

Abstract
Nine-membered enediyne antitumor antibiotics C-1027, neocarzinostatin (NCS), and kedarcidin (KED) possess enediyne cores to which activity-modulating peripheral moieties are attached via (R)- or (S)-vicinal diols. We have previously shown that this stereochemical difference arises from hydrolysis of epoxide precursors by epoxide hydrolases (EHs) with different regioselectivities – the “inverting” EH, such as SgcF, hydrolyzes an (S)-epoxide substrate to yield an (R)-diol in C-1027 biosynthesis, while the “retaining” EHs, such as NcsF2 and KedF, hydrolyze an (S)-epoxide substrate to yield an (S)-diol in NCS and KED biosynthesis. We now report the characterization of a series of EH mutants and provide a predictive model for EH regioselectivity in the biosynthesis of the 9-membered enediyne antitumor antibiotics. A W236Y mutation in SgcF increased the retaining activity towards (S)-styrene oxide 3-fold, and a W236Y/Q237M double mutation in SgcF, mimicking NcsF2 and KedF, resulted in a 20-fold increase in the retaining activity. To test the predictive utility of these mutations, two putative enediyne biosynthesis-associated EHs were identified by genome mining and confirmed as inverting enzymes – SpoF from Salinospora tropica CNB-440 and SgrF (SGR_625) from Streptomyces griseus IFO 13350. Finally, phylogenetic analysis of EHs revealed a familial classification according to inverting versus retaining activity. Taken together, these results provide a predictive model for the vicinal diol stereochemistry in enediyne biosynthesis and set the stage for further elucidating the origins of EH regioselectivity.
The exponential growth of genomic data in the last decade has provided a great deal of information about microbial natural product biosynthesis and ushered in an era of genome mining for new molecules of potential therapeutic value.1,2 However, it has also highlighted challenges associated with accurately predicting chemical structure from genetic information.3 For example, many biosynthetic clusters possess unknown genes, and even genes with high sequence similarity may encode enzymes catalyzing unexpected reactions.4,5
The enediyne antitumor antibiotics are a family of microbial natural products that exemplify this information gap between genes and chemical structure. Enediynes are potent cytotoxic compounds in clinical use as anticancer agents that function by a unique DNA cleavage mechanism.6–9 This unusual biological activity originates from the fascinating chemical structure comprised of a central enediyne core and activity-modulating peripheral moieties (Figure 1A). Several biosynthetic gene clusters have been sequenced and characterized, leading to a proposed convergent biosynthesis in which each of the peripheral moieties is assembled prior to attachment to the central core.10–14 Although these clusters have revealed a great deal about peripheral group biosynthesis,15,16 the assembly and modification of the enediyne core itself remains poorly understood. For instance, three of the five genes that comprise the conserved “enediyne cassette” are of unknown function, and most of the genes that modify the core have not been identified.17,18
Figure 1.

(A) Structures of the enediyne chromophores of chromoproteins C-1027 (1), neocarzinostatin (NCS, 2), and kedarcidin (KED, 3), with respective (R)- and (S)-vicinal diol stereochemistry highlighted. (B) The stereochemical outcome from hydrolysis of (S)-4 catalyzed by SgcF, NcsF2, and KedF, the respective EHs from the 1, 2, and 3 biosynthetic machinery. Arrows indicate the regioselectivity of epoxide ring opening in each enzyme reaction.
Despite the difficulties associated with characterizing the enediyne core biosynthetic machinery, we have successfully identified genes involved in modifying a putative 9-membered enediyne core intermediate. Each of the three 9-membered biosynthetic gene clusters [C-1027 (1), neocarzinostatin (NCS, 2), and kedarcidin (KED, 3)] encode an epoxide hydrolase (EH), SgcF, NcsF2, and KedF, respectively, proposed to generate a vicinal diol intermediate from an epoxide precursor.19–21 Intriguingly, the diol stereochemistry differs among these enediynes – an (R)-diol for C-1027 in comparison to the (S)-diols for NCS and KED (Figure 1A). Biochemical characterization using styrene oxide (4) as a substrate mimic demonstrated catalytic activities consistent with a biosynthetic scheme in which an (S)-epoxide is directed to different stereochemical outcomes by EHs with differing regioselectivities (Figure 1B). Specifically, SgcF acts as an “inverting” enzyme by regioselectively catalyzing the net addition of water at the more hindered carbon of (S)-4 to invert the stereochemistry, yielding (R)-1-phenyl-1,2-ethanediol [(R)-5]. In contrast, the “retaining” enzymes NcsF2 and KedF attack at the less hindered carbon of (S)-4 to afford (S)-1-phenyl-1,2-ethanediol [(S)-5], retaining the (S)-configuration.
These three highly related EHs present an outstanding opportunity to probe the basis for regioselectivity in enediyne biosynthesis and bioinformatically assign the stereochemical configuration of new enediynes. Herein we identify a rare EH tyrosine-to-tryptophan mutation in SgcF that influences regioselectivity. Genome mining for this mutation in enediyne biosynthesis-associated EHs identified two additional enzymes (SpoF from Salinospora tropica CNB-44022 and SgrF from Streptomyces griseus IFO 1335023) that were biochemically confirmed as inverting EHs. The predictive utility of this model was expanded by the observed phylogenetic separation of enediyne biosynthesis-associated EHs into inverting and retaining groups. In addition, the location of spoF within the gene cluster encoding the biosynthesis of the (R)-diol-containing sporolides suggests that this sequence-based model may be used to predict the stereochemical configuration of proposed 9-membered enediynes identified from genome sequencing projects.
MATERIALS AND METHODS
General
DNA sequencing and oligonucleotide synthesis was performed by the University of Wisconsin-Madison Biotechnology Center. Chemicals including racemic, (R)-, and (S)-styrene oxide, and racemic, (R)- and (S)-1-phenyl-1,2-ethanediol were purchased from Sigma-Aldrich (St. Louis, MO). Dithiothreitiol (DTT) was purchased from Research Products International (Mt. Prospect, IL) and Complete Protease Inhibitor was purchased from Roche Applied Science (Indianapolis, IN). Media components and buffers were from Fisher Scientific (Pittsburgh, PA). PCR amplification used Pfx polymerase (Invitrogen, Carlsbad, CA), and 3′ A-overhangs were added with Taq polymerase from Invitrogen for 10 min at 72 ºC prior to subcloning into pGEM-T Easy (Promega, Madison, WI). Sequence alignments and phylogenetic analyses were done with ClustalX, and phylogenetic trees were drawn using Hypertree or MEGA.24 The homology models were constructed using SWISS-MODEL.25 Primers (Table S1), plasmids (Table S2) and sequence comparison of the five EHs (Table S3) are summarized. Sequence alignment of EHs (Fig. S1), SDS-PAGE analysis of the purified EHs (Fig. S2), and curve-fitted steady-state kinetic data (Fig. S3) are provided.
Cloning of the sgrF and spoF genes that encode putative EHs
The sgrF gene was PCR-amplified from plasmid TGL_028_D08 using primers SGR625-NdeI-F and SGR625-XhoI-R (Table S1). The plasmid TGL_028_D08 contains ~12 kb of Streptomyces griseus IFO 13350 genomic DNA23 inserted into the HincII site of the pTS1 vector (Nippon Genetech, Tokyo, Japan), and includes the EH gene SGR625 that we have renamed sgrF. The sgrF PCR product possessed an added 3′ XhoI restriction site and an added 5′ NdeI restriction site together with the DNA encoding the N-terminal amino acid sequence MAH6VD4K of the pCDF-2 Ek/LIC vector (Novagen, Madison WI) in order to generate protein with N-terminal features identical to SgcF produced from pBS1096.19 This PCR product was then cloned into pGEM-T Easy (Promega, Madison WI) to yield pBS1118 and confirmed by DNA sequencing. The sgrF gene was then cloned as an NdeI-XhoI fragment into pET29 (Novagen, Madison WI) to afford the sgrF expression construct pBS1119. The spoF gene was cloned using the identical strategy by PCR-amplification from Salinospora tropica CNB-440 genomic DNA22 using primers SpoF-NdeI-F and SpoF-XhoI-R (Table S1). The PCR product was cloned into pGEM-T Easy to make pBS1120, from which the NdeI-XhoI fragment was transferred into pET29 to produce the SpoF expression construct pBS1121.
Mutant SgcF (W236Y)
Site-directed mutagenesis was carried out by PCR overlap extension from the pBS1096 template.19 To generate the SgcF (W236Y) mutant, two sets of primers (SgcF-W236Y-F + SgcF-W236Y-mutR; SgcF-W236Y-mutF + SgcF-W236Y-R) (Table S1) were used to PCR-amplify two SgcF fragments of 417 and 170 bp, respectively, that overlapped at the mutation site located in the primers SgcF-W236Y-mutF and SgcF-W236Y-mutR. The two PCR fragments were then used as template to amplify the entire 566 bp fragment containing the mutation using the flanking primers SgcF-W236Y-F and SgcF-W236Y-R (Table S1). This fragment was cloned into pGEM-T Easy to afford pBS1122, which was confirmed by DNA sequencing. A 510 bp AatII-SmaI fragment containing the mutation was then cloned from pBS1122 back into pBS1096 to produce the expression construct pBS1123 (Table S2).
Mutant SgcF (W176L)
The same mutagenic strategy as above was employed but using the mutagenic primers SgcF-W176L-mutF and SgcF-W176L-mutR (Table S1). The resulting 566 bp product of PCR overlap extension was cloned into pGEM-T Easy to yield pBS1124 and confirmed by sequencing. The 510 bp fragment possessing the W176L mutation was cloned back into pBS1096 to afford the expression construct pBS1125 (Table S2).
Mutant SgcF (Q237M)
The above strategy was used with primers SgcF-Q237M-mutF and SgcF-Q237M-mutR (Table S1), and the 566 bp product was cloned into pGEM-T Easy to create pBS1126. The 510 bp AatII-SmaI fragment with the Q237M mutation was cloned back into pBS1096 to generate the expression construct pBS1127 (Table S2).
Mutant SgcF (W236Y/Q237M)
The above PCR overlap extension strategy was employed but using pBS1123 as template and primers SgcF-W236Y/Q237M-mutF and SgcF-W236Y/Q237M-mutR (Table S1). The mutated PCR product was cloned into pGEM-T Easy to make pBS1128, from which the AatII-SmaI fragment was moved back into pBS1096 to afford the expression construct pBS1129 (Table S2).
Mutants SgcF (W176L/Q237M) and SgcF (W176L/W236Y/Q237M)
The 315 bp AatII-XcmI fragment from the W176L-containing pBS1124 was cloned into the same sites of the Q237M-containing pBS1126 and the W236Y/Q237M-containing pBS1128 to afford pBS1130 and pBS1131, respectively. The AatII-SmaI fragments from the W176L/Q237M-containing pBS1130 and the W176L/W236Y/Q237M-containing pBS1131 were cloned back into pBS1096 to yield the expression constructs pBS1132 and pBS1133, respectively (Table S2).
Mutant NcsF2 (Y235W)
PCR overlap extension was performed as above but using the template pBS504220 and the two primer sets (NcsF2-Y235W-F + NcsF2-Y235W-mutR; NcsF2-Y235W-mutF + NcsF2-Y235W-R) (Table S1) to yield respective overlapping PCR fragments of 410 and 470 bp. These fragments were used as template to amplify the entire 860 bp mutated fragment using the flanking primers NcsF2-Y235W-F and NcsF2-Y235W-R (Table S1), which was then cloned into pGEM-T Easy to make pBS5043. After confirmation by DNA sequencing, a 490 bp PstI-AatII fragment was cloned from pBS5043 back into the ncsF2-containing expression plasmid pBS5042 to afford pBS5044 (Table S2).
Mutant NcsF2 (L176W)
The same mutagenic strategy as for NcsF2 (Y235W) was employed but with mutagenic primers NcsF2-L176W-mutF and NcsF2-L176W-mutR (Table S1). The resulting 860 bp product of PCR overlap extension was cloned into pGEM-T Easy to yield pBS5045 and confirmed by sequencing. The 490 bp PstI-AatII fragment possessing the L176W mutation was cloned back into pBS5042 to afford the expression construct pBS5046 (Table S2).
Mutant NcsF2 (M236Q)
The above strategy was used with primers NcsF2-M236Q-mutF and NcsF2-M236Q-mutR (Table S1), and the 860 bp product was cloned into pGEM-T Easy to create pBS5047. The 490 bp PstI-AatII fragment with the M236Q mutation was cloned back into pBS5042 to generate the expression construct pBS5048 (Table S2).
Mutant NcsF2 (Y235W/M236Q)
The above PCR overlap extension strategy was employed but using pBS5044 as template and primers NcsF2-Y235W/M236Q-mutF and NcsF2-Y235W/M236Q-mutR (Table S1). The mutated PCR product was cloned into pGEM-T Easy to make pBS5049, from which the PstI-AatII fragment was moved back into pBS5042 to afford the expression construct pBS5050 (Table S2).
Mutants NcsF2 (L176W/M236Q) and NcsF2 (L176W/Y235W/M236Q)
The 490 bp PstI-AatII fragment from either the M236Q-containing pBS5047 or Y235W/M236Q-containing pBS5049 was cloned into the same site of L176W-containing pBS5046 to afford the expression constructs pBS5051 and pBS5052, respectively (Table S2).
Overproduction and purification of EHs
Introduction of the various constructs expressing wild-type EHs and their variants (Table S2) into E. coli BL21(DE3) and overproduction and purification of the resultant EHs were performed as previously described.19
EH activity assays towards styrene oxide
HPLC assays were performed in 200 μL reaction mixtures containing 2 mM styrene oxide and 50 mM potassium phosphate buffer, pH 8.0, following the previously described procedure.19 Thus, the reaction was initiated by adding 50 μM enzyme and incubated at 25 ºC for 1 h. The reaction was quenched by extraction with ethyl acetate (3 x 200 μL) and the organic extract was evaporated to dryness in a speed-vac. The resulting residue was dissolved in 50 μL of acetonitrile and 25 μL was analyzed by HPLC. Control reactions without enzyme were carried out in parallel. For general activity assays, the substrate used was racemic styrene oxide and the HPLC was performed on a Varian HPLC system equipped with Prostar 210 pumps, a photodiode array detector, and an Alltech Alltima C18 column (5 μm, 4.6 x 250 mm, Grace Davison Discovery Sciences, Deerfield, IL) using a 12 min linear gradient from 10 to 50% acetonitrile in water. Chiral HPLC was performed on an Agilent 1260 HPLC system equipped with a Chiralcel OD-H column (5 μm, 4.6 x 250 mm, Grace Davison Discovery Sciences) using a 70 min isocratic elution with 2.5% isopropanol in n-hexane.
Steady-state kinetics of EHs
Enzyme kinetic assays were performed as previously described.19 Briefly, the hydrolysis of each enantiomer of styrene oxide with each enzyme was kinetically characterized at by adding an appropriate amount of enzyme to a 1 mL reaction mixture containing 10 μL of 300 mM sodium periodate in DMF, 20 μL of an appropriate concentration of styrene oxide, and 50 mM sodium phosphate buffer at pH 8.0. Product formation was monitored by the increase in absorbance at 290 nm over time at 25 ºC. Equations for Michaelis-Menten kinetics with or without substrate inhibition were fit to the data by nonlinear regression analysis of initial velocity versus substrate concentration using the online curve-fitting tools at http://zunzun.com.
RESULTS AND DISCUSSION
Sequence comparisons to identify regioselectivity determinants
Sequence alignments between the three characterized enediyne biosynthesis-associated EHs (SgcF, NcsF2, and KedF) revealed highly similar enzymes with 62–64% identity and 74–75% similarity (Table S3). These enzymes possess features typical of canonical EHs of the α/β-hydrolase fold family,26 most notably the presence of two tyrosine residues that serve to anchor and activate the epoxide towards nucleophilic attack (Figure 2, Figure S1). Strikingly, although the retaining enzymes NcsF2 and KedF possess both tyrosine residues (Y235 and Y304, Figure 2A), one of these tyrosines has been replaced by tryptophan (W236) in the inverting enzyme SgcF (Figure 2B, Figure S1). The substitution of this key conserved residue in an inverting enzyme of otherwise high sequence similarity with retaining enzymes suggests that this substitution may influence regioselectivity of the enzyme reaction (Figure 2). Intriguingly, a role for tyrosine residues in directing regioselective epoxide ring opening by EHs has been proposed based on molecular dynamics simulations.27 We were therefore motivated to study the role of the SgcF Y236W substitution in directing EH regioselectivity (i.e. inversion versus retention).
Figure 2.

A comparison of the proposed reactions catalyzed by (A) the retaining EHs NcsF2 and KedF, which possess a full complement of two conserved tyrosine residues, and (B) the inverting EH SgcF, in which one of the tyrosines is replaced by tryptophan. The square parentheses denote that the reaction is hypothetical.
Characterization of the regioselectivity of the SgcF (W236Y) variant
To assess the effect of the tryptophan substitution on regioselectivity we constructed the SgcF (W236Y) variant possessing the full complement of two tyrosine residues. If these two tyrosines are important for retaining activity then installation of this canonical active site feature should increase this activity. Similarly, the NcsF2 (Y235W) variant was constructed to determine if this mutation could increase inverting activity of NcsF2. The His6-tagged variant enzymes were purified and their activity towards (±)-4 was assessed by HPLC. Unexpectedly, although both variants were soluble, only SgcF (W236Y) was active, demonstrating that the Y235W mutation severely disrupted NcsF2 catalysis. The regioselectivity of SgcF (W236Y)-catalyzed hydrolysis of (R)- and (S)-4 was then characterized by chiral HPLC analysis of the diol products (Figure 3). Consistent with a role for this residue in directing regioselectivity, the retaining activity of the SgcF (W236Y) variant towards (S)-4 increased 3-fold relative to wild-type SgcF. Specifically, whereas only 1.7% of SgcF catalytic turnovers (~1 in 60) produced (S)-5 from (S)-4, this was increased to 4.6% of turnovers (~1 in 20) for SgcF (W236Y) (Table 1, Figure 4). Similarly, the W236Y mutation increased retaining activity towards the (R)-4 substrate to obtain (R)-5 from 83% of turnovers relative to 70% in wild-type SgcF. Thus, although the SgcF (W236Y) variant was only modestly more retaining towards (S)-4, this mutation clearly affects regioselectivity.
Figure 3.
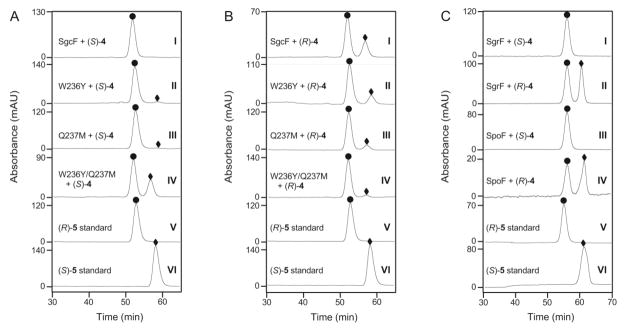
Chiral HPLC analysis of the stereochemistry of 5 generated by EHs. (A) Products of (S)-4 hydrolysis catalyzed by SgcF: wild-type (I), W236Y (II), Q237M (III), W236Y/Q237M (IV). (B) Products of (R)-4 hydrolysis catalyzed by SgcF: wild-type (I), W236Y (II), Q237M (III), W236Y/Q237M (IV). (C) Products of hydrolysis of: (S)-4 by SgrF (I), (R)-4 by SgrF (II), (S)-4 by SpoF (III), (R)-4 by SpoF (IV). In panels A–C, traces V and VI represent (R)-5 (●) and (S)-5 (◆) standards, respectively.
Table 1.
Regioselectivity of SgcF wild-type and variants towards (R)- and (S)-4 reported as percent enantiomeric excess (% ee) of the product (R)-5, % retaining activity, and the difference in free energy of activation between hydrolysis at each epoxide carbon (ΔΔG‡).a
| (R)-4 substrate | (S)-4 substrate | |||||
|---|---|---|---|---|---|---|
| EH | % ee of (R)-5 | % retain | ΔΔG‡ (kJ/mol)b | % ee of (R)-5 | % retain | ΔΔG‡ (kJ/mol)b |
| SgcF wild-type | 40.1 ± 2.5 | 70 | −2.10 | 96.6 ± 0.1 | 1.7 | −10.03 |
| SgcF (W236Y) | 65.6 ± 3.6 | 83 | −3.90 | 90.8 ± 3.4 | 4.6 | −7.62 |
| SgcF (Q237M) | 79.6 ± 5.2 | 90 | −5.44 | 95.9 ± 0.6 | 2.1 | −9.58 |
| SgcF (W236Y/Q237M) | 88.5 ± 0.5 | 94 | −6.93 | 28.4 ± 4.9 | 36 | −1.45 |
Figure 4.

Comparison of regio- and enantioselectivities of EHs. Regioselectivity is depicted as % of turnovers that result in retention of stereochemistry of the substrate (S)-4. Enantioselectivity describes the ratio of specificity constants for (R)-4 versus (S)-4. Positive values represent an enzyme preference for (R)-4 [(kcat/Km)R/(kcat/Km)S], and negative values denote a preference for (S)-4 [-(kcat/Km)S/(kcat/Km)R].
Structure-based assessment of additional potential regioselectivity determinants
The modest change in regioselectivity accompanying the W236Y mutation suggests that other mutations act in concert with W236 to repurpose the SgcF active site for inversion. To search for such determinants we revisited the sequence comparisons looking for residues that were conserved only among the retaining enzymes NcsF2 and KedF but not in the inverting enzyme SgcF. We elected to limit our search to amino acids in the active site by using a homology model based on the crystal structure of the EH from Aspergillus niger (AnEH), which is 30% identical to SgcF and NcsF2 (Figure 5). This served to focus our efforts on two additional active site residue positions occupied by similar amino acids in the retaining enzymes compared to the inverting enzyme SgcF: (i) the residue adjacent to W236 in SgcF is Q237, while the residue adjacent to Y235 in NcsF2 and KedF is M236 and (ii) position 176 is occupied by the aromatic amino acid W in SgcF, but by hydrophobic residues L and A in NcsF2 and KedF, respectively (Figure 5, Figure S1).
Figure 5.

Comparison of NcsF2 (magenta CPK colors) and SgcF (green CPK colors) active sites based on a homology model with AnEH (PDB ID 1qo7).
Combinatorial libraries of SgcF and NcsF2 variants were prepared and tested for regioselectivity. Mutagenesis at each of these three residues individually and in combination were prepared to afford libraries of (i) SgcF variants possessing NcsF2-like amino acid substitutions and (ii) NcsF2 variants possessing SgcF-like substitutions. Thus a total of seven variants for each enzyme were created in order to explore all possible combinations. Although the additional NcsF2 variants and the SgcF (W176L) variant were insoluble, two SgcF variants, Q237M and W236Y/Q237M, could be produced and purified as soluble enzymes (Figure S2A). The regioselectivity towards both (±)-4 was then assessed using chiral HPLC (Figure 3). Although the single mutation Q237M had almost no effect on regioselectivity towards (S)-4, the combination of substitutions in the W236Y/Q237M variant produced a 21-fold increase in retaining activity to 36%, or more than 1 in 3 enzyme turnovers (Table 1, Figure 3A panel IV, Figure 4). In summary, the W236Y/Q237M mutation significantly increased the retaining activity of SgcF towards both (R)- and (S)-4 such that it becomes more “NcsF2-like” (Figure 4). Interestingly, the inability of NcsF2 to accept SgcF-like mutations, in contrast to the relative ease with which SgcF can accommodate NcsF2-like mutations, suggests that SgcF evolved from an NcsF2-like ancestor.
We next kinetically characterized each enzyme to characterize the effects of mutations in more detail (Table 2, Figure S3). Steady-state kinetic parameters of each variant towards(R)- and (S)-4 were determined using a previously described colorimetric assay19 and are reported in Table 2. Although the W236Y mutation had little effect on the kinetic parameters towards (S)-4, it significantly increased kcat (2-fold) and decreased Km (70-fold) to produce a 150-fold increase in kcat/Km towards (R)-4. Thus, this single mutation switched the enantioselectivity of SgcF from an S- to R-preference (Figure 4). In contrast, the Q237M mutation depleted activity towards both enantiomers to maintain the overall enantioselectivity of the wild-type enzyme. Interestingly, the W236Y/Q237M variant maintained the lower specificity of Q237M towards (S)-4 and the elevated specificity of W236Y towards (R)-4 to elicit a further 60-fold increase in R-selectivity relative to the W236Y enzyme. Taken together, the results of the kinetic analyses demonstrate a trend towards increasing “NcsF2-like” behavior as NcsF2-like mutations are acquired in SgcF (Figure 5) and reveal the influence on substrate selectivity that these mutations impart on enediyne biosynthesis-associated EHs.
Table 2.
Steady-state kinetic parameters for EHs towards (R)- and (S)-4 as substrates.
| (R)-4 substrate | (S)-4 substrate | ||||||
|---|---|---|---|---|---|---|---|
| EH | kcat (min−1) | Km (mM) | kcat/Km | kcat (min−1) | Km (mM) | kcat/Km | Eg |
| SgcF wild-type | 7.5 ± 0.7 | 2.8 ± 1.1 | 2.68 | 48 ± 3 | 0.89 ± 0.41 | 53.9 | 20.1 |
| SgcF (W236Y)a | 16 ± 1 | 0.041 ± 0.063 | 400 | 35 ± 10 | 0.92 ± 0.85 | 38.0 | −10.5 |
| SgcF (Q237M) | NDb | NDb | ~0.04c | 3.2 ± 0.2 | 4.1 ± 0.8 | 0.78 | 19.5 |
| SgcF (W236Y/Q237M)d | 11.3 ± 0.6 | 0.031 ± 0.034 | 355 | 3.3 ± 1 | 6.3 ± 3.1 | 0.52 | −677 |
| NcsF2e | 133 ± 4 | 0.5 ± 0.1 | 266 | 31 ± 2 | 5.0 ± 0.6 | 6.0 | −44 |
| KedFf | 35 ± 2.4 | 3.5 ±0.64 | 10.0 | 36.6 ± 1.1 | 0.91 ± 0.1 | 40.2 | 4.0 |
| SpoF | 3.4 ± 0.4 | 2.8 ± 0.9 | 1.25 | 34 ± 3 | 1.1 ± 0.6 | 32.3 | 25.8 |
| SgrF | 7.9 ± 0.6 | 2.6 ± 0.6 | 3.11 | 43 ± 4 | 0.88 ± 0.45 | 48.9 | 15.7 |
Best fit parameters to a modified Michaelis-Menten equation including substrate inhibition: for (R)-4, Ki = 38 mM; for (S)-4, Ki = 26 mM.
Not determined (ND).
Estimate based on apparent linearity of initial rate versus substrate concentration plot.
Best fit parameters to a modified Michaelis-Menten equation including substrate inhibition: for (R)-4, Ki = 20 mM; for (S)-4, Ki = 18 mM.
Values obtained from reference.20
Values obtained from reference.21
Refers to the enantioselectivity [kcat/Km(fast)]/[kcat/Km(slow)] where for positive values (S)-4 is the preferred (fast) substrate and for negative values (R)-4 is faster.
Despite the clear effects of these mutations on regio- and enantioselectivity, our results show that they are only partially responsible and the complete suite of mutations capable of refashioning the active site remain to be identified. For example, although the SgcF (W236Y) mutation imparts NcsF2-like enantioselectivity, KedF possesses the same YM sequence as NcsF2 but opposite enantioselectivity (Table 2). Thus active site features other than these residues are responsible for directing enantioselectivity in KedF versus NcsF2, and the unusual (R)-4 preference of NcsF2 may reflect the different shape of the putative NcsF2 enediyne substrate resulting from the additional adjacent epoxide20 (Figure 2).
The tryptophan residue can predict EH regioselectivity and enediyne stereochemistry
The above biochemical characterization revealed that the W236Y and W236Y/Q237M variants possessed the most perturbed regioselectivity towards (S)-4, the enantiomer proposed to be the most biosynthetically relevant as a substrate mimic. We therefore set out to search for enediyne biosynthesis-associated EHs possessing this tyrosine-to-tryptophan substitution in order to characterize their regioselectivity and test if this mutation can predict inverting activity. A BLAST search of the NCBI database produced many (~20) hits with high sequence identity (>50%) to SgcF, and the top 3 hits were EHs from strains known to possess enediyne biosynthetic genes: Streptomyces griseus IFO 13350 (SgrF, 72% identity),23 Salinospora tropica CNB-440 (SpoF, 70% identity),22 as well as NcsF2 (62% identity). Interestingly, both SgrF and SpoF possess the Y-to-W mutation: SgrF has the identical WQ motif observed in SgcF, and SpoF has WR (Figure S1).
The regioselectivities of the predicted inverting enzymes SgrF and SpoF were then assessed biochemically. The genes encoding SgrF and SpoF were cloned from S. griseus IFO 13350 and S. tropica CNB-440, respectively, and purified His6-tagged enzymes were obtained by Ni-affinity chromatography (Figure S2B). As expected, both enzymes catalyzed stereochemical inversion of (S)-4 to afford (R)-5 in very high enantiomeric purity (Figure 3C). Moreover, steady-state kinetic parameters of both enzymes were very similar to those obtained for SgcF, with ~20-fold preference for the (S)-enantiomer of 4 (Table 2). In summary, we have correctly predicted EH regioselectivity based on the presence of a single mutation.
Because vicinal diol stereochemistry of 9-membered enediyne cores is a consequence of EH regioselectivity, our predictive model for the latter can be extended to predict the former. Specifically, our current data indicates that inverting EHs (SgcF, SgrF, SpoF) generate enediyne cores featuring a (R)-vicinal diol and the more typical (i.e. containing two tyrosines) retaining EHs (NcsF2, KedF) yield (S)-vicinal diols. Fortuitously, the gene for the inverting SpoF enzyme is located within the biosynthetic gene cluster encoding sporolides A and B, polycyclic macrolides proposed to arise from a 9-membered enediyne precursor containing an (R)-vicinal diol.22, 28 This further supports our predictive model linking EH gene sequence to the vicinal diol stereochemistry of 9-membered enediyne cores.
Phylogenetic analysis provides a familial classification of inverting and retaining EHs
Although the tyrosine-to-tryptophan substitution appears to play a role in directing regioselectivity, it is clearly not the only determinant. To assess if the overall EH protein sequences can be classified as inverting or retaining without specific knowledge of this mutation, we employed phylogenetic analysis on bona fide inverting (SgcF, SgrF, SpoF) and retaining (NcsF2, KedF) enzymes. The results demonstrate that (i) EHs associated with enediyne biosynthesis cluster together within the larger family of EHs for which extensive phylogenetic analysis has been done26 (Figure 6B), and (ii) these enediyne biosynthesis-associated EHs can be classified according to inverting or retaining activity (Figure 6A). Taken together with the biochemical data, this strongly suggests that EH sequences can be used to predict inverting or retaining activity, and therefore respective (R)- or (S)-vicinal diol stereochemistry of 9-membered enediyne cores.
Figure 6.
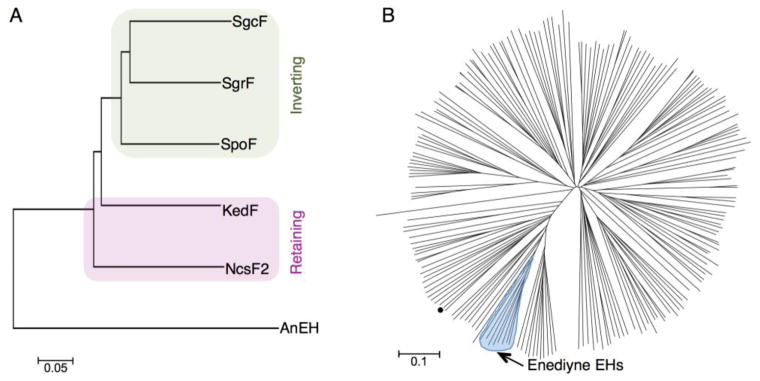
Phylogenetic analyses of EHs. (A) The biochemically characterized enediyne-associated EHs in a phylogram rooted with AnEH. (B) An unrooted phylogram of diverse EHs, including enediyne-associated EHs, constructed from sequences used previously.26 AnEH is denoted by the black-filled circle. A complete list of sequences can be found in SI.
Implications for unknown enediynes
The S. griseus IFO 13350 genome23 possesses a cluster of enediyne biosynthetic genes including the five-gene enediyne cassette (pksE, E3, E4, E5, E10) that is absolutely conserved among all enediyne clusters sequenced to date.10–14,17,18,21 This cluster most probably encodes for production of a 9-membered enediyne because it harbors genes that are apparently conserved only among 9-membered enediynes (e.g. E2, E6–E9, E11), and the SgrF enzyme is phylogenetically clustered with other EHs that are all associated with 9-membered enediyne biosynthesis. The location of the sgrF gene, ~15 kb from pksE, is consistent with the location of SgcF, NcsF2, and KedF within their respective gene clusters, and sgrF is flanked by nearby genes with homology to those from 9-membered enediyne clusters such as mdpL and mdpD2. Thus, together with our results showing that sgrF is biochemically and phylogenetically an inverting EH, we predict that the enediyne product from S. griseus IFO 13350 is a 9-membered enediyne possessing an (R)-vicinal diol.
CONCLUSIONS
We have presented a predictive model that links gene sequence to molecular structure in 9-membered enediyne biosynthetic machinery. EHs from enediyne biosynthetic gene clusters can be phylogenetically classified as retaining or inverting (Figure 6A), and the latter activity originates in part from the unusual loss of one of the two tyrosines that are conserved among EHs of the α/β-hydrolase fold family. In addition, for all EHs characterized that are associated with production of a known compound, inverting and retaining EHs generate the (R)- and (S)-vicinal diols of enediyne cores, respectively. This model allows us to predict that S. griseus IFO 13350 may produce an enediyne core featuring a (R)-vicinal diol, and will no doubt aid structure prediction of enediynes discovered from future genome sequencing projects. Finally, these studies provide a model system with which to study the molecular determinants of EH regioselectivity in greater detail.
Supplementary Material
Acknowledgments
Funding
This work was supported in part by the National Institutes of Health grants GM085770 (to B.S.M) and CA78747 (to B.S.). G.P.H. is a Natural Sciences and Engineering Research Council of Canada (NSERC) postdoctoral fellow.
ABBREVIATIONS
- EH
epoxide hydrolase
- KED
kedarcidin
- NCS
neocarzinostatin
Footnotes
The authors declare no competing financial interests.
Included in SI are Tables S1–S4 and Figs. S1–S3. Table S1 summarizes all the primers, Table S2 contains all the plasmids, Table S3 highlights the sequence homology among the five enediyne biosynthesis-associated EHs described in this study, and Table S4 lists the EH sequences used to construct the phylogram in Fig. 6B. Fig. S1 shows sequence alignment of the five enediyne biosynthesis-associated EHs, highlighting the catalytic triad residues and the oxirane-binding and other active site residues that were mutated in this study. Fig. S2 confirmed the homogeneity of the EHs used in this study. Fig. S3 summarized steady-state-kinetic analyses of the EHs with (R)- and (S)-styrene oxide as substrates. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bode HB, Müller R. The impact of bacterial genomics on natural product research. Angew Chem Int Ed. 2005;44:6828–6846. doi: 10.1002/anie.200501080. [DOI] [PubMed] [Google Scholar]
- 2.Cox R, Piel J, Moore BS, Weissman KJ. Editorial: genomics themed issue. Nat Prod Rep. 2009;26:1353–1508. [Google Scholar]
- 3.Walsh CT, Fischbach MA. Natural products version 2.0: connecting genes to molecules. J Am Chem Soc. 2010;132:2469–2493. doi: 10.1021/ja909118a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palmer DRJ, Garrett JB, Sharma V, Meganathan R, Babbitt PC, Gerlt JA. Unexpected divergence of enzyme function and sequence: “N-acylamino acid racemase” is o-succinylbenzoate synthase. Biochemistry. 1999;38:4252–4258. doi: 10.1021/bi990140p. [DOI] [PubMed] [Google Scholar]
- 5.Van Lanen SG, Lin S, Shen B. Biosynthesis of the enediyne antitumor antibiotic C-1027 involves a new branching point in chorismate metabolism. Proc Natl Acad Sci USA. 2008;105:494–499. doi: 10.1073/pnas.0708750105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galm U, Hager MH, Van Lanen SG, Ju J, Thorson JS, Shen B. Antitumor antibiotics: bleomycin, enediynes, and mitomycin. Chem Rev. 2005;105:739–758. doi: 10.1021/cr030117g. [DOI] [PubMed] [Google Scholar]
- 7.Maeda H. SMANCS and polymer-conjugated macromolecular drugs: advantages in cancer chemotherapy. Adv Drug Deliv Rev. 2001;46:169–185. doi: 10.1016/s0169-409x(00)00134-4. [DOI] [PubMed] [Google Scholar]
- 8.Nicolaou KC, Dai WM. Chemistry and biology of the enediyne anticancer antibiotics. Angew Chem Int Ed. 1991;30:1387–1416. [Google Scholar]
- 9.Sievers EL, Linenberger M. Mylotarg: antibody-targeted chemotherapy comes of age. Curr Opin Oncol. 2001;13:522–527. doi: 10.1097/00001622-200111000-00016. [DOI] [PubMed] [Google Scholar]
- 10.Gao Q, Thorson JS. The biosynthetic genes encoding for the production of the dynemicin enediyne core in Micromonospora chersina ATCC53710. FEMS Microbiol Lett. 2008;282:105–114. doi: 10.1111/j.1574-6968.2008.01112.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahlert J, Shepard E, Lomovskaya N, Zazopoulos E, Stafa A, Bachmann BO, Huang K, Fonstein L, Czisny A, Whitwam RE, Farnet CM, Thorson JS. The calicheamicin gene cluster and its iterative type I enediyne PKS. Science. 2002;297:1173–1176. doi: 10.1126/science.1072105. [DOI] [PubMed] [Google Scholar]
- 12.Liu W, Christenson SD, Standage S, Shen B. Biosynthesis of the enediyne antitumor antibiotic C-1027. Science. 2002;297:1170–1173. doi: 10.1126/science.1072110. [DOI] [PubMed] [Google Scholar]
- 13.Liu W, Nonaka K, Nie L, Zhang J, Christenson SD, Bae J, Van Lanen SG, Zazopoulos E, Farnet CM, Yang CF, Shen B. The neocarzinostatin biosynthetic gene cluster from Streptomyces carzinostaticus ATCC 15944 involving two iterative type I polyketide synthases. Chem Biol. 2005;12:293–302. doi: 10.1016/j.chembiol.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 14.Van Lanen SG, Oh TJ, Liu W, Wendt-Pienkowski E, Shen B. Characterization of the maduropeptin biosynthetic gene cluster from Actinomadura madurae ATCC 39144 supporting a unifying paradigm for enediyne biosynthesis. J Am Chem Soc. 2007;129:13082–13094. doi: 10.1021/ja073275o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liang ZX. Complexity and simplicity in the biosynthesis of enediyne natural products. Nat Prod Rep. 2010;27:499–528. doi: 10.1039/b908165h. [DOI] [PubMed] [Google Scholar]
- 16.Van Lanen SG, Shen B. Biosynthesis of enediyne antitumor antibiotics. Curr Top Med Chem. 2008;8:448–459. doi: 10.2174/156802608783955656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horsman GP, Chen Y, Thorson JS, Shen B. Polyketide synthase chemistry does not direct biosynthetic divergence between 9- and 10-membered enediynes. Proc Natl Acad Sci USA. 2010;107:11331–11335. doi: 10.1073/pnas.1003442107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu W, Ahlert J, Gao Q, Wendt-Pienkowski E, Shen B, Thorson JS. Rapid PCR amplification of minimal enediyne polyketide synthase cassettes leads to a predictive familial classification model. Proc Natl Acad Sci USA. 2003;100:11959–11963. doi: 10.1073/pnas.2034291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin S, Horsman GP, Chen Y, Li W, Shen B. Characterization of the SgcF epoxide hydrolase supporting an (R)-vicinal diol intermediate for enediyne antitumor antibiotic C-1027 biosynthesis. J Am Chem Soc. 2009;131:16410–16417. doi: 10.1021/ja901242s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin S, Horsman GP, Shen B. Characterization of the epoxide hydrolase NcsF2 from the neocarzinostatin biosynthetic gene cluster. Org Lett. 2010;12:3816–3819. doi: 10.1021/ol101473t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lohman JR, Huang S-X, Horsman GP, Dilfer PE, Huang T, Chen Y, Wendt-Pienkowski E, Shen B. Cloning and sequencing of the kedarcidin biosynthetic gene cluster from Streptoalloteichus sp. ATCC 53650 revealing new insights into biosynthesis of the enediyne family of antitumor antibiotics. Mol BioSyst. 2013;9:478–491. doi: 10.1039/c3mb25523a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Udwary DW, Zeigler L, Asolkar RN, Singan V, Lapidus A, Fenical W, Jensen PR, Moore BS. Genome sequencing reveals complex secondary metabolome in the marine actinomycete Salinispora tropica. Proc Natl Acad Sci USA. 2007;104:10376–10381. doi: 10.1073/pnas.0700962104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ohnishi Y, Ishikawa J, Hara H, Suzuki H, Ikenoya M, Ikeda H, Yamashita A, Horinouchi S. Genome sequence of the streptomycin-producing microorganism Streptomyces griseus IFO 13350. J Bacteriol. 2008;190:4050–4060. doi: 10.1128/JB.00204-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hall BG. Building phylogenetic trees from molecular data with MEGA. Mol Biol Evol. 2013;30:1229–1235. doi: 10.1093/molbev/mst012. [DOI] [PubMed] [Google Scholar]
- 25.Kiefer F, Arnold K, Künzli M, Bordoli L, Schwede T. The SWISS-MODEL repository and associated resources. Nucleic Acids Res. 2009;37:D387–392. doi: 10.1093/nar/gkn750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Loo B, Kingma J, Arand M, Wubbolts MG, Janssen DB. Diversity and biocatalytic potential of epoxide hydrolases identified by genome analysis. Appl Environ Microbiol. 2006;72:2905–2917. doi: 10.1128/AEM.72.4.2905-2917.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schiøtt B, Bruice TC. Reaction mechanism of soluble epoxide hydrolase: insights from molecular dynamics simulations. J Am Chem Soc. 2002;124:14558–14570. doi: 10.1021/ja021021r. [DOI] [PubMed] [Google Scholar]
- 28.McGlinchey RP, Nett M, Moore BS. Unraveling the biosynthesis of the sporolide cyclohexenone building block. J Am Chem Soc. 2008;130:2406–2407. doi: 10.1021/ja710488m. [DOI] [PubMed] [Google Scholar]
- 29.Gawley RE. Do the terms “% ee” and “% de” make sense as expressions of stereoisomer composition or stereoselectivity? J Org Chem. 2006;71:2411–2416. doi: 10.1021/jo052554w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lindberg D, Ahmad S, Widersten M. Mutations in salt-bridging residues at the interface of the core and lid domains of epoxide hydrolase StEH1 affect regioselectivity, protein stability and hysteresis. Arch Biochem Biophys. 2010;495:165–173. doi: 10.1016/j.abb.2010.01.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.