Slatkin M, Excoffier L (1996) Testing for linkage disequilibrium in genotypic data using the expectation-maximization algorithm. Heredity 76:377–383 [DOI] [PubMed] [Google Scholar]
- Stephens J, Schneider J, Tanguay D, Choi J, Acharya T, Stanley S, Jiang R, et al (2001a) Haplotype variation and linkage disequilibrium in 313 human genes. Science 293:489–493 [DOI] [PubMed] [Google Scholar]
- Stephens M, Smith N, Donnelly P (2001b) A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 68:978–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavtigian S, Simard J, Teng D, Abtin V, Baumgard M, Beck A, Camp N, et al (2001) A candidate prostate cancer susceptibility gene at chromosome 17p. Nat Genet 27:172–180 [DOI] [PubMed] [Google Scholar]
- Terwilliger JD (1995) A powerful likelihood method for the analysis of linkage disequilibrium between trait loci and one or more polymorphic marker loci. Am J Hum Genet 56:777–787 [PMC free article] [PubMed] [Google Scholar]
- Terwilliger JD, Ott J (1994) Handbook of human genetic linkage. Johns Hopkins University Press, Baltimore [Google Scholar]
- Tishkoff S, Pakstis A, Ruano G, Kidd K (2000) The accuracy of statistical methods for estimation of haplotype frequencies: an example from the CD4 locus. Am J Hum Genet 67:518–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venter J, Adams M, Myers E, Li P, Mural R, Sutton G, Smith H, et al (2001) The sequence of the human genome. Science 291:1304–1351 [DOI] [PubMed] [Google Scholar]
- Xiong M, Guo SW (1997) Fine-scale genetic mapping based on linkage disequilibrium: theory and applications. Am J Hum Genet 60:1513–1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Papadopoulos N, Marra G, Perrera C, Jiricny J, Boland CR, Lynch HT, et al (2000) Conversion of diploidy to haploidy. Nature 403:723–724 [DOI] [PubMed] [Google Scholar]
- Zhang S, Pakstis A, Kidd K, Zhao H (2001) Comparisons of two methods for haplotype reconstruction and haplotype frequency estimation from population data. Am J Hum Genet 69:906–912 [DOI] [PMC free article] [PubMed] [Google Scholar]