

Abstract
Biomaterial-induced tissue responses in patients with total joint replacement are associated with the generation of wear particles, which may lead to chronic inflammation and local bone destruction (periprosthetic osteolysis). Inflammatory reactions associated with wear particles are mediated by several important signaling pathways, the most important of which involves the transcription factor NF-κB. NF-κB activation is essential for macrophage recruitment and maturation, as well as the production of pro-inflammatory cytokines and chemokines such as TNF-α, IL-1β, IL-6, MCP1, etc. In addition, NF-κB activation contributes to osteoclast differentiation and maturation via RANK/RANKL signaling, which increases bone destruction and reduces bone formation. Targeting individual downstream cytokines directly (such as TNF-α or IL-1β) may not effectively prevent wear particle induced osteolysis. A more logical upstream therapeutic approach may be provided by direct modulation of the core IκB/IKKα/β/NF-κB signaling pathway in the local environment, however, the timing, dose, and strategy for administration should be considered. Suppression of chronic inflammation via inhibition of NF-κB activity in patients with malfunctioning joint replacements may be an effective strategy to mitigate wear particle induced periprosthetic osteolysis.
Keywords: Biomaterials, periprosthetic osteolysis, chronic inflammation, NF-κB
Introduction
Total Joint replacement (TJR) is a surgical procedure for end-stage arthritis that involves the implantation of permanent biomaterials. The increasing number of TJR cases has been extended to younger and middle-age patients. Therefore, long-term biocompatibility, durability and functionality are important goals for these implants. Despite many recent advances, revision of TJRs is still a major concern. The tissue response induced by implanted biomaterials, as well as the potential for loosening and periprosthetic osteolysis still remain significant challenges [1].
Despite the introduction of novel bearing surfaces, the continued generation of wear particles is still expected, and together with increased intracapsular pressure, is considered to be a key factor resulting in inflammation and bone destruction [2]. Protein bound wear particles interact with surface receptors on macrophages, resulting in activation of the NF-κB pathway. This induces the expression of chemokines and cytokines, the recruitment of more macrophages, and stimulation of the osteoclastogenesis pathway. The downstream network of the NF-κB pathway facilitates periprosthetic osteolysis.
The NF-κB pathway can be activated by many diverse stimuli and signaling mechanisms (including several different cytokines), which makes it challenging to block these signals from upstream. In this review, we summarize basic concepts involved in wear particle induced chronic inflammation and periprosthetic osteolysis, and the central role of NF-κB. We also address the potential value and current approaches to blocking the NF-κB pathway and the potential advantages and limitations relevant to wear particle disease.
Currently used biomaterials and the generation of wear particles
Most joint replacements consist of metal or ceramic articulating with polyethylene/ ceramic/metallic components. Continued wear of the bearing surfaces throughout the lifetime of an implant leads to the generation and release of particulate debris. These particles activate macrophages, leading to a local inflammatory response, and potentially, periprosthetic osteolysis [3, 4].
The physical, material, topographic and chemical properties of an implant and its byproducts are the main factors that determine the subsequent biological response [4–7]. Fibrous tissue may form and encapsulate an implant, and in so doing, isolate it from surrounding tissues [4]. With regards to total hip replacement, polyethylene wear rates exceeding 0.15 mm/year have been shown to significantly enhance the risk of aseptic loosening [8], whereas wear rates below 0.05 mm/year are otherwise associated with better function and implant stability [9]. The traditional polymers, such as ultra high molecular weight polyethylene (UHMWPE) have had wear rates as high as 0.4 mm/year [8]. The development of highly cross-link polyethylene has reduced the production of wear particles, and studies after one decade of use show great promise [10–12]. Polymethylmethacrylate (PMMA) bone cement particles appear to be less inflammatory compared to polyethylene particles [13]. Metallic wear particles from cobalt chrome metal-on-metal hip replacements are much smaller than polymeric particles but much greater in number [14]. Bulk titanium facilitates osseointegration, however, titanium debris is associated with localized osteolysis [15]. Protein coatings are being developed to minimize the inflammatory response to these wear particles. Modern ceramic bearing surfaces are very tough and wear resistant [16]. Compared to standard metal heads, the ceramic wear rate of the ceramic-on-polyethylene is reduced by 10–50% [16].
Biomaterial induced inflammation
Acute inflammation is the initial response of the human body to harmful stimuli in order to maintain tissue homeostasis. This is initiated by macrophage release of cytokines and chemokines that cause increased movement of leukocytes from the intravascular space directly into the injured tissues. This process lasts only from minutes to days, depending on the extent of injury. Neutrophils and other leukocytes migrate from adjacent blood vessels to the perivascular tissues and the injury site [17–19], which is followed by a cascade of events involving macrophages and other cells in the acute inflammatory response.
Chronic inflammation is a state in which acute inflammation, fibrosis and repair are occurring simultaneously. It is characterized by the presence of monocyte/macrophages, lymphocytes, fibroblasts and other cells, the proliferation of blood vessels and connective tissue remodeling [20–22]. This reaction is accompanied by simultaneous destruction and healing of the tissues at the injury site.
Macrophages play a prominent role in the development of immune responses to biomaterials. Although most materials evoke an innate non-specific, non-antigenic associated host immune response, macrophages and dendritic cells may process and present potential antigenic stimuli to cells of the adaptive immune system. Whether innate or adaptive (antigenic) immune responses are activated, the macrophage is a key cell because of the great number of biologically active products (cytokines, chemokines, reactive oxygen species, prostanoids etc) it produces [21]. In this review, we especially focus on aseptic periprosthetic osteolysis as one example of particle-associated chronic inflammation.
Pathogenesis of biomaterials induced inflammation
Implantation of an orthopaedic device initiates a host response that under favorable conditions leads to peri-implant tissue regeneration and formation of a stable implant-tissue interface [4, 23]. This host response largely follows the general principles of wound healing with activation of the innate immune system, in which cells of the monocyte-macrophage lineage orchestrate acute inflammation, resolution and repair. In contrast, the role of adaptive immunity in orthopaedic biomaterial-induced-inflammation is less clear, and is limited to special cases of metal hypersensitivity, implant infection and host response against composite grafts containing cells of non-autologous origin. Typically the host response to implant biomaterial is divided into stages that follow each other in continuous manner and that can be recognized by typical peri-implant histology [24, 25].
Acute phase response
The host response to an orthopaedic device is initiated by trauma to the local tissues and vasculature, unavoidably caused by the initial surgery. This stage, referred to as provisional matrix formation, is characterized by local peri-implant hematoma formation, and the rapid activation of major cascade systems of blood, namely the coagulation and the complement systems [26–28]. These processes are activated by contact of blood with exposed extracellular matrix proteins, and by the biomaterial surface itself, which activates the classical and alternative pathways of the complement system and the intrinsic system of the coagulation cascade. Activation of these systems leads to deposition of C3b complement fragments onto the material surface, release of chemotactic C3a and C5a complement fragments and, with the conjoint action of the blood coagulation system and the subsequent activation of thrombocytes, to the formation of a peri-implant fibrin meshwork [26, 29]. In addition to damage to the local vasculature with subsequent release and activation of plasma protein cascades, initial surgical trauma causes mast cell degranulation and, presumably, release of damage-associated molecular patterns (DAMPs) from necrotic cells and damaged extracellular matrix (ECM) [30, 31].
The immediate outcome of these events is formation of a three-dimensional fibrin matrix that contains many thrombocyte-, mast cell-, ECM- and complement derived- chemoattractants, cytokines and growth factors [4, 23, 25]. These factors increase the permeability of the microvasculature, and cause the activation of local endothelium with recruitment of innate immune cells to the local tissue. Furthermore, the surface of the biomaterial becomes coated with a layer of plasma-, interstitial fluid- and ECM-derived proteins, the most important of which are complement components, fibrinogen, fibrin, immunoglobulin G, albumin, fibronectin, vitronectin and various DAMPs (Fig. 1A) [32–35]. This coating of the implant surface with proteins takes place immediately; this layer of various host-derived proteins mediates subsequent biomaterial-cell interactions [36].
Fig 1. Tissue response and generation of wear particles to implanted biomaterials.
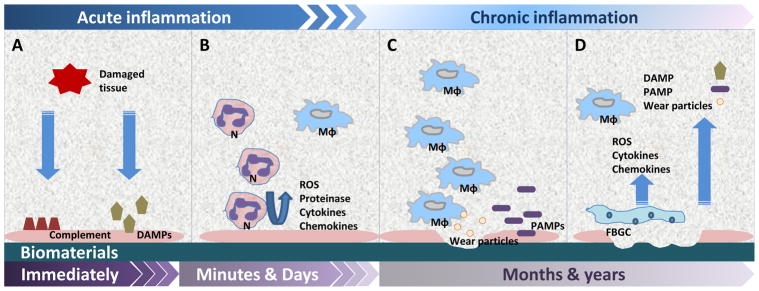
A) Tissue damage caused by the initial surgery activate the complement system and release DAMP, forming a protein layer (pink) coated on the implanted device immediately. B) Acute phase inflammation involves the efflux of neutrophils (N) and macrophages (Mϕ), which produce reactive oxygen species (ROS), proteinase, cytokines, and chemokines. C) Macrophage infiltration are dominant in the chronic inflammation stage. PAMP can be recognized by PRR that results in macrophage activation and generation of excess wear particles. D) Macrophages may undergo cell fusion to form FBGC, resulting in ongoing inflammation and tissue damage.
The formation of a provisional matrix is rapidly followed by an efflux of neutrophils and onocyte/macrophages to the peri-implant tissues due to local endothelial activation and production of chemotactic factors. Neutrophils and macrophages phagocytose cell- and ECM debris and, when encountering the protein covered implant surface which is non-phagocytosable, secrete reactive oxygen and nitrogen species, proteinases, cytokines and chemokines (Fig. 1B) that cause local tissue damage and subsequent further recruitment of monocyte/macrophages [37–41].
Chronic inflammation
The stage of acute inflammation is typically short-lived, lasting only few days, and the neutrophil-dominated acute phase is followed by further infiltration of monocyte/macrophages to the peri-implant tissues [4, 23, 25]. Monocytes migrate to the area of inflammation by following concentration gradients of chemotactic factors; they then differentiate into mature macrophages. Macrophages remove the provisional matrix and apoptotic cells by means of phagocytosis. In the context of implanted biomaterials, the extent to which macrophages become activated by the implant surface or byproducts is dependent on the material, size, surface topology and surface chemistry of the device that, in turn, regulates the composition of the protein coating forming on the material surface [42–47]. Other important factors include mechanical forces and the release of degradation products from the implant [48]. The cytokine microenvironment which is dictated by the state of macrophage activation further modulates the outcome of the macrophage-biomaterial interaction [49].
There has been increasing interest in the role of DAMPs, Pathogen-Associated Molecular Patterns (PAMPs), and pattern-recognition receptors (PRRs), such as Toll-like receptors (TLRs), in the detection of host protein-coated biomaterials and biomaterial induced inflammation (Fig. 1C). Initial surgical trauma and subsequent peri-implant inflammation releases reactive oxygen species, matrix metalloproteinase (MMPs) and other substances associated with the production of endogenous DAMP molecules, which could adhere to biomaterial surfaces exacerbating inflammation [50–52]. In addition to host-derived DAMPs, bacterial derived PAMPs such as lipopolysaccharide can adhere to the implant surface as a subclinical bacterial biofilm or circulate within blood to land on to implant surfaces [53–56]. These DAMPs and PAMPs are recognized by TLRs and other PRRs on the cell surface and within the cytoplasm (see below).
Besides these “natural” TLR ligands, there is evidence that some synthetic materials, such as oxidized polymers released from UHMWPE can act directly as TLR-ligands and induce macrophage activation [57]. In addition, cobalt ions released from metal-on-metal joint replacements can directly bind to TLR4 and initiate an inflammatory response [58–60]. Phagocytosed biomaterial wear particles can cause endosomal damage and activate inflammatory pathways via intracellular release and subsequent recognition of endolysosomal constituents via PRRs [61, 62]. In vivo models of orthopedic wear particle induced osteolysis have also demonstrated the importance of TLRs in biomaterial-induced inflammation [63–65].
Host macrophages attempt to remove and degrade biomaterial particles by phagocytosis, but if the implant or byproducts are large, macrophages undergo cell fusion to form multinucleated foreign body giant cells (FBGC) [4]. FBGCs are able to engulf somewhat larger foreign bodies and secrete reactive oxygen and nitrogen radicals, proteinases and other lysosomal enzymes directly in some cases (frustrated phagocytosis) [37, 38, 66, 67]. The extracellular secretions can cause direct damage and degradation of the implant. Thus, there are complex interactions between macrophages, the protein-coated biomaterial surface and the multitude of auto- and paracrine signals and factors derived from cells that regulate the microenvironment surrounding the implant (Fig. 1D).
FBGCs attempt to encapsulate the biomaterial device to contain and resolve the acute inflammatory response. If this is successful, the acute (and chronic) inflammatory reactions are short lived, and subsequent release of macrophage-derived growth factors leads to the ingress of fibroblasts, mesenchymal stromal cells (MSC) and cells capable of neovascularization [4, 23]. This granulation tissue encapsulates the biomaterial implant in a fibrous layer, or in the case of stable cementless joint replacements, integration of the prosthesis with bone to form a functional construct.
If the inflammatory stimulus and response are overwhelming, chronic inflammation persists leading to, in the case of cementless joint replacements, continued production of pro-inflammatory cytokines, failure of integration, loosening and osteolysis [48, 68–73]. Such adverse conditions might result from continued implant micromotion and tissue damage with continued release of DAMPs, ongoing low-grade implant infection with periodic release of PAMPs and DAMPs, and release of wear-products from the implant. These factors cause chronic macrophage activation and production of inflammatory mediators and continued recruitment of inflammatory cells.
NF-κB signaling in chronic inflammation
NF-κB is a transcription factor that broadly influences gene expression of factors controlling the survival, differentiation, and proliferation of cells. Furthermore, NF-κB regulates pro-inflammatory cytokine release and is closely linked to both innate and adaptive immunity [74–76]. There are five NF-κB family members in mammals, including RelA/p65, RelB, c-Rel, p50, and p52. These proteins have a structurally conserved amino-terminal 300 amino acid region, which contains the dimerization, nuclear-localization and DNA-binding domains. The c-Rel, RelB and RelA proteins also have a carboxy-terminal non-homologous transactivation domain, which strongly activates transcription from NF-κB-binding sites in target genes. The other Rel proteins, such as p50 homo-dimers, lack the transactivation domain, but they still bind to NF-κB consensus sites in DNA and, therefore, function as transcriptional repressors. The p50 and p52 proteins are generated by proteolytic processing of precursor p105 and p100 proteins, respectively.
In the “resting” state, NF-κB dimers are held inactive in the cytoplasm through association with IκB proteins. Inducing stimuli trigger activation of the IκB kinase complex, leading to phosphorylation, ubiquitination, and degradation of IκB proteins. Released NF-κB dimers translocate to the nucleus, bind specific DNA sequences, and promote transcription of target genes. NF-κB proteins bind to κB sites as dimers, either homo-dimers or heterodimers, and can exert both positive and negative effects on target gene transcription.
The NF-κB signaling pathways have been broadly classified into two types: classical (canonical) and alternative (non-canonical). The canonical pathway is representative of IκB degradation dependent pathway. Many receptors such as TNF-α Receptor, IL-1β Receptor and TLR, representing pattern recognition receptors trigger signaling cascades which culminate in the activation of IKKβ (inhibitor of κB kinase β, also known as IKK2). IKKβ exists in a complex with the closely related kinase IKKα (inhibitor of κB kinase α, also known as IKK1) and the regulatory protein NEMO (NF-κB essential modulator, also known as IKKγ). In contrast, the non-canonical pathway depends on p100 processing induced by IKKα and NF-κB inducing kinase activation. In both pathways, proteins translocate from the cytoplasm into the nucleus to regulate the transcription of numerous target genes for pro-inflammatory cytokines, chemokines, cell-adhesion molecules, acute phase response proteins, immune-regulatory molecules and transcription factors.
Up-stream—NF-κB stimulators
A wide range of soluble and membrane-bound extracellular ligands activate the NF-κB pathway, most notably through members of TNF-α Receptor, TLR, IL-1Receptor, and antigen receptor super families (Fig. 2). It was also recently reported that the regulation of NF-κB activity is determined by changes in the intracellular environment [77]. These intracellular NF-κB activating pathways include the responses to DNA damage and reactive oxygen species, as well as recognition of intracellular pathogens mediated by the oligomerization domain like receptor and retinoic acid-inducible gene I family of proteins.
Fig 2. The central role of NF-κB signaling in wear-particle induced periprosthetic osteolysis.

Wear particles directly or indirectly activate NF-κB upstream including the TNF-α receptor (TNFR), IL-1 receptor (IL1R), and toll-like receptor (TLR) in macrophages. The activation of NF-κB enhances the expression of chemokines, RANKL, and proteinases that lead to osteolysis via different mechanisms (green boxes). Chemokines recruit macrophages, osteoprogenitor cells, and mesenchymal stem cells (MSC), whereas RANKL induces the maturation of osteoclasts. Suppression of NF-κB activation can be achieved by targeting 1) upstream activators; 2) IκB; 3) IKK kinase; 4) the core component of NF-κB (RelA/p50); or 5) nuclear translocation and DNA binding ability of NF-κB. Notably, the NF-κB downstream target genes may also affect osteoblasts and MSCs (pink box).
TNF-α
In 1999, Nakashima et al. first described that NF-κB was activated and translocated from the cytoplasm to the nucleus in human primary macrophages exposed to titanium (Ti) particles [78]. Exposure of macrophages derived from human peripheral blood monocytes/macrophages to these particles for only thirty minutes activated NF-κB and NF-IL-6, followed by increased production of TNF-α and IL-6 protein. This phenomena was also seen when particle phagocytosis was inhibited, suggesting that phagocytosis was not necessary for activation of NF-κB in macrophage. In other words, cell contact with wear particles from orthopaedic implants was sufficient to induce the activation of NF-κB. They also suggested that other signaling pathways in addition to NF-κB are involved in particle-induced activation of transcription factors and the release of cytokines. Titanium particles have also been shown to activate the NF-κB and NF-IL-6 pathways in osteoblast-like cell MG-63, whereas the activation and consequent cytokine production was suppressed when particle phagocytosis was inhibited [79]. This suggests that particle phagocytosis may be important for NF-κB activation in osteoblasts; further investigation using primary osteoblasts is essential to clarify this point.
Schwarz et al showed that exposure of J774 murine macrophages to pure Ti particles (1–3 μm in diameter) lead to increased TNF-α production similar to that by human peripheral blood monocytes. The production of TNF-α was preceded by a drop in cellular levels of IκBα protein and translocation of p50/p65 NF-κB to the nucleus 30 minutes after stimulation, consistent with the activation of NF-κB. IL-6 mRNA was first seen 4 hours after the addition of the Ti, indicating that the production of this cytokine is secondary to the immediate NF-κB response. Using the mouse calvarial model, they also showed a dramatic inflammatory response and extensive bone resorption to Ti particles that was suppressed in p50−/− mice [80].
LPS and LPS-treated Ti particles but not particles alone stimulated NF-κB activation in differentiated THP-1 cells [81]. In addition, NF-κB is rapidly induced by Ti in ANA-1 cells via the p105 degradation pathway, and it has been suggested that the TNF-α induction is mediated in part through NF-κB binding to the κB2α site of the TNF-α promoter [82]. In another study, macrophages of the THP-1 cell line were exposed to particles of polyethylene- and titanium alloy, which resulted in significant activation of both NF-κB and TNF-α promoters and TNF-α release in a dose dependent manner. [83].
Toll like Receptors (TLRs)
Activation of the NF-κB signal pathway is primarily initiated by TLRs (Fig. 1). Takagi et al. first reported that TLRs were expressed at the interfacial membrane around aseptically loosened implants [84]. Double staining techniques showed that the TLR positive cells in aseptic loosening were mostly macrophages and FBGC. Macrophages derived from rat bone marrow were then used to study TLR4 and TLR9 mRNA levels by quantitative polymerase chain reaction. Unexpectedly, endotoxin free Ti particles decreased macrophage TLR4 and TLR9 mRNA significantly, although MMP-9 mRNA levels used as macrophage activation marker were significantly increased. The reduced expression of TLR4 mRNA was also found in murine RAW 264.7 macrophage cells exposed to Ti particles [85]. It was postulated that auto- and/or paracrine inflammatory cytokines down-regulated the expression of TLR to avoid damage and harmful effects caused by excessive inflammatory responses [84, 85].
Hirayama et al. stimulated bone marrow macrophages with pure Ti particles with/without LPS coating (Ti/LPS+ and Ti/LPS− respectively). The mRNA levels of pro-inflammatory cytokines, TLRs and their adaptor molecules were measured by real time polymerase chain reaction method. Whereas levels of pro-inflammatory cytokines significantly increased, the expression of TLR4, TLR5, and TLR9 mRNA decreased with Ti/LPS+ particle exposure. The mRNA levels of MyD88, IRAK1, IRAK4 decreased gradually, and TRAF6 underwent an initial transient increase, followed by suppression. The mRNA levels of TLR2 and IRAK2 increased after phagocytosis of Ti/LPS+. This suggested that self-protective mechanisms regulate excessive host responses in macrophages exposed to inflammatory stimuli [86], and implied possible strategies to regulate the NF-κB signaling pathway.
Greenfield et al. [64] reported decreased osteolysis in TLR2−/− mice compared to wild type mice using the murine calvarial model of particle-induced osteolysis. TLR2−/− but not TLR4−/− macrophages demonstrated decreased production of TNF-α when exposed with Ti particles in culture. These in vitro and in vivo data strongly support the critical role of TLR1/2 in aseptic loosening and osteolysis of implants. Interestingly, they also found that TNF-α expression was completely neutralized when wear particles were delivered to macrophages that lack TLR4 and TLR2, however osteolysis was only partially inhibited in vitro by these knockouts [64]. They postulated that the acute particle induced inflammatory response was TLR dependent, while later inflammatory responses and osteolysis were only partially TLR dependent.
Ligand-TLR binding induces rearrangements of TLR domains and recruitment of their adaptors, triggering the activation of NF-κB. Pearl et al. reported that the inflammatory responses induced by PMMA particles were mitigated by a MyD88 inhibitor. Similar results were found in MyD88−/− macrophages stimulated with PMMA[87]. These findings suggest that particles are recognized by TLR, which is dependent in part by the MyD88 signaling pathway. Maitra R et al. also reported that UHMWPE particles activated TLR1/2, leading to an inflammatory reaction, which is mediated by the NF-κB signal pathway[88].
Osteopontin
When bone marrow macrophages were exposed to Ti particles, the expression of pro-inflammatory cytokines and chemotactic factors in the conditioned medium were significantly reduced by osteopontin (OPN) deficiency. Although, phagocytosis of Ti particles by bone marrow-derived macrophages was not attenuated in OPN deficient mice compared to wild type mice, phagocytosis-mediated NF-κB activation was impaired [64].
Downstream – NK-κB Target genes
Production of pro-inflammatory cytokines, extra-cellular matrix degrading proteinases and certain chemokines by macrophages, fibroblasts and osteoblasts exposed to wear particles are regulated by intracellular systems [2], one of which is NF-κB. This NF-κB directed protein expression significantly impacts the interfacial tissues around orthopaedic implants, and the development of periprosthetic loosening and osteolysis [89–94]. Hundreds of NF-κB target genes have been reported, that govern inflammatory factors, apoptosis, cell stress, growth and transcriptional factors. The list of reported target gene has been well-documented by Gilmore’s laboratory website (http://www.bu.edu/nf-kb/gene-resources/target-genes/). Here we summarize the inflammatory genes that have been correlated with particle induced periprosthetic osteolysis (Table 1).
Table 1.
Major NF-κB target genes involved in wear particle induced periprosthetic osteolysis
| Name | Involved cell(s) | Function | References |
|---|---|---|---|
| TNF-α | Macrophage | Inflammation, osteolysis | [78, 80–83, 104–107, 121–123] |
| IL-1β | Macrophage | Inflammation, osteolysis | [105, 107, 108, 124, 125] |
| IL-6 | Macrophage Osteoprogenitors |
Inflammation Anti-osteoclastogenesis |
[103–106, 108, 118] |
| MCP1 | Macrophage | Recruit macrophage | [108, 109] |
| MIP1α | Macrophage/ MSC | Recruit macrophage and MSC | [106, 109] |
| Cox-2/Prostaglandin E2 | Macrophage/fibroblast | Inflammation | [103, 107, 108] |
| RANKL | Macrophage/Osteoclast | Osteoclastogenesis Bone resorption | [95–101, 106, 108] |
| MMPs | Macrophage | Tissue destruction | [52] |
| Type I collagen | Osteoblast | Bone formation | [112, 113] |
| IL-10 | MSC/ macrophage | Immune modulation | [114, 115, 124] |
| IFN-γ | Osteoprogenitors T cells | Anti-osteoclastogenesis | [118] |
| Cathepsin K | Osteoclast | Degrade bone matrix | [97, 106] |
| TRAP | Osteoclast | Degrade bone matrix | [97, 106, 107] |
| Smurf1/2 | Osteoprogenitors | Suppress bone formation via degrading β-catenin | [116] |
Pro-inflammatory cytokines and the activation of osteoclasts
Many inflammatory cytokine activate NF-κB, and downstream target genes. These latter cytokines, including TNF-α and IL-1β, also induce receptor activator of NF-kB ligand (RANKL) expression by several cell types including macrophages [95]. The interaction of RANKL with RANK receptor on the osteoclast induces osteoclastogenesis and bone resorption, and regulates calcium homeostasis [96–98]. Increased expression of RANKL has been noted in peri-implant tissues from patients with prosthetic loosening, giant cell tumor of bone and non-unions of fractures [99–101]. Osteoclasts are multinucleated cells of monocyte/macrophage origin that degrade bone matrix; they are characterized by high expression of tartrate resistant acid phosphatase (TRAP) and cathepsin K. Mice deficient in the RANKL gene exhibit severe osteopetrosis, defective tooth eruption, and completely lack osteoclasts because of impaired osteoclastogenesis [97]. Osteoclast formation induced by pro-inflammatory cytokines requires NF-κB p50 and p52, c-Fos, and NFATc1 (nuclear factor of activated T-cells, cytoplasmic, calcineurin-dependent 1) expression in osteoclast precursors. NF-κB controls early osteoclast differentiation from precursors induced directly by RANKL and TNF, leading to activation of c-Fos followed by NFATc1. Inhibition of NF-κB prevents RANKL- and TNF- induced bone resorption [102].
Chemokines
NF-κB regulates both the activation and recruitment of osteoclasts and their progenitors thereby modulating osteolysis [2]. Many important chemokines are downstream from NF-κB, including IL-6, MCP1, and MIP1α. Wear particles increase the production of chemokines from macrophages, fibroblasts, and other cell types, which enhances osteoclastogenesis [103–108]. MSC are also recruited via MIP1α production induced by PMMA wear particles [109]. However, the specific interactions and biological mechanisms among cells of the monocyte/macrophage lineage and MSCs in particle induced periprosthetic osteolysis are still being elucidated [110, 111].
Potential compensated role of NF-kappa;B in the osteolytic process
NF-κB regulates other genes that have protective effects on osteoclastogenesis. For example, the gene regulating Type I collagen is a target gene for NF-κB [112]. Secretion of collagen by osteoblasts induces bone formation [113]. In addition, MSCs have an immune modulating role via secretion of anti-inflammatory cytokines such as IL-10 [114], which is also targeted by NF-κB [115]. Notably, recent findings indicated that suppression of NF-κB activity can enhance the osteogenesis ability of MSCs via blocking β-catenin degradation[116, 117]. Thus, activation of NF-κB in osteoblasts and MSCs by wear particles could be a “double-edge sword” for bone. Furthermore, interferon γ and IL-6 both are also downstream target genes for NF-κB, and demonstrate anti-osteoclastogenesis properties. Interestingly, the expression of interferon γ and IL-6 is reduced by Ti stimulation [118], suggesting that the particles may induce another dominant signal to override the regulation by NF-κB activation.
Although all biological functions of NF-κB are still not completely known, it is generally accepted that interference with NF-κB will mitigate the bone destruction associated with wear particles from joint replacements. Previous studies have used transformed cell lines or leukemia cells for wear particle-related experiments [80, 81, 83, 85]. Further studies using untransformed, freshly isolated myeloid cells are essential to validate these concepts and provide more clinically relevant information[78, 86].
Modulation of NF-κB activity
Suppression of NF-kB activity can be achieved via several different strategies. Many clinically used anti-inflammatory drugs are able to mitigate NF-κB signaling [119]. However, high doses are required to achieve the inhibitory effects, which might prove to be toxic clinically. Specific suppression of NF-kB activity may be achieved by targeting 1) points upstream of the NF-kB activating signal; 2) IKKα/β kinase activity; 3) IκB protein stability; and 4) NF-κB protein functions; i.e. nuclear translocation, DNA binding ability, or transactivation inhibition (Fig. 1). Most of the identified NF-κB inhibitors are synthetic compounds or natural products [120]. Protein derived antibodies and other peptides, as well as decoy oligodeoxynucleotide (ODN) were also developed and show promising results in blocking NF-κB activity [120]. Gene therapy is an alternative strategy to target NF-kB signaling. Currently, a viral vector based delivery system is the most efficient way to transduce the therapeutic gene expression in vivo. With designed promoter-expression systems, purified high titer viral vectors can selectively express the desired protein or siRNA in specific cell types or inflammation-associated environments.
Targeting NF-kB upstream
TNF-α is one of the major upstream cytokines that can activate the NF-κB pathway. Because of the central role of TNF-α in wear particle induced osteolysis, several studies have focused on blocking, neutralizing or silencing the TNF-α ligand [121, 122] in experimental models of osteolysis. Etanercept is a dimeric fusion protein that interacts with the TNF-α receptor and the Fc region of human immunoglobulin. Etanercept neutralizes TNF-α by competitive ligand binding to the TNF-α cell receptor. Etanercept is used clinically for the treatment of rheumatoid arthritis and other immune disorders; a murine wear particle induced osteolysis model using Etanercept showed great promise [121, 123]. However, in a prospective study with 20 patients with acetabular loosening, no difference was observed in periprosthetic bone resorption between Etanercept and placebo control patients. Additional signal pathways, other than TNF-α, may have compensatory roles in particle induced inflammation and osteolysis. For example, by using IL-1 receptor deficient mice or IL-1 receptor antagonist treatment was shown to prevent osteoclastogenesis induced by TNF-α and RANKL in in vitro and in vivo models [124, 125]. The use of siRNAs to knock down pro-inflammatory genes is another possible approach. siRNA allows the silencing of genes involved in the NF-κB pathway such as genes for TNF-α or IL-1β that have critical roles in bone resorption. These could be delivered locally in select situations [126]. However, siRNAs are costly and must be given frequently, and both in vivo and clinical translation become more difficult due to issues related to toxicity and instability of the complexes.
Direct targeting the core component in the NF-kB pathway
IKKα/β is one of the primary targets of NF-κB inhibitors. Several IKKα/β specific kinase inhibitors have been developed, such as imatinib/Gleevec [127]. These synthetic compounds may function as ATP analogs that bind to IKKα/β specifically [120]. The dominant negative form of IKKα/β has also been used to inhibit NF-κB activation. Interestingly, over-expression of IKKβ, but not IKKα, can suppress lipopolysaccharide induced cytokine production effectively [128]. Alternatively, blocking the IKKα/β interaction with IKKγ by using the NEMO-binding domain peptide can also suppress NF-κB activation, which was shown to block PMMA-induced osteoclastogenesis and inflammatory osteolysis using a murine calvarial model [129]. Stabilization and prevention of the degradation of IκB using a proteasome inhibitor is another strategy to modulate NF-κB activation. Bortezomib, a proteasome inhibitor used to treat multiple myeloma has been shown to ameliorate titanium particle induced inflammation [130].
There are several reports using the adenovirus system (Ad-IKKβdn & Ad-IκBαSR) and adeno-associated virus (AAV5-IKKβdn) to suppress NF-kb activity. By over-expression of the dominant negative IKKβ, or direct expression of IκBα by the viral vectors, these studies showed promising results in animal models of rheumatoid arthritis and chronic inflammatory diseases. Suppression of NF-κB activity via viral vector based gene therapy has been reviewed in detail elsewhere [131]. However, viral vectors alone may cause an adverse immunogenic response, including the induction of bone resorption [132]. The potential application of viral vector systems is therefore controversial.
The use of a decoy ODN is an efficient strategy to specifically suppress transcription factor activity [133] These short synthesized duplex DNAs are designed to mimic the transcription response element. The idea of competitive binding with transcription factors can be potent and specific, with potentially, a better safety profile. However, their clinical application may be limited due to issues related to low bioavailability and a short half-life. Recently, some chemical modifications have been developed to overcome these limitations. For example, by using a phosphothioate bond instead of a phosphodiester bond with sulfur, the ODN is more resistant to DNAase and showed increased stability in serum. Decoy ODNs have been shown to prevent NF-κB transactivation of cytokine genes by binding to free NF-κB [134]. In vivo and in vitro studies have shown that by applying ODN locally, bone loss is prevented and tissue healing is promoted in rheumatoid arthritis [134]. NF-κB ODN has been used in animal models of several inflammatory diseases, such as chronic obstructive pulmonary disease [135], vascular and cardiovascular disease [136–139], liver injury[140], atopic dermatitis[141], and periodontal disease [142].
Conclusions
Current translational research to modulate chronic inflammation and osteoclastogenesis non-operatively has met with limited success [143]. The potential targets of treatment include TNF-α, RANK/RANKL, COX-2, and others [143], all of which are correlated with the NF-κB activation. With regards to aseptic loosening wear particle-induced peri-prosthetic osteolysis, modulation of NF-κB could provide a non-surgical therapeutic target [144, 145].
Despite the fact that there are more than one thousand compounds capable of suppressing NF-kB activity, these compounds are generally only functional under specific conditions (cellular models and stimulatory agents), or require very high doses to achieve the intended suppressive effects [120]. As a consequence, currently, only a few compounds have found their way into clinical trials.
If modulation of NF-kB activity were to be implemented clinically in regards to biomaterials, critical issues would have to be tackled including the location and mode of delivery, the timing and profile for delivery, the dosage, and issues related to adverse events and potential toxicity. Ideally, the agent could be applied locally, thus minimizing systemic effects [146]. Systemic blocking or interference with NF-κB activity would undoubtedly impair the normal host immune system. Hepatic toxicity was observed at the embryonic stage in the compromised NF-κB activation mouse model [147]. In transgenic knockout animal studies, developmental defects or embryonic lethal effects were reported in NF-κB signal deficient mice. Since biomaterial associated chronic inflammation is generally confined, local rather than systemic delivery may have significant benefit and easier selective targeting to the desired cell types.
Optimal dosage and temporal delivery profiles are also critical factors in regulation of NF-κB activation. Suppression of the canonical NF-κB pathway may induce the activation of the non-canonical NF-κB pathway. It is unclear as to whether the canonical and non-canonical NF-κB pathways have similar or diverse target genes; studies on the biomaterial induced non-canonical pathway are also limited. However, recent reports showed promising therapeutic effects when co-targeting both pathways in several diseases [148]. In addition, cross-talk between the NF-κB pathway and other transcriptional factors may be enhanced with the treatment of NF-κB inhibitors. For example, the AP-1 binding sequence was found to be correlated with the NF-κB binding element [149]. Suppression of NF-κB activity may enhance AP-1 transactivation and enhance cytokine expression. Finally, inhibition of NF-κB activity may also block the negative feedback regulator, TNF-α induced protein 3. Loss of TNF-α induced protein 3 in transgenic mouse has lead to excessive cytokine production and death in cases of septic shock [150].
Previous studies have confirmed that macrophages and osteoclasts become activated by exposure to wear particles through the NF-κB signaling pathway [151]. Therefore the regulation of NF-κB activity is a logical therapeutic strategy to reduce tissue damage due to chronic inflammation associated with wear particles. However, potential adverse effects on other cell populations, including osteoblasts and their precursors are still largely unknown. Selective targeting of NF-κB activity in desired cell populations may further improve the therapeutic efficiency and safety of treatment.
The chronic inflammatory reaction due to excessive production of wear particles may lead to aseptic loosening and osteolysis in some patients with malfunctioning joint replacements. Local direct targeting of NF-κB may potentially suppress the chronic inflammatory response and reduce periprosthetic osteolysis, thereby improving the longevity of joint replacements.
Abbreviation
- DAMP
damage associated molecular pattern
- ECM
extracellular matrix
- FBGC
foreign body giant cells
- IKK
inhibitor of κB kinase
- MMP
matrix metalloproteinase
- MSC
mesenchymal stromal cells
- NEMO
NF-κB essential modulator
- ODN
oligodeoxynucleotide
- NFATc1
nuclear factor of activated T-cells, cytoplasmic, calcineurin-dependent 1
- OPN
Osteopontin
- PAMP
pathogen associated molecular pattern
- PMMA
polymethymethacrylate
- RANKL
receptor activator of NF-κB ligand
- Ti
Titanium
- TJR
total joint replacement
- TLR
toll-like receptor
- TRAP
tartrate resistant acid phosphatase
- UHMWPE
Ultra high molecular weight polyethylene
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tsao A, Jones L, Lewallen D Implant Wear Symposium Clinical Work G. What patient and surgical factors contribute to implant wear and osteolysis in total joint arthroplasty? The Journal of the American Academy of Orthopaedic Surgeons. 2008;16 (Suppl 1):13. doi: 10.5435/00124635-200800001-00004. [DOI] [PubMed] [Google Scholar]
- 2.Purdue P, Koulouvaris P, Nestor B, Sculco T. The central role of wear debris in periprosthetic osteolysis. HSS journal: the musculoskeletal journal of Hospital for Special Surgery. 2006;2:102–13. doi: 10.1007/s11420-006-9003-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Long M, Rack HJ. Titanium alloys in total joint replacement - a materials science perspective. Biomaterials. 1998;19:1621. doi: 10.1016/s0142-9612(97)00146-4. [DOI] [PubMed] [Google Scholar]
- 4.Anderson JM, Rodriguez A, Chang DT. Foreign body reaction to biomaterials. Semin Immunol. 2008;20:86–100. doi: 10.1016/j.smim.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee HS, Stachelek SJ, Tomczyk N, Finley MJ, Composto RJ, Eckmann DM. Correlating macrophage morphology and cytokine production resulting from biomaterial contact. Journal of biomedical materials research Part A. 2013;101:203–12. doi: 10.1002/jbm.a.34309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Navarro M, Michiardi A, CastaÒo O, Planell JA. Biomaterials in orthopaedics. Journal of the Royal Society, Interface / the Royal Society. 2008;5:1137–58. doi: 10.1098/rsif.2008.0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Love RJ, Jones KS. 4.404 - Adaptive Immune Responses to Biomaterials. In: Paul D, editor. Comprehensive Biomaterials. Oxford: Elsevier; 2011. pp. 37–47. [Google Scholar]
- 8.Sochart D. Relationship of acetabular wear to osteolysis and loosening in total hip arthroplasty. Clinical orthopaedics and related research. 1999:135–50. [PubMed] [Google Scholar]
- 9.Ilchmann T, Markovic L, Joshi A, Hardinge K, Murphy J, Wingstrand H. Migration and wear of long-term successful Charnley total hip replacements. The Journal of bone and joint surgery British volume. 1998;80:377–81. doi: 10.1302/0301-620x.80b3.8455. [DOI] [PubMed] [Google Scholar]
- 10.Thomas G, Simpson D, Mehmood S, Taylor A, McLardy-Smith P, Gill H, et al. The seven-year wear of highly cross-linked polyethylene in total hip arthroplasty: a double-blind, randomized controlled trial using radiostereometric analysis. The Journal of bone and joint surgery American volume. 2011;93:716–22. doi: 10.2106/JBJS.J.00287. [DOI] [PubMed] [Google Scholar]
- 11.Engh C, Hopper R, Huynh C, Ho H, Sritulanondha S. A prospective, randomized study of cross-linked and non-cross-linked polyethylene for total hip arthroplasty at 10-year follow-up. The Journal of arthroplasty. 2012;27:2–70. doi: 10.1016/j.arth.2012.03.048. [DOI] [PubMed] [Google Scholar]
- 12.Reynolds S, Malkani A, Ramakrishnan R, Yakkanti M. Wear analysis of first-generation highly cross-linked polyethylene in primary total hip arthroplasty: an average 9-year follow-up. The Journal of arthroplasty. 2012;27:1064–8. doi: 10.1016/j.arth.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 13.Goodman S. Wear particulate and osteolysis. The Orthopedic clinics of North America. 2005;36:41. doi: 10.1016/j.ocl.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 14.MacDonald S. Metal-on-metal total hip arthroplasty: the concerns. Clinical orthopaedics and related research. 2004:86–93. doi: 10.1097/01.blo.0000150309.48474.8b. [DOI] [PubMed] [Google Scholar]
- 15.Joon P, Young Kon K. Biomaterials. CRC Press; 2002. Metallic Biomaterials; pp. 1–20. [Google Scholar]
- 16.Jazrawi L, Kummer F, DiCesare P. Alternative bearing surfaces for total joint arthroplasty. The Journal of the American Academy of Orthopaedic Surgeons. 1998;6:198–203. doi: 10.5435/00124635-199807000-00001. [DOI] [PubMed] [Google Scholar]
- 17.Henson P, Johnston R. Tissue injury in inflammation. Oxidants, proteinases, and cationic proteins The Journal of clinical investigation. 1987;79:669–74. doi: 10.1172/JCI112869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Malech H, Gallin J. Current concepts: immunology. Neutrophils in human diseases The New England journal of medicine. 1987;317:687–94. doi: 10.1056/NEJM198709103171107. [DOI] [PubMed] [Google Scholar]
- 19.James MA. Chapter 4 Mechanisms of inflammation and infection with implanted devices. Cardiovascular Pathology. 1993:2. [Google Scholar]
- 20.Williams G, Williams W. Granulomatous inflammation--a review. Journal of clinical pathology. 1983;36:723–33. doi: 10.1136/jcp.36.7.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnston R. Current concepts: immunology. Monocytes and macrophages. The New England journal of medicine. 1988;318:747–52. doi: 10.1056/NEJM198803243181205. [DOI] [PubMed] [Google Scholar]
- 22.Browder T, Folkman J, Pirie-Shepherd S. The hemostatic system as a regulator of angiogenesis. The Journal of biological chemistry. 2000;275:1521–4. doi: 10.1074/jbc.275.3.1521. [DOI] [PubMed] [Google Scholar]
- 23.Franz S, Rammelt S, Scharnweber D, Simon JC. Immune responses to implants - A review of the implications for the design of immunomodulatory biomaterials. Biomaterials. 2011;32:6692–709. doi: 10.1016/j.biomaterials.2011.05.078. [DOI] [PubMed] [Google Scholar]
- 24.Anderson JM. Mechanisms of Inflammation and Infection with Implanted Devices. Cardiovasc Pathol. 1993;2:S33–S41. [Google Scholar]
- 25.Luttikhuizen DT, Harmsen MC, Van Luyn MJA. Cellular and molecular dynamics in the foreign body reaction. Tissue Eng. 2006;12:1955–70. doi: 10.1089/ten.2006.12.1955. [DOI] [PubMed] [Google Scholar]
- 26.Gorbet MB, Sefton MV. Biomaterial-associated thrombosis: roles of coagulation factors, complement, platelets and leukocytes. Biomaterials. 2004;25:5681–703. doi: 10.1016/j.biomaterials.2004.01.023. [DOI] [PubMed] [Google Scholar]
- 27.Ekdahl KN, Lambris JD, Elwing H, Ricklin D, Nilsson PH, Teramura Y, et al. Innate immunity activation on biomaterial surfaces: A mechanistic model and coping strategies. Adv Drug Deliver Rev. 2011;63:1042–50. doi: 10.1016/j.addr.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nilsson B, Ekdahl KN, Mollnes TE, Lambris JD. The role of complement in biomaterial-induced inflammation. Mol Immunol. 2007;44:82–94. doi: 10.1016/j.molimm.2006.06.020. [DOI] [PubMed] [Google Scholar]
- 29.Andersson J, Ekdahl KN, Lambris JD, Nilsson B. Binding of C3 fragments on top of adsorbed plasma proteins during complement activation on a model biomaterial surface. Biomaterials. 2005;26:1477–85. doi: 10.1016/j.biomaterials.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 30.Tang LP, Jennings TA, Eaton JW. Mast cells mediate acute inflammatory responses to implanted biomaterials. P Natl Acad Sci USA. 1998;95:8841–6. doi: 10.1073/pnas.95.15.8841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zdolsek J, Eaton JW, Tang L. Histamine release and fibrinogen adsorption mediate acute inflammatory responses to biomaterial implants in humans. J Transl Med. 2007:5. doi: 10.1186/1479-5876-5-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McFarland CD, Thomas CH, DeFilippis C, Steele JG, Healy KE. Protein adsorption and cell attachment to patterned surfaces. J Biomed Mater Res. 2000;49:200–10. doi: 10.1002/(sici)1097-4636(200002)49:2<200::aid-jbm7>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 33.Brodbeck WG, Colton E, Anderson JM. Effects of adsorbed heat labile serum proteins and fibrinogen on adhesion and apoptosis of monocytes/macrophages on biomaterials. J Mater Sci-Mater M. 2003;14:671–5. doi: 10.1023/a:1024951330265. [DOI] [PubMed] [Google Scholar]
- 34.Keselowsky BG, Bridges AW, Burns KL, Tate CC, Babensee JE, LaPlaca MC, et al. Role of plasma fibronectin in the foreign body response to biomaterials. Biomaterials. 2007;28:3626–31. doi: 10.1016/j.biomaterials.2007.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McNally AK, Jones JA, MacEwan SR, Colton E, Anderson JM. Vitronectin is a critical protein adhesion substrate for IL-4-induced foreign body giant cell formation. J Biomed Mater Res A. 2008;86A:535–43. doi: 10.1002/jbm.a.31658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilson CJ, Clegg RE, Leavesley DI, Pearcy MJ. Mediation of biomaterial-cell interactions by adsorbed proteins: A review. Tissue Eng. 2005;11:1–18. doi: 10.1089/ten.2005.11.1. [DOI] [PubMed] [Google Scholar]
- 37.Henson PM. The immunologic release of constituents from neutrophil leukocytes. I. The role of antibody and complement on nonphagocytosable surfaces or phagocytosable particles. Journal of immunology. 1971;107:1535–46. [PubMed] [Google Scholar]
- 38.Henson PM. The immunologic release of constituents from neutrophil leukocytes. II. Mechanisms of release during phagocytosis, and adherence to nonphagocytosable surfaces. Journal of immunology. 1971;107:1547–57. [PubMed] [Google Scholar]
- 39.Nimeri G, Ohman L, Elwing H, Wettero J, Bengtsson T. The influence of plasma proteins and platelets on oxygen radical production and F-actin distribution in neutrophils adhering to polymer surfaces. Biomaterials. 2002;23:1785–95. doi: 10.1016/s0142-9612(01)00305-2. [DOI] [PubMed] [Google Scholar]
- 40.Nimeri G, Majeed M, Elwing H, Ohman L, Wettero J, Bengtsson T. Oxygen radical production in neutrophils interacting with platelets and surface-immobilized plasma proteins: role of tyrosine phosphorylation. J Biomed Mater Res A. 2003;67:439–47. doi: 10.1002/jbm.a.10081. [DOI] [PubMed] [Google Scholar]
- 41.Patel JD, Krupka T, Anderson JM. iNOS-mediated generation of reactive oxygen and nitrogen species by biomaterial-adherent neutrophils. J Biomed Mater Res A. 2007;80:381–90. doi: 10.1002/jbm.a.30907. [DOI] [PubMed] [Google Scholar]
- 42.Brodbeck WG, Nakayama Y, Matsuda T, Colton E, Ziats NP, Anderson JM. Biomaterial surface chemistry dictates adherent monocyte/macrophage cytokine expression in vitro. Cytokine. 2002;18:311–9. doi: 10.1006/cyto.2002.1048. [DOI] [PubMed] [Google Scholar]
- 43.Xing S, Santerre JP, Labow RS, Boynton EL. Differential response to chemically altered polyethylene by activated mature human monocyte-derived macrophages. Biomaterials. 2002;23:3595–602. doi: 10.1016/s0142-9612(02)00088-1. [DOI] [PubMed] [Google Scholar]
- 44.Brodbeck WG, Voskerician G, Ziats NP, Nakayama Y, Matsuda T, Anderson JM. In vivo leukocyte cytokine mRNA responses to biomaterials are dependent on surface chemistry. J Biomed Mater Res A. 2003;64A:320–9. doi: 10.1002/jbm.a.10425. [DOI] [PubMed] [Google Scholar]
- 45.Jones JA, Dadsetan M, Collier TO, Ebert M, Stokes KS, Ward RS, et al. Macrophage behavior on surface-modified polyurethanes. J Biomat Sci-Polym E. 2004;15:567–84. doi: 10.1163/156856204323046843. [DOI] [PubMed] [Google Scholar]
- 46.Refai AK, Textor M, Brunette DM, Waterfield JD. Effect of titanium surface topography on macrophage activation and secretion of proinflammatory cytokines and chemokines. J Biomed Mater Res A. 2004;70A:194–205. doi: 10.1002/jbm.a.30075. [DOI] [PubMed] [Google Scholar]
- 47.Chen SL, Jones JA, Xu YG, Low HY, Anderson JM, Leong KW. Characterization of topographical effects on macrophage behavior in a foreign body response model. Biomaterials. 2010;31:3479–91. doi: 10.1016/j.biomaterials.2010.01.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nich C, Takakubo Y, Pajarinen J, Ainola M, Salem A, Sillat T, et al. Macrophages-Key cells in the response to wear debris from joint replacements. J Biomed Mater Res A. 2013 doi: 10.1002/jbm.a.34599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pajarinen J, Kouri VP, Jamsen E, Li TF, Mandelin J, Konttinen YT. Macrophages response to titanium particles is determined by macrophage polarization. Acta Biomater. doi: 10.1016/j.actbio.2013.06.027. [DOI] [PubMed] [Google Scholar]
- 50.Bennewitz NL, Babensee JE. The effect of the physical form of poly(lactic-co-glycolic acid) carriers on the humoral immune response to co-delivered antigen. Biomaterials. 2005;26:2991–9. doi: 10.1016/j.biomaterials.2004.08.023. [DOI] [PubMed] [Google Scholar]
- 51.Rogers TH, Babensee JE. Altered adherent leukocyte profile on biomaterials in Toll-like receptor 4 deficient mice. Biomaterials. 2010;31:594–601. doi: 10.1016/j.biomaterials.2009.09.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kido A, Pap G, Nägler D, Ziomek E, Menard R, Neumann H, et al. Protease expression in interface tissues around loose arthroplasties. Clinical orthopaedics and related research. 2004:230–6. doi: 10.1097/01.blo.0000136650.33036.fd. [DOI] [PubMed] [Google Scholar]
- 53.Greenfield EM, Bi YM, Ragab AA, Goldberg VM, Nalepka JL, Seabold JM. Does endotoxin contribute to aseptic loosening of orthopedic implants? J Biomed Mater Res B. 2005;72B:179–85. doi: 10.1002/jbm.b.30150. [DOI] [PubMed] [Google Scholar]
- 54.Nalepka JL, Lee MJ, Kraay MJ, Marcus RE, Goldberg VM, Chen X, et al. Lipopolysaccharide found in aseptic loosening of patients with inflammatory arthritis. Clin Orthop Relat R. 2006:229–35. doi: 10.1097/01.blo.0000224050.94248.38. [DOI] [PubMed] [Google Scholar]
- 55.Xing ZQ, Pabst MJ, Hasty KA, Smith RA. Accumulation of LPS by polyethylene particles decreases bone attachment to implants. J Orthop Res. 2006;24:959–66. doi: 10.1002/jor.20038. [DOI] [PubMed] [Google Scholar]
- 56.Tatro JM, Taki N, Islam AS, Goldberg VM, Rimnac CM, Doerschuk CM, et al. The balance between endotoxin accumulation and clearance during particle-induced osteolysis in murine calvaria. J Orthop Res. 2007;25:361–9. doi: 10.1002/jor.20289. [DOI] [PubMed] [Google Scholar]
- 57.Maitra R, Clement CC, Crisi GM, Cobelli N, Santambrogio L. Immunogenecity of Modified Alkane Polymers Is Mediated through TLR1/2 Activation. PloS one. 2008:3. doi: 10.1371/journal.pone.0002438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tyson-Capper AJ, Lawrence H, Holland JP, Deehan DJ, Kirby JA. Metal-on-metal hips: cobalt can induce an endotoxin-like response. Annals of the rheumatic diseases. 2013;72:460–1. doi: 10.1136/annrheumdis-2012-202468. [DOI] [PubMed] [Google Scholar]
- 59.Konttinen YT, Pajarinen J. Adverse reactions to metal-on-metal implants. Nature reviews Rheumatology. 2013;9:5–6. doi: 10.1038/nrrheum.2012.218. [DOI] [PubMed] [Google Scholar]
- 60.Potnis PA, Dutta DK, Wood SC. Toll-like receptor 4 signaling pathway mediates proinflammatory immune response to cobalt-alloy particles. Cellular immunology. 2013;282:53–65. doi: 10.1016/j.cellimm.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 61.Maitra R, Clement CC, Scharf B, Crisi GM, Chitta S, Paget D, et al. Endosomal damage and TLR2 mediated inflammasome activation by alkane particles in the generation of aseptic osteolysis. Mol Immunol. 2009;47:175–84. doi: 10.1016/j.molimm.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Burton L, Paget D, Binder NB, Bohnert K, Nestor BJ, Sculco TP, et al. Orthopedic wear debris mediated inflammatory osteolysis is mediated in part by NALP3 inflammasome activation. J Orthop Res. 2013;31:73–80. doi: 10.1002/jor.22190. [DOI] [PubMed] [Google Scholar]
- 63.Grandjean-Laquerriere A, Tabary O, Jacquot J, Richard D, Frayssinet P, Guenounou M, et al. Involvement of toll-like receptor 4 in the inflammatory reaction induced by hydroxyapatite particles. Biomaterials. 2007;28:400–4. doi: 10.1016/j.biomaterials.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 64.Greenfield EM, Beidelschies MA, Tatro JM, Goldberg VM, Hise AG. Bacterial Pathogen-associated Molecular Patterns Stimulate Biological Activity of Orthopaedic Wear Particles by Activating Cognate Toll-like Receptors. Journal of Biological Chemistry. 2010;285:32378–84. doi: 10.1074/jbc.M110.136895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pearl JI, Ma T, Irani AR, Huang ZN, Robinson WH, Smith RL, et al. Role of the Toll-like receptor pathway in the recognition of orthopedic implant wear-debris particles. Biomaterials. 2011;32:5535–42. doi: 10.1016/j.biomaterials.2011.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xia ZD, Zhu TB, Du JY, Zheng QX, Wang L, Li SP, et al. Macrophages in Degradation of Collagen Hydroxylapatite(Cha), Beta-Tricalcium Phosphate Ceramics (Tcp) Artificial Bone-Graft an in-Vivo Study. Chinese Med J-Peking. 1994;107:845–9. [PubMed] [Google Scholar]
- 67.Christenson EM, Anderson JM, Hiltner A. Oxidative mechanisms of poly(carbonate urethane) and poly(ether urethane) biodegradation: In vivo and in vitro correlations. J Biomed Mater Res A. 2004;70A:245–55. doi: 10.1002/jbm.a.30067. [DOI] [PubMed] [Google Scholar]
- 68.Gallo J, Goodman SB, Konttinen YT, Raska M. Particle disease: biologic mechanisms of periprosthetic osteolysis in total hip arthroplasty. Innate Immun. 19:213–24. doi: 10.1177/1753425912451779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Goodman SB, Gibon E, Yao Z. The basic science of periprosthetic osteolysis. Instr Course Lect. 62:201–6. [PMC free article] [PubMed] [Google Scholar]
- 70.Greenfield EM, Bi Y, Ragab AA, Goldberg VM, Nalepka JL, Seabold JM. Does endotoxin contribute to aseptic loosening of orthopedic implants? J Biomed Mater Res B Appl Biomater. 2005;72:179–85. doi: 10.1002/jbm.b.30150. [DOI] [PubMed] [Google Scholar]
- 71.Holt G, Murnaghan C, Reilly J, Meek RM. The biology of aseptic osteolysis. Clin Orthop Relat Res. 2007;460:240–52. doi: 10.1097/BLO.0b013e31804b4147. [DOI] [PubMed] [Google Scholar]
- 72.Purdue PE, Koulouvaris P, Potter HG, Nestor BJ, Sculco TP. The cellular and molecular biology of periprosthetic osteolysis. Clin Orthop Relat Res. 2007;454:251–61. doi: 10.1097/01.blo.0000238813.95035.1b. [DOI] [PubMed] [Google Scholar]
- 73.Sundfeldt M, Carlsson LV, Johansson CB, Thomsen P, Gretzer C. Aseptic loosening, not only a question of wear: a review of different theories. Acta Orthop. 2006;77:177–97. doi: 10.1080/17453670610045902. [DOI] [PubMed] [Google Scholar]
- 74.Baeuerle P, Henkel T. Function and activation of NF-kappa B in the immune system. Annual review of immunology. 1994;12:141–79. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 75.Thanos D, Maniatis T. NF-kappa B: a lesson in family values. Cell. 1995;80:529–32. doi: 10.1016/0092-8674(95)90506-5. [DOI] [PubMed] [Google Scholar]
- 76.Baldwin A. The NF-kappa B and I kappa B proteins: new discoveries and insights. Annual review of immunology. 1996;14:649–83. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 77.Chen F, Bower J, Demers LM, Shi X. Upstream signal transduction of NF-kB activation. Atlas of Genetics and Cytogenetics in Oncology and Haematology. 2002 [Google Scholar]
- 78.Nakashima Y, Sun D, Trindade M, Maloney W, Goodman S, Schurman D, et al. Signaling pathways for tumor necrosis factor-alpha and interleukin-6 expression in human macrophages exposed to titanium-alloy particulate debris in vitro. The Journal of bone and joint surgery American volume. 1999;81:603–15. doi: 10.2106/00004623-199905000-00002. [DOI] [PubMed] [Google Scholar]
- 79.Shida J, Trindade MC, Goodman SB, Schurman DJ, Smith RL. Induction of interleukin-6 release in human osteoblast-like cells exposed to titanium particles in vitro. Calcif Tissue Int. 2000;67:151–5. doi: 10.1007/s00223001125. [DOI] [PubMed] [Google Scholar]
- 80.Schwarz E, Lu A, Goater J, Benz E, Kollias G, Rosier R, et al. Tumor necrosis factor-alpha/nuclear transcription factor-kappaB signaling in periprosthetic osteolysis. Journal of orthopaedic research: official publication of the Orthopaedic Research Society. 2000;18:472–80. doi: 10.1002/jor.1100180321. [DOI] [PubMed] [Google Scholar]
- 81.Akisue T, Bauer T, Farver C, Mochida Y. The effect of particle wear debris on NFkappaB activation and pro-inflammatory cytokine release in differentiated THP-1 cells. Journal of biomedical materials research. 2002;59:507–15. doi: 10.1002/jbm.1264. [DOI] [PubMed] [Google Scholar]
- 82.Soloviev A, Schwarz E, Kuprash D, Nedospasov S, Puzas J, Rosier R, et al. The role of p105 protein in NFkappaB activation in ANA-1 murine macrophages following stimulation with titanium particles. Journal of orthopaedic research: official publication of the Orthopaedic Research Society. 2002;20:714–22. doi: 10.1016/S0736-0266(01)00180-2. [DOI] [PubMed] [Google Scholar]
- 83.Baumann B, Seufert J, Jakob F, Nöth U, Rolf O, Eulert J, et al. Activation of NF-kappaB signalling and TNFalpha-expression in THP-1 macrophages by TiAlV- and polyethylene-wear particles. Journal of orthopaedic research: official publication of the Orthopaedic Research Society. 2005;23:1241–8. doi: 10.1016/j.orthres.2005.02.017.1100230602. [DOI] [PubMed] [Google Scholar]
- 84.Takagi M, Tamaki Y, Hasegawa H, Takakubo Y, Konttinen L, Tiainen V-M, et al. Toll-like receptors in the interface membrane around loosening total hip replacement implants. Journal of biomedical materials research Part A. 2007;81:1017–26. doi: 10.1002/jbm.a.31235. [DOI] [PubMed] [Google Scholar]
- 85.Pajarinen J, Mackiewicz Z, Pöllänen R, Takagi M, Epstein N, Ma T, et al. Titanium particles modulate expression of Toll-like receptor proteins. Journal of biomedical materials research Part A. 2010;92:1528–37. doi: 10.1002/jbm.a.32495. [DOI] [PubMed] [Google Scholar]
- 86.Hirayama T, Tamaki Y, Takakubo Y, Iwazaki K, Sasaki K, Ogino T, et al. Toll-like receptors and their adaptors are regulated in macrophages after phagocytosis of lipopolysaccharide-coated titanium particles. Journal of orthopaedic research: official publication of the Orthopaedic Research Society. 2011;29:984–92. doi: 10.1002/jor.21369. [DOI] [PubMed] [Google Scholar]
- 87.Pearl J, Ma T, Irani A, Huang Z, Robinson W, Smith R, et al. Role of the Toll-like receptor pathway in the recognition of orthopedic implant wear-debris particles. Biomaterials. 2011;32:5535–42. doi: 10.1016/j.biomaterials.2011.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Maitra R, Clement C, Scharf B, Crisi G, Chitta S, Paget D, et al. Endosomal damage and TLR2 mediated inflammasome activation by alkane particles in the generation of aseptic osteolysis. Molecular immunology. 2009;47:175–84. doi: 10.1016/j.molimm.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Santavirta S, Konttinen Y, Bergroth V, Eskola A, Tallroth K, Lindholm T. Aggressive granulomatous lesions associated with hip arthroplasty. Immunopathological studies The Journal of bone and joint surgery American volume. 1990;72:252–8. [PubMed] [Google Scholar]
- 90.Kim K, Rubash H, Wilson S, D’Antonio J, McClain E. A histologic and biochemical comparison of the interface tissues in cementless and cemented hip prostheses. Clinical orthopaedics and related research. 1993:142–52. [PubMed] [Google Scholar]
- 91.Takagi M, Konttinen Y, Santavirta S, Sorsa T, Eisen A, Nordsletten L, et al. Extracellular matrix metalloproteinases around loose total hip prostheses. Acta orthopaedica Scandinavica. 1994;65:281–6. doi: 10.3109/17453679408995454. [DOI] [PubMed] [Google Scholar]
- 92.Takagi M, Konttinen Y, Kemppinen P, Sorsa T, Tschesche H, Bläser J, et al. Tissue inhibitor of metalloproteinase 1, collagenolytic and gelatinolytic activity in loose hip endoprostheses. The Journal of rheumatology. 1995;22:2285–90. [PubMed] [Google Scholar]
- 93.Takei I, Takagi M, Ida H, Ogino T, Santavirta S, Konttinen Y. High macrophage-colony stimulating factor levels in synovial fluid of loose artificial hip joints. The Journal of rheumatology. 2000;27:894–9. [PubMed] [Google Scholar]
- 94.Konttinen Y, Takagi M, Mandelin J, Lassus J, Salo J, Ainola M, et al. Acid attack and cathepsin K in bone resorption around total hip replacement prosthesis. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2001;16:1780–6. doi: 10.1359/jbmr.2001.16.10.1780. [DOI] [PubMed] [Google Scholar]
- 95.Hofbauer L, Schoppet M. Clinical implications of the osteoprotegerin/RANKL/RANK system for bone and vascular diseases. JAMA: the journal of the American Medical Association. 2004;292:490–5. doi: 10.1001/jama.292.4.490. [DOI] [PubMed] [Google Scholar]
- 96.Lacey D, Timms E, Tan H, Kelley M, Dunstan C, Burgess T, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–76. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- 97.Kong Y, Yoshida H, Sarosi I, Tan H, Timms E, Capparelli C, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397:315–23. doi: 10.1038/16852. [DOI] [PubMed] [Google Scholar]
- 98.Xu J, Tan J, Huang L, Gao X, Laird R, Liu D, et al. Cloning, sequencing, and functional characterization of the rat homologue of receptor activator of NF-kappaB ligand. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2000;15:2178–86. doi: 10.1359/jbmr.2000.15.11.2178. [DOI] [PubMed] [Google Scholar]
- 99.Crotti T, Smith M, Findlay D, Zreiqat H, Ahern M, Weedon H, et al. Factors regulating osteoclast formation in human tissues adjacent to peri-implant bone loss: expression of receptor activator NFkappaB, RANK ligand and osteoprotegerin. Biomaterials. 2004;25:565–73. doi: 10.1016/s0142-9612(03)00556-8. [DOI] [PubMed] [Google Scholar]
- 100.Huang L, Xu J, Wood D, Zheng M. Gene expression of osteoprotegerin ligand, osteoprotegerin, and receptor activator of NF-kappaB in giant cell tumor of bone: possible involvement in tumor cell-induced osteoclast-like cell formation. The American journal of pathology. 2000;156:761–7. doi: 10.1016/s0002-9440(10)64942-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Laird R, Pavlos N, Xu J, Brankov B, White B, Fan Y, et al. Bone allograft non-union is related to excessive osteoclastic bone resorption: a sheep model study. Histology and histopathology. 2006;21:1277–85. doi: 10.14670/HH-21.1277. [DOI] [PubMed] [Google Scholar]
- 102.Yamashita T, Yao Z, Li F, Zhang Q, Badell I, Schwarz E, et al. NF-kappaB p50 and p52 regulate receptor activator of NF-kappaB ligand (RANKL) and tumor necrosis factor-induced osteoclast precursor differentiation by activating c-Fos and NFATc1. The Journal of biological chemistry. 2007;282:18245–53. doi: 10.1074/jbc.M610701200. [DOI] [PubMed] [Google Scholar]
- 103.Bukata S, Gelinas J, Wei X, Rosier R, Puzas J, Zhang X, et al. PGE2 and IL-6 production by fibroblasts in response to titanium wear debris particles is mediated through a Cox-2 dependent pathway. Journal of orthopaedic research: official publication of the Orthopaedic Research Society. 2004;22:6–12. doi: 10.1016/S0736-0266(03)00153-0. [DOI] [PubMed] [Google Scholar]
- 104.Punt I, Cleutjens J, de Bruin T, Willems P, Kurtz S, van Rhijn L, et al. Periprosthetic tissue reactions observed at revision of total intervertebral disc arthroplasty. Biomaterials. 2009;30:2079–84. doi: 10.1016/j.biomaterials.2008.12.071. [DOI] [PubMed] [Google Scholar]
- 105.Warashina H, Sakano S, Kitamura S, Yamauchi K, Yamaguchi J, Ishiguro N, et al. Biological reaction to alumina, zirconia, titanium and polyethylene particles implanted onto murine calvaria. Biomaterials. 2003;24:3655–61. doi: 10.1016/s0142-9612(03)00120-0. [DOI] [PubMed] [Google Scholar]
- 106.Koulouvaris P, Ly K, Ivashkiv L, Bostrom M, Nestor B, Sculco T, et al. Expression profiling reveals alternative macrophage activation and impaired osteogenesis in periprosthetic osteolysis. Journal of orthopaedic research: official publication of the Orthopaedic Research Society. 2008;26:106–16. doi: 10.1002/jor.20486. [DOI] [PubMed] [Google Scholar]
- 107.Fujii J, Yasunaga Y, Yamasaki A, Ochi M. Wear debris stimulates bone-resorbing factor expression in the fibroblasts and osteoblasts. Hip international: the journal of clinical and experimental research on hip pathology and therapy. 2011 doi: 10.5301/hip.2011.7977. [DOI] [PubMed] [Google Scholar]
- 108.Tunyogi-Csapo M, Koreny T, Vermes C, Galante J, Jacobs J, Glant T. Role of fibroblasts and fibroblast-derived growth factors in periprosthetic angiogenesis. Journal of orthopaedic research: official publication of the Orthopaedic Research Society. 2007;25:1378–88. doi: 10.1002/jor.20449. [DOI] [PubMed] [Google Scholar]
- 109.Huang Z, Ma T, Ren P-G, Smith R, Goodman S. Effects of orthopedic polymer particles on chemotaxis of macrophages and mesenchymal stem cells. Journal of biomedical materials research Part A. 2010;94:1264–9. doi: 10.1002/jbm.a.32803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gibon E, Batke B, Jawad M, Fritton K, Rao A, Yao Z, et al. MC3T3-E1 osteoprogenitor cells systemically migrate to a bone defect and enhance bone healing. Tissue engineering Part A. 2011;18:968–73. doi: 10.1089/ten.tea.2011.0545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Haleem-Smith H, Argintar E, Bush C, Hampton D, Postma W, Chen F, et al. Biological responses of human mesenchymal stem cells to titanium wear debris particles. Journal of orthopaedic research: official publication of the Orthopaedic Research Society. 2012;30:853–63. doi: 10.1002/jor.22002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Novitskiy G, Potter J, Rennie-Tankersley L, Mezey E. Identification of a novel NF-kappaB-binding site with regulation of the murine alpha2(I) collagen promoter. The Journal of biological chemistry. 2004;279:15639–44. doi: 10.1074/jbc.M311499200. [DOI] [PubMed] [Google Scholar]
- 113.Ollivere B, Wimhurst J, Clark I, Donell S. Current concepts in osteolysis. The Journal of bone and joint surgery British. 94:10–5. doi: 10.1302/0301-620X.94B1.28047. [DOI] [PubMed] [Google Scholar]
- 114.Shi Y, Hu G, Su J, Li W, Chen Q, Shou P, et al. Mesenchymal stem cells: a new strategy for immunosuppression and tissue repair. Cell research. 20:510–8. doi: 10.1038/cr.2010.44. [DOI] [PubMed] [Google Scholar]
- 115.Cao S, Zhang X, Edwards J, Mosser D. NF-kappaB1 (p50) homodimers differentially regulate pro- and anti-inflammatory cytokines in macrophages. The Journal of biological chemistry. 2006;281:26041–50. doi: 10.1074/jbc.M602222200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chang J, Liu F, Lee M, Wu B, Ting K, Zara JN, et al. NF-kappaB inhibits osteogenic differentiation of mesenchymal stem cells by promoting beta-catenin degradation. Proc Natl Acad Sci U S A. 2013;110:9469–74. doi: 10.1073/pnas.1300532110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chang J, Wang Z, Tang E, Fan Z, McCauley L, Franceschi R, et al. Inhibition of osteoblastic bone formation by nuclear factor-kappaB. Nat Med. 2009;15:682–9. doi: 10.1038/nm.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Rakshit D, Ly K, Sengupta T, Nestor B, Sculco T, Ivashkiv L, et al. Wear debris inhibition of anti-osteoclastogenic signaling by interleukin-6 and interferon-gamma. Mechanistic insights and implications for periprosthetic osteolysis. The Journal of bone and joint surgery American volume. 2006;88:788–99. doi: 10.2106/JBJS.E.00711. [DOI] [PubMed] [Google Scholar]
- 119.Yamamoto Y, Gaynor R. Therapeutic potential of inhibition of the NF-kappaB pathway in the treatment of inflammation and cancer. The Journal of clinical investigation. 2001;107:135–42. doi: 10.1172/JCI11914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gilmore T, Herscovitch M. Inhibitors of NF-kappaB signaling: 785 and counting. Oncogene. 2006;25:6887–99. doi: 10.1038/sj.onc.1209982. [DOI] [PubMed] [Google Scholar]
- 121.Schwarz E, Campbell D, Totterman S, Boyd A, O’Keefe R, Looney R. Use of volumetric computerized tomography as a primary outcome measure to evaluate drug efficacy in the prevention of peri-prosthetic osteolysis: a 1-year clinical pilot of etanercept vs. placebo. Journal of orthopaedic research: official publication of the Orthopaedic Research Society. 2003;21:1049–55. doi: 10.1016/S0736-0266(03)00093-7. [DOI] [PubMed] [Google Scholar]
- 122.Sun K, Li Y, Lu Z, Zhang L, Gao Z, Jin Q. Suppression of titanium particle-induced TNF-alpha expression and apoptosis in human U937 macrophages by siRNA silencing. The International journal of artificial organs. 2013 doi: 10.5301/ijao.5000218. [DOI] [PubMed] [Google Scholar]
- 123.Childs L, Goater J, O’Keefe R, Schwarz E. Efficacy of etanercept for wear debris-induced osteolysis. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2001;16:338–47. doi: 10.1359/jbmr.2001.16.2.338. [DOI] [PubMed] [Google Scholar]
- 124.Yang SY, Wu B, Mayton L, Mukherjee P, Robbins P, Evans C, et al. Protective effects of IL-1Ra or vIL-10 gene transfer on a murine model of wear debris-induced osteolysis. Gene therapy. 2004;11:483–91. doi: 10.1038/sj.gt.3302192. [DOI] [PubMed] [Google Scholar]
- 125.Wei S, Kitaura H, Zhou P, Ross F, Teitelbaum S. IL-1 mediates TNF-induced osteoclastogenesis. The Journal of clinical investigation. 2005;115:282–90. doi: 10.1172/JCI23394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cheng T, Zhang X. NFkB gene silencing inhibits wear particles-induced inflammatory osteolysis. Journal of Medical Hypotheses. 2008;71:727–9. doi: 10.1016/j.mehy.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 127.O’Hare T, Corbin A, Druker B. Targeted CML therapy: controlling drug resistance, seeking cure. Current opinion in genetics & development. 2006;16:92–9. doi: 10.1016/j.gde.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 128.O’Connell M, Bennett B, Mercurio F, Manning A, Mackman N. Role of IKK1 and IKK2 in lipopolysaccharide signaling in human monocytic cells. The Journal of biological chemistry. 1998;273:30410–4. doi: 10.1074/jbc.273.46.30410. [DOI] [PubMed] [Google Scholar]
- 129.Clohisy J, Yamanaka Y, Faccio R, Abu-Amer Y. Inhibition of IKK activation, through sequestering NEMO, blocks PMMA-induced osteoclastogenesis and calvarial inflammatory osteolysis. Journal of orthopaedic research: official publication of the Orthopaedic Research Society. 2006;24:1358–65. doi: 10.1002/jor.20184. [DOI] [PubMed] [Google Scholar]
- 130.Mao X, Pan X, Cheng T, Zhang X. Therapeutic potential of the proteasome inhibitor Bortezomib on titanium particle-induced inflammation in a murine model. Inflammation. 2012;35:905–12. doi: 10.1007/s10753-011-9392-7. [DOI] [PubMed] [Google Scholar]
- 131.Tas S, Vervoordeldonk M, Tak P. Gene therapy targeting nuclear factor-kappaB: towards clinical application in inflammatory diseases and cancer. Current gene therapy. 2009;9:160–70. doi: 10.2174/156652309788488569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Childs L, Goater J, O’Keefe R, Schwarz E. Effect of anti-tumor necrosis factor-alpha gene therapy on wear debris-induced osteolysis. The Journal of bone and joint surgery American volume. 2001;83-A:1789–97. doi: 10.2106/00004623-200112000-00004. [DOI] [PubMed] [Google Scholar]
- 133.Osako M, Nakagami H, Morishita R. Modification of decoy oligodeoxynucleotides to achieve the stability and therapeutic efficacy. Current topics in medicinal chemistry. 2012;12:1603–7. doi: 10.2174/156802612803531397. [DOI] [PubMed] [Google Scholar]
- 134.Dinh TD, Higuchi Y, Kawakami S, Yamashita F, Hashida M. Evaluation of osteoclastogenesis via NFkappaB decoy/mannosylated cationic liposome-mediated inhibition of pro-inflammatory cytokine production from primary cultured macrophages. Pharmaceutical research. 2011;28:742–51. doi: 10.1007/s11095-011-0366-0. [DOI] [PubMed] [Google Scholar]
- 135.Desmet C, Gosset P, Pajak B, Cataldo D, Bentires-Alj M, Lekeux P, et al. Selective blockade of NF-kappa B activity in airway immune cells inhibits the effector phase of experimental asthma. Journal of immunology (Baltimore, Md: 1950) 2004;173:5766–75. doi: 10.4049/jimmunol.173.9.5766. [DOI] [PubMed] [Google Scholar]
- 136.Miyake T, Aoki M, Osako M, Shimamura M, Nakagami H, Morishita R. Systemic administration of ribbon-type decoy oligodeoxynucleotide against nuclear factor kappaB and ets prevents abdominal aortic aneurysm in rat model. Molecular therapy: the journal of the American Society of Gene Therapy. 2010;19:181–7. doi: 10.1038/mt.2010.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Miyake T, Aoki M, Morishita R. Inhibition of anastomotic intimal hyperplasia using a chimeric decoy strategy against NFkappaB and E2F in a rabbit model. Cardiovascular research. 2008;79:706–14. doi: 10.1093/cvr/cvn139. [DOI] [PubMed] [Google Scholar]
- 138.Miyake T, Aoki M, Nakashima H, Kawasaki T, Oishi M, Kataoka K, et al. Prevention of abdominal aortic aneurysms by simultaneous inhibition of NFkappaB and ets using chimeric decoy oligonucleotides in a rabbit model. Gene therapy. 2006;13:695–704. doi: 10.1038/sj.gt.3302704. [DOI] [PubMed] [Google Scholar]
- 139.Nakashima H, Aoki M, Miyake T, Kawasaki T, Iwai M, Jo N, et al. Inhibition of experimental abdominal aortic aneurysm in the rat by use of decoy oligodeoxynucleotides suppressing activity of nuclear factor kappaB and ets transcription factors. Circulation. 2004;109:132–8. doi: 10.1161/01.CIR.0000105725.61763.A2. [DOI] [PubMed] [Google Scholar]
- 140.Ogushi I, Iimuro Y, Seki E, Son G, Hirano T, Hada T, et al. Nuclear factor kappa B decoy oligodeoxynucleotides prevent endotoxin-induced fatal liver failure in a murine model. Hepatology (Baltimore, Md) 2003;38:335–44. doi: 10.1053/jhep.2003.50298. [DOI] [PubMed] [Google Scholar]
- 141.Nakamura H, Aoki M, Tamai K, Oishi M, Ogihara T, Kaneda Y, et al. Prevention and regression of atopic dermatitis by ointment containing NF-kB decoy oligodeoxynucleotides in NC/Nga atopic mouse model. Gene therapy. 2002;9:1221–9. doi: 10.1038/sj.gt.3301724. [DOI] [PubMed] [Google Scholar]
- 142.Shimizu H, Nakagami H, Morita S, Tsukamoto I, Osako M, Nakagami F, et al. New treatment of periodontal diseases by using NF-kappaB decoy oligodeoxynucleotides via prevention of bone resorption and promotion of wound healing. Antioxidants & redox signaling. 2009;11:2065–75. doi: 10.1089/ars.2008.2355. [DOI] [PubMed] [Google Scholar]
- 143.Talmo C, Shanbhag A, Rubash H. Nonsurgical management of osteolysis: challenges and opportunities. Clinical orthopaedics and related research. 2006;453:254–64. doi: 10.1097/01.blo.0000246531.59876.a8. [DOI] [PubMed] [Google Scholar]
- 144.Dinh T, Higuchi Y, Kawakami S, Yamashita F, Hashida M. Evaluation of osteoclastogenesis via NFkappaB decoy/mannosylated cationic liposome-mediated inhibition of pro-inflammatory cytokine production from primary cultured macrophages. Pharmaceutical research. 2011;28:742–51. doi: 10.1007/s11095-011-0366-0. [DOI] [PubMed] [Google Scholar]
- 145.Clohisy J, Hirayama T, Frazier E, Han S-K, Abu-Amer Y. NF-kB signaling blockade abolishes implant particle-induced osteoclastogenesis. Journal of orthopaedic research: official publication of the Orthopaedic Research Society. 2004;22:13–20. doi: 10.1016/S0736-0266(03)00156-6. [DOI] [PubMed] [Google Scholar]
- 146.de Poorter J, Tolboom T, Rabelink M, Pieterman E, Hoeben R, Nelissen R, et al. Towards gene therapy in prosthesis loosening: efficient killing of interface cells by gene-directed enzyme prodrug therapy with nitroreductase and the prodrug CB1954. The journal of gene medicine. 2005;7:1421–8. doi: 10.1002/jgm.795. [DOI] [PubMed] [Google Scholar]
- 147.Lavon I, Goldberg I, Amit S, Landsman L, Jung S, Tsuberi B, et al. High susceptibility to bacterial infection, but no liver dysfunction, in mice compromised for hepatocyte NF-kappaB activation. Nature medicine. 2000;6:573–7. doi: 10.1038/75057. [DOI] [PubMed] [Google Scholar]
- 148.Fabre C, Mimura N, Bobb K, Kong S-Y, Gorgun G, Cirstea D, et al. Dual inhibition of canonical and noncanonical NF-kappaB pathways demonstrates significant antitumor activities in multiple myeloma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18:4669–81. doi: 10.1158/1078-0432.CCR-12-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Stein B, Baldwin A, Ballard D, Greene W, Angel P, Herrlich P. Cross-coupling of the NF-kappa B p65 and Fos/Jun transcription factors produces potentiated biological function. The EMBO journal. 1993;12:3879–91. doi: 10.1002/j.1460-2075.1993.tb06066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289:2350–4. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Rao AJ, Gibon E, Ma T, Yao Z, Smith RL, Goodman SB. Revision joint replacement, wear particles, and macrophage polarization. Acta biomaterialia. 2012;8:2815–23. doi: 10.1016/j.actbio.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]