

Abstract
Polo-like kinase 1 (Plk1) is a well-established mitotic regulator with a diverse range of biological functions continually being identified throughout the cell cycle. Pre-clinical evidence suggests that the molecular targeting of Plk1 could be an effective therapeutic strategy in a wide range of cancers, however, that success has yet to be translated to the clinical level. The lack of clinical success has raised the question of whether there is a true oncogenic addiction to Plk1 or if its overexpression in tumors is solely an artifact of increased cellular proliferation. In this review, we address Plk1’s role in carcinogenesis by discussing the cell cycle and DNA damage response with respect to their associations with classic oncogenic and tumor suppressor pathways that contribute to the transcriptional regulation of Plk1. A thorough examination of the available literature suggests that Plk1 activity can be dysregulated through key transformative pathways including both p53, and pRb. Based on available literature, it may be somewhat premature to make a definitive conclusion on Plk1’s role in carcinogenesis. However, evidence support the notion that oncogene dependence on Plk1 is not a late occurrence in carcinogenesis and it is likely that Plk1 plays an active role in carcinogenic transformation.
Introduction
Over the past two decades, since the advent of mammalian polo-like kinase 1 (Plk1), the scientific community has witnessed a tremendous surge of studies aimed at defining the biological functions of this mitotic kinase. We now have a wealth of information available regarding the spectacular role of Plk1 in mitosis and throughout the cell cycle. However, Plk1’s role and functional significance in carcinogenesis and tumor progression is not well understood. The question has been raised whether the consistently observed overexpression of Plk1 in a variety of cancers is due to its direct involvement in the neoplastic transformation of cells or if it is solely the result of increased proliferation due to the transformative process. Plk1 expression being cell cycle dependent with a peak in mitosis, which is evident in mitotically active normal tissues, and the lack of chromosomal translocations or mutations found in the Plk1 gene is in support of the latter(1, 2). On the other hand, it has been shown that forced overexpression of Plk1 results in a malignant transformation of normal human fibroblasts in vitro that are capable of producing tumors when xenografted into nude mice (3), suggesting that Plk1 is capable of directly contributing to carcinogenesis. However, it is not clear how and when Plk1 overexpression occurs during tumor formation. The answer to this critical question most likely lies within one or more of Plk1’s many regulatory loops or protein interactions that have been identified in multiple oncogenes including Akt, Myc, Mdm2, beta-catenin and tumor suppressors such as p53, pRb, Brca2, and Pten (4–11). In fact, a recent review of the cancer genome by Vogelstein et al categorized twelve signaling pathways that regulate three core cellular processes whose understanding is of the most pressing need in basic cancer research, and Plk1 has direct interaction with all core processes and 75% of the signaling pathways (Figure 1.) (12). Further, current evidence suggests that the dysregulation of Plk1 occurs early in carcinogenesis as observed in hepato-, papillary, and pancreatic-carcinomas (13–15). In this review, we are attempting to highlight components of transcriptional regulation and the resulting overexpression of Plk1 in the context of classical oncogenic models involving the cell cycle and tumor suppressors p53 and pRb to illustrate how the dysregulation of these pathways may contribute to early events in carcinogenesis.
Figure 1. Plk1 interacting pathways.
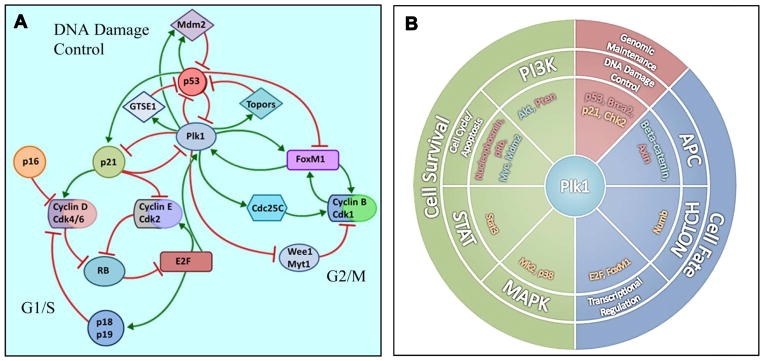
Plk1 overexpression and the resulting contribution to aberrant DNA damage control and genomic instability is accentuated by A) multiple feedback loops highlighting Plk1’s interaction with several pathways involved in cell cycle progression and the DNA damage response; and B) eight of the twelve signaling pathways (middle ring) and all three core cellular processes (outer ring) identified by Vogelstein et al (12) to confer a selective growth advantage in cancer. Plk1 has a direct interaction with a wide range of proteins (inner ring) that are transcribed by oncogenic (green) or tumor suppressive (red) driver genes. Further, Plk1 directly interacts with several proteins that are integral to the described pathways (orange). (4–12, 32, 70, 72, 99–104)
Polo-like kinase 1: A controller of mitotic orchestra and beyond
The discovery of the Polo kinase in Drosophila was made by Sunkel and Glover in 1988 following the observation that mutant polo results in abnormal spindle formation (16). In 1993, Clay and colleagues determined the nucleotide sequence of a cDNA encoding the mammalian protein kinase that was closely related to the enzyme encoded by the Drosophila mutant polo and designated it as polo-like kinase (Plk) (17). Today, the mammalian homologue family of polo-like kinases, consists of five described members, polo-like kinases 1–5 (Plk1-5), that are characterized by the presence of an N-terminal kinase and C-terminal polo-box domain(17, 18). The Plk family is a group of highly conserved serine/threonine kinases that is typically associated with cell cycle progression and mitosis, however, recent studies have suggested involvement of this kinase family in cancer (reviewed in (19)). Plk1 has emerged as a key mitotic regulator and is most commonly known for being a critical component of centrosome maturation, kinetochore-microtubule attachment, bipolar spindle formation and cytokinesis (20–23). However, studies have revealed a diverse range of biological functions beyond typical mitotic events including Plk1’s involvement with p27 and RhoA, regulatory loops with the transcription factors FoxM1 and Stat3, extensive interplay with Cdk1, phosphorylation of p53 family members, p63 and p73, as well as a recent implication in DNA replication (24–32). Further, the overexpression of Plk1 has been linked to poor disease prognosis and a decreased survival rate, while the inhibition of Plk1 activity has emerged as a promising therapeutic target for cancer management in the clinical setting (33, 34). The exact role of Plk1 in carcinogenesis has yet to be determined, however, pre-clinical evidence suggests that Plk1 expression may be necessary for cancer cell survival and is overexpressed in a variety of cancers, including melanoma, breast, ovarian, thyroid, colon, prostate, pancreatic, head and neck, non-small cell lung and non-Hodgkin’s lymphomas (35–43).
The Cell Cycle: Filling in the Gaps
The cell cycle is a highly orchestrated progression of events that culminates in cellular division and the production of two daughter cells. Progression through each of the four main phases, G1 (Gap 1), S (Synthesis), G2 (Gap 2), and M (mitosis), is tightly regulated through phosphorylation and ubiquitination events primarily driven by the “master cell cycle regulators”, cyclins and cyclin-dependent kinases (cdks). Plk1 expression is directly associated with the progression of these phases, where it begins to accumulate during S phase, peaks at the G2/M transition, plateaus throughout mitosis and has a sharp reduction upon mitotic exit (1). Of note, the expression of Plk1 in cancer cells differs from that of non-transformed cells in that it localizes to the nucleus prior to G2/M and can be easily detected even in G1/S phase, suggesting that Plk1 must have cancer cell-specific functions in interphase. This notion is supported by the observation that Plk1 is indeed involved in the G1/S transition and DNA replication in cancer cells (44–46). The nuclear localization of Plk1 during the interphase of cancer cells has been neglected for a long time in the field as one tends to focus on its classical mitotic functions, but this important observation has been independently confirmed by others (47).
Cdks and their endogenous inhibitors have gained a lot of attention as potential therapeutic targets due to their frequent dysregulation in cancer. They can be found both up and downstream of Plk1, and offer insight into how Plk1 expression may be altered absent of direct mutation. The positive regulation of cdk’s has been associated with carcinogenesis and forced progression through the cell cycle, as demonstrated by the proto-oncogene Cyclin D1 in a variety of cancers (48–50). However, the cdk activation process can be negatively regulated by two distinct families of cdk inhibitors, the inhibitor of Cdk4 (INK4) and cdk-interacting protein (CIP)/kinase inhibitor protein (KIP) families. The members of the INK4 family (p16 INK4a, p15 INK4b, p18 INK4c and p19 INK4d) are specific inhibitors of Cdk4 and Cdk6 (hereafter, we will only reference Cdk4 due to the redundancy of Cdk6 function.), whereas the CIP/KIP family of proteins (p21 Cip1/Waf1, p27 Kip1 and p57 Kip2), have a broader inhibitory profile. The INK4 proteins inhibit G1 progression through formation of a catalytically inactive INK4-Cdk4 complex thus competitively displacing cyclin D leading to rapid ubiquitin-dependent proteasomal degradation and preventing the first step of cdk activation (51). Considering the fact that Plk1 is essential for G1/S transition in cancer cells, it will be of great interest to test the hypothesis that Plk1 enhances cyclin D stability via deregulation of INK4 in cancer cells. If this is true, this will be another piece of strong evidence that Plk1 is a bona fide oncogene. The CIP/KIP family of proteins inhibit Cdk2 bound to cyclin E and A, and to a lesser extent cyclin B-Cdk1, by binding to the formed complex and obscuring the catalytic cleft which prevents the loading of ATP (52, 53). Of note, the formation of the INK4-Cdk4 but not the binding of CIP/KIP proteins to the cyclin D-Cdk4 complex prevents phosphorylation of pRb which is a critical component for synthesis of cell cycle machinery, including Plk1, by E2F dependent transcription (discussed below). Further, all the CKI’s appear to be at least tenuously tied to Plk1, and when considering the frequent dysregulation of p16 and p21 in cancer, their involvement in regulatory loops that both directly and indirectly affect the expression and activity of Plk1 are of a particular interest.
p16 is the most prominent member of the INK4 family proteins and is routinely associated with cellular senescence and tumor suppression. Induction of p16 has been reported as a tumor suppressive response to oncogenic stress such as constitutive activation of oncogenic Ras (54). Dysregulation of p16 can occur by deletion of the INK4a/ARF locus or mutation to the INK4a gene resulting in a loss of p16, found in a large percentage of cancers, and additionally, a Cdk4 mutation that prevents binding of p16 has been identified in human melanomas (54–57). These observations have been substantiated in vivo where the deletion of Ink4a in mice results in spontaneous tumorigenesis and accelerates formation of carcinogen-induced cancers (58). Further, the loss of somatic p16 significantly accelerates melanomagenesis following the activation of a melanocyte-specific oncogenic K-Ras allele, indicating p16’s role as a transformative factor (59). Recent studies have linked the down-regulation of Plk1 with the induction of senescence in primary cells, however, this mechanism appears to be independent of p16 expression (60). While this data potentially removes a link between p16 induced senescence and Plk1, a second association involving tumor suppression through p16’s namesake, inhibition of Cdk4, lies upstream of Plk1 in a RB-E2F transcriptional pathway.
RB-E2F: Factoring in transcription
As previously discussed, p16 is capable of binding to Cdk4 and preventing the initial activation that is typically conveyed by D-type cyclins. The active cyclin D-Cdk4 complex phosphorylates pRb which contributes to the disassociation of pRb from E2F, thus increasing E2F’s promoter activity (61). Aside from Cdk4 inhibition, in cancers such as retinoblastoma and small cell lung cancer, loss of pRb expression has been attributed to direct mutation (62). Further, a mechanism that targets the three members of the RB family (a.k.a. pocket proteins), pRb, p107, and p130, involves a conserved domain known as the small pocket that interacts with the viral LXCXE motif, thus making them a target for viral oncogenes such as human papilloma virus E7, and adenovirus E1A (63–65). The interaction of viral oncoproteins and E2F with RB does not occur at the same site, however the juxtaposition of the domains allows a separate region of the oncoprotein to disrupt E2F binding (66). At a superficial level, E2F unrepressed by RB binding recruits co-activators such as histone acetyltransferases (HAT) that leads to an open chromatin configuration which provides accessibility to transcriptional machinery and subsequent transcription of E2F responsive genes (reviewed in (67)). While recent studies have demonstrated that E2F is involved in the transcription of genes with a diverse range of function, the majority of identified targets are involved in cell cycle control and/or DNA synthesis, including Plk1 (68). In support, Plk1 is indeed involved in DNA replication in cancer cells. Specifically, Plk1 phosphorylation of Orc2, a member of DNA replication machinery, promotes DNA replication under conditions of stress (45). In other words, Plk1 overrides DNA damage-induced intra-S-phase checkpoint arrest and promotes DNA synthesis in the presence of unrepaired DNA, eventually contributing to genomic instability.
E2F transcription factors are a major contributor to the typically irreversible progression of the mammalian cell cycle. One example of this feed forward loop can be seen in late G1 progression following initial disassociation of Cdk4 phosphorylated RB from E2F which results in increasing transcriptional activity and correlating expression of cyclin E. Increased availability of cyclin E binds to and activates Cdk2 in a complex that further phosphorylates RB, thereby increasing E2F promoter activity. However, to ensure ordered progression through the cell cycle, there are counterbalances to E2F self-promotion. Among the E2F targets are the CKIs p18, p19 and p57, as well as RB proteins pRb and p107. It would stand to reason that the increased expression of these proteins would contribute to the decreased activity of E2F in late stages of S phase. Although, considering the amount of mutations that can occur upstream of E2F, it is not surprising that the dysregulation of E2F has been correlated to carcinogenesis, but whether or not the tumor-promoting activity of deregulated E2F is confined to cell cycle regulation is still under investigation (reviewed in (69)). However, it’s not a stretch to imagine how the large number of E2F targets may act in concert with one another to overcome the cell cycle checkpoints and contribute to genomic instability and it is our assertion that Plk1 is a key player in that process.
There is some evidence that the deletion of the E2F element in the Plk1 promoter does not significantly affect Plk1 promoter function, however that observation does not appear to be the consensus and other studies have shown both repression and activation of Plk1 promoter activity in the presence of E2F (70). One such study indicates that when associated to pRb, E2F acts as a repressor of Plk1 in the presence of the nucleosome remodeling complex, SWI/SNF (Switch/Sucrose NonFermentable), through histone modification (6). In contrast, when unbound by RB, E2F activators have been correlated with an increase in Plk1 promoter activity, which has been supported by an observation of decreased Plk1 expression following E2F knockdown in cancer cells (71, 72). However, it is important to further test this observation in normal cells. While E2F may play a secondary role to the better defined CDE/CHR (cell cycle-dependent element/cell cycle genes homology region) and CCAAT elements found in the Plk1 promoter, this data suggests that E2F may be a contributor and therefore a necessary consideration when developing models for the overexpression of Plk1 that is associated with a large number of cancers.
Plk1 and p53: Elements of tumor suppression
Given the amount of biological pathways that are associated with p53, it is of no surprise that this classical tumor suppressor overlaps with Plk1 in multiple seemingly independent mechanisms. Using the most basic description, p53 is a transcription factor induced by cellular stress that routinely leads to apoptosis or cell cycle arrest through direct protein-protein interaction, repression and/or transactivation of critical pro-apoptotic or cell cycle regulatory genes (73–75). Detailed reviews of p53 and its biological importance can be found elsewhere (76, 77). As mentioned above, the CCAAT and CDE/CHR elements have been identified as major contributors to Plk1 transcriptional activity (78). The CCAAT box is a target of the transcription factor NF-Y and is a common promoter element of G2/M associated genes including Plk1. While it seems uncontested that p53 confers a level of direct negative regulation to Plk1, there is a conflict in the literature as to whether the regulation is achieved through binding to p53 responsive elements or through the CCAAT element in the Plk1 promoter (70, 79). Although, McKenzie et al eloquently present their case, we will reserve the subject for future discussion when more evidence is available. Yet, because p53 is an antagonist of Plk1, one would predict that loss-of-function p53 mutations, which occur in 50% of cancers, could be largely responsible for Plk1 elevation in these cancers, however, we acknowledge that this notion needs further experimentation.
To briefly expound on the CDE/CHR element of the Plk1 promoter, repression of the Plk1 gene in G0/G1 is conferred by the CHR site while mutation of the CDE sequence found four nucleotides upstream had no significant effect on Plk1 transcription (78). Using Muller and Engeland’s classification, the lack of a functional CDE site typically seen in the CDE/CHR motif found in other cell cycle genes such as Cdc2 and Cdc25C defines Plk1 as a class II gene (80–82). However, repression through the CDE element can be, at least in part, conferred by the p53 transcriptional target p21, as seen by release of p21-dependent repression following CDE mutation (70, 83). In addition, it has been shown that high levels of p53 may also cause repression of cell cycle genes through a p21-dependent switch from a MMB to a DREAM complex binding at CHR elements through cyclin-cdk inhibition and subsequent hypophosphorylation of RB, however, this process has yet to be directly correlated to Plk1 (84). Alternatively to the RB pathway, other studies have suggested in the case of oncogene-induced senescence, p21 is capable of preventing Plk1 expression through direct binding to the promoter of Plk1 which may be part of an adaptive response to stress and provide insight into p53-dependent senescence induced in primary cells following knockdown of Plk1 (60, 85). To add a layer of complexity to the pathway, it also appears that long-term downregulation of Plk1 stabilizes p21 expression in both normal diploid and p53-defective cancer cells, presumably through both p53-dependent and -independent mechanisms, suggesting a double negative feedback loop involving p21 and Plk1 (86, 87).
There seems to be little doubt that p53 has the ability to exert some level of regulation on Plk1, but keeping true to the reoccurring theme of Plk1 being embedded into regulatory loops, it appears that Plk1 has the ability to control p53 activity through multiple pathways as well. This is of particular importance since dysregulation of p53 cripples a primary line of cellular defense against proliferation under aberrant conditions. For example, it has been shown that Plk1 inhibits p53 transcriptional activity and pro-apoptotic function through direct binding to a sequence-specific DNA-binding domain of p53 (88). Further, the depletion of Plk1 has been correlated to reduced levels of Mdm2, an E3 ubiquitin ligase and critical regulator of p53, which was corroborated with the observation that phosphorylation of Mdm2 at Ser260 by Plk1 stimulates Mdm2-mediated turnover of p53 (5, 86). Additionally, a recent study has shown that Plk1 in vivo phosphorylation of Ser718 on Topors, an ubiquitin and SUMO E3 ligase, inhibits Topors-mediated sumylation of p53 and enhances p53 degradation through ubiquitination (89). Interestingly, aside from ubiquitination and signaling, Plk1-associated kinase activity also directly influences the spatial regulation of p53 through phosphorylation of G2 and S-phase expressed 1 (GTSE1), resulting in the constitutive shuttling of p53 from the nucleus to the cytoplasm thus facilitating the proteasomal degradation of p53 during cellular recovery from DNA damage (90). Of note, while p21 typically falls downstream of p53, it has been suggested that p21 mediated suppression of Plk1 is responsible for maintaining p53 expression and activation during the stress response (91). These data provide evidence that tight regulation of Plk1 may be necessary for p53 and p21 to adequately perform their tumor suppressive functions and that overexpression of Plk1 may negatively regulate both pathways in concert and independently from one another.
Conclusion and Outlook
Based on the information and discussion provided in this review, we believe that Plk1 potentially plays a significant role in carcinogenic transformation. Indeed, Plk1 is not overexpressed due to a direct mutation or translocation, however this is possibly not the sole criterion for being a transformative factor. We feel the existing data provides a strong foundation for a p53-p21-Plk1 axis that can significantly promote aneuploidy and genomic instability. Considering all three proteins are integrated into the DNA damage response, it is reasonable to suspect that abrogated DNA damage checkpoints may play a significant role in this process (79, 92–94). Support for this comes from the study showing that Plk1 phosphorylation of GTSE1 is essential for cellular recovery from G2 DNA damage checkpoints, suggesting that elevated Plk1-mediated p53 inactivation is one mechanism of premature checkpoint termination, eventually leading to aneuploidy and chromosome instability (90). There is evidence of a similar mechanism in Xenopus where Plx1 (Plk1 homologue) has been linked to checkpoint adaptation via phosphorylation of the checkpoint mediator Claspin, a protein that promotes cell cycle arrest by facilitating the ATR-dependent phosphorylation of BRCA1 and CHEK1 in response to DNA damage (95).
When considering the INK4-Cdk-RB pathway, E2F targeting of Plk1 provides a potential mechanism for overexpression of Plk1 to be an early occurrence in carcinogenesis based on upstream mutations. Further, it offers a tentative connection and level of regulation between the p53 and RB pathways. Other studies have suggested a similar relationship involving the INK4-ARF locus, where loss of RB results in E2F activation of the ARF promoter, invoking a p14-Mdm2-p53 pathway (96, 97). Further, studies have also shown that enhanced Mdm2 activity is capable of inhibiting RB function (98). It is interesting to postulate that overexpression of Plk1 may then contribute to mitigating this endogenous defense mechanism through enhancement of Mdm2 and inhibition of both RB and p53 or its downstream target p21 under the same conditions.
Given the lack of translocation or direct mutation found in the Plk1 gene, we understand and respect the notion that Plk1 overexpression may be an artifact of the increased cellular proliferation that is associated with cancer. However, considering the information presented in this review, we believe that it is far too early to make any reasonable conclusions on Plk1’s role in carcinogenesis. Pre-clinical experiments have demonstrated that Plk1 is an effective therapeutic target in many cancers, and we find it reasonable to suspect that oncogene dependence on Plk1 is not a late occurrence in cancer formation. We feel it is far more likely that Plk1 plays an active role in genomic instability and aberrant cell survival, ultimately driving the cell into carcinogenic transformation.
Acknowledgments
This work was partially supported by funding from the NIH (T32 ES007015-35 to BDC; R01AR059130, R01CA176748 to NA; R01CA157429 to XL) and the Department of Veterans Affairs (VA Merit Review Award 1I01BX001008 to NA).
References
- 1.Golsteyn RM, Schultz SJ, Bartek J, Ziemiecki A, Ried T, Nigg EA. Cell cycle analysis and chromosomal localization of human Plk1, a putative homologue of the mitotic kinases Drosophila polo and Saccharomyces cerevisiae Cdc5. J Cell Sci. 1994;107 (Pt 6):1509–17. doi: 10.1242/jcs.107.6.1509. [DOI] [PubMed] [Google Scholar]
- 2.Holtrich U, Wolf G, Brauninger A, Karn T, Bohme B, Rubsamen-Waigmann H, et al. Induction and down-regulation of PLK, a human serine/threonine kinase expressed in proliferating cells and tumors. Proc Natl Acad Sci U S A. 1994;91:1736–40. doi: 10.1073/pnas.91.5.1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith MR, Wilson ML, Hamanaka R, Chase D, Kung H, Longo DL, et al. Malignant transformation of mammalian cells initiated by constitutive expression of the polo-like kinase. Biochem Biophys Res Commun. 1997;234:397–405. doi: 10.1006/bbrc.1997.6633. [DOI] [PubMed] [Google Scholar]
- 4.Arai T, Haze K, Iimura-Morita Y, Machida T, Iida M, Tanaka K, et al. Identification of beta-catenin as a novel substrate of Polo-like kinase 1. Cell Cycle. 2008;7:3556–63. doi: 10.4161/cc.7.22.7072. [DOI] [PubMed] [Google Scholar]
- 5.Dias SS, Hogan C, Ochocka AM, Meek DW. Polo-like kinase-1 phosphorylates MDM2 at Ser260 and stimulates MDM2-mediated p53 turnover. FEBS Lett. 2009;583:3543–8. doi: 10.1016/j.febslet.2009.09.057. [DOI] [PubMed] [Google Scholar]
- 6.Gunawardena RW, Siddiqui H, Solomon DA, Mayhew CN, Held J, Angus SP, et al. Hierarchical requirement of SWI/SNF in retinoblastoma tumor suppressor-mediated repression of Plk1. J Biol Chem. 2004;279:29278–85. doi: 10.1074/jbc.M400395200. [DOI] [PubMed] [Google Scholar]
- 7.Incassati A, Patel D, McCance DJ. Induction of tetraploidy through loss of p53 and upregulation of Plk1 by human papillomavirus type-16 E6. Oncogene. 2006;25:2444–51. doi: 10.1038/sj.onc.1209276. [DOI] [PubMed] [Google Scholar]
- 8.Kasahara K, Goto H, Izawa I, Kiyono T, Watanabe N, Elowe S, et al. PI 3-kinase-dependent phosphorylation of Plk1-Ser99 promotes association with 14-3-3gamma and is required for metaphase-anaphase transition. Nat Commun. 2013;4:1882. doi: 10.1038/ncomms2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee M, Daniels MJ, Venkitaraman AR. Phosphorylation of BRCA2 by the Polo-like kinase Plk1 is regulated by DNA damage and mitotic progression. Oncogene. 2004;23:865–72. doi: 10.1038/sj.onc.1207223. [DOI] [PubMed] [Google Scholar]
- 10.Liu XS, Song B, Elzey BD, Ratliff TL, Konieczny SF, Cheng L, et al. Polo-like kinase 1 facilitates loss of Pten tumor suppressor-induced prostate cancer formation. J Biol Chem. 2011;286:35795–800. doi: 10.1074/jbc.C111.269050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Padmanabhan A, Li X, Bieberich CJ. Protein Kinase A Regulates MYC Protein through Transcriptional and Post-translational Mechanisms in a Catalytic Subunit Isoform-specific Manner. J Biol Chem. 2013;288:14158–69. doi: 10.1074/jbc.M112.432377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–58. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ito Y, Miyoshi E, Sasaki N, Kakudo K, Yoshida H, Tomoda C, et al. Polo-like kinase 1 overexpression is an early event in the progression of papillary carcinoma. Br J Cancer. 2004;90:414–8. doi: 10.1038/sj.bjc.6601540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Petrelli A, Perra A, Schernhuber K, Cargnelutti M, Salvi A, Migliore C, et al. Sequential analysis of multistage hepatocarcinogenesis reveals that miR-100 and PLK1 dysregulation is an early event maintained along tumor progression. Oncogene. 2012;31:4517–26. doi: 10.1038/onc.2011.631. [DOI] [PubMed] [Google Scholar]
- 15.Weichert W, Schmidt M, Jacob J, Gekeler V, Langrehr J, Neuhaus P, et al. Overexpression of Polo-like kinase 1 is a common and early event in pancreatic cancer. Pancreatology. 2005;5:259–65. doi: 10.1159/000085280. [DOI] [PubMed] [Google Scholar]
- 16.Sunkel CE, Glover DM. polo, a mitotic mutant of Drosophila displaying abnormal spindle poles. J Cell Sci. 1988;89 (Pt 1):25–38. doi: 10.1242/jcs.89.1.25. [DOI] [PubMed] [Google Scholar]
- 17.Clay FJ, McEwen SJ, Bertoncello I, Wilks AF, Dunn AR. Identification and cloning of a protein kinase-encoding mouse gene, Plk, related to the polo gene of Drosophila. Proc Natl Acad Sci U S A. 1993;90:4882–6. doi: 10.1073/pnas.90.11.4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lane HA, Nigg EA. Cell-cycle control: POLO-like kinases join the outer circle. Trends Cell Biol. 1997;7:63–8. doi: 10.1016/S0962-8924(96)10051-9. [DOI] [PubMed] [Google Scholar]
- 19.Strebhardt K. Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nat Rev Drug Discov. 2010;9:643–60. doi: 10.1038/nrd3184. [DOI] [PubMed] [Google Scholar]
- 20.Lane HA, Nigg EA. Antibody microinjection reveals an essential role for human polo-like kinase 1 (Plk1) in the functional maturation of mitotic centrosomes. J Cell Biol. 1996;135:1701–13. doi: 10.1083/jcb.135.6.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seong YS, Kamijo K, Lee JS, Fernandez E, Kuriyama R, Miki T, et al. A spindle checkpoint arrest and a cytokinesis failure by the dominant-negative polo-box domain of Plk1 in U-2 OS cells. J Biol Chem. 2002;277:32282–93. doi: 10.1074/jbc.M202602200. [DOI] [PubMed] [Google Scholar]
- 22.Sumara I, Gimenez-Abian JF, Gerlich D, Hirota T, Kraft C, de la Torre C, et al. Roles of polo-like kinase 1 in the assembly of functional mitotic spindles. Curr Biol. 2004;14:1712–22. doi: 10.1016/j.cub.2004.09.049. [DOI] [PubMed] [Google Scholar]
- 23.van Vugt MA, van de Weerdt BC, Vader G, Janssen H, Calafat J, Klompmaker R, et al. Polo-like kinase-1 is required for bipolar spindle formation but is dispensable for anaphase promoting complex/Cdc20 activation and initiation of cytokinesis. J Biol Chem. 2004;279:36841–54. doi: 10.1074/jbc.M313681200. [DOI] [PubMed] [Google Scholar]
- 24.Dibb M, Han N, Choudhury J, Hayes S, Valentine H, West C, et al. The FOXM1-PLK1 axis is commonly upregulated in oesophageal adenocarcinoma. Br J Cancer. 2012;107:1766–75. doi: 10.1038/bjc.2012.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koida N, Ozaki T, Yamamoto H, Ono S, Koda T, Ando K, et al. Inhibitory role of Plk1 in the regulation of p73-dependent apoptosis through physical interaction and phosphorylation. J Biol Chem. 2008;283:8555–63. doi: 10.1074/jbc.M710608200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Komatsu S, Takenobu H, Ozaki T, Ando K, Koida N, Suenaga Y, et al. Plk1 regulates liver tumor cell death by phosphorylation of TAp63. Oncogene. 2009;28:3631–41. doi: 10.1038/onc.2009.216. [DOI] [PubMed] [Google Scholar]
- 27.Krajewska M, Heijink AM, Bisselink YJ, Seinstra RI, Sillje HH, de Vries EG, et al. Forced activation of Cdk1 via wee1 inhibition impairs homologous recombination. Oncogene. 2012 doi: 10.1038/onc.2012.296. [DOI] [PubMed]
- 28.Li J, Wang J, Jiao H, Liao J, Xu X. Cytokinesis and cancer: Polo loves ROCK‘n’ Rho(A) J Genet Genomics. 2010;37:159–72. doi: 10.1016/S1673-8527(09)60034-5. [DOI] [PubMed] [Google Scholar]
- 29.Maia AR, Garcia Z, Kabeche L, Barisic M, Maffini S, Macedo-Ribeiro S, et al. Cdk1 and Plk1 mediate a CLASP2 phospho-switch that stabilizes kinetochore-microtubule attachments. J Cell Biol. 2012;199:285–301. doi: 10.1083/jcb.201203091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Donovan DS, MacFhearraigh S, Whitfield J, Swigart LB, Evan GI, Mc Gee MM. Sequential Cdk1 and Plk1 phosphorylation of protein tyrosine phosphatase 1B promotes mitotic cell death. Cell Death Dis. 2013;4:e468. doi: 10.1038/cddis.2012.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Song B, Liu XS, Liu X. Polo-like kinase 1 (Plk1): an Unexpected Player in DNA Replication. Cell Div. 2012;7:3. doi: 10.1186/1747-1028-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Du XL, Wang CJ, Lin DC, Ruan X, Feng YB, et al. Reciprocal activation between PLK1 and Stat3 contributes to survival and proliferation of esophageal cancer cells. Gastroenterology. 2012;142:521–30. e3. doi: 10.1053/j.gastro.2011.11.023. [DOI] [PubMed] [Google Scholar]
- 33.Strebhardt K, Kneisel L, Linhart C, Bernd A, Kaufmann R. Prognostic value of pololike kinase expression in melanomas. Jama. 2000;283:479–80. doi: 10.1001/jama.283.4.479. [DOI] [PubMed] [Google Scholar]
- 34.Weichert W, Schmidt M, Gekeler V, Denkert C, Stephan C, Jung K, et al. Polo-like kinase 1 is overexpressed in prostate cancer and linked to higher tumor grades. Prostate. 2004;60:240–5. doi: 10.1002/pros.20050. [DOI] [PubMed] [Google Scholar]
- 35.Gray PJ, Jr, Bearss DJ, Han H, Nagle R, Tsao MS, Dean N, et al. Identification of human polo-like kinase 1 as a potential therapeutic target in pancreatic cancer. Mol Cancer Ther. 2004;3:641–6. [PubMed] [Google Scholar]
- 36.Knecht R, Elez R, Oechler M, Solbach C, von Ilberg C, Strebhardt K. Prognostic significance of polo-like kinase (PLK) expression in squamous cell carcinomas of the head and neck. Cancer Res. 1999;59:2794–7. [PubMed] [Google Scholar]
- 37.Mito K, Kashima K, Kikuchi H, Daa T, Nakayama I, Yokoyama S. Expression of Polo-Like Kinase (PLK1) in non-Hodgkin’s lymphomas. Leuk Lymphoma. 2005;46:225–31. doi: 10.1080/10428190400015709. [DOI] [PubMed] [Google Scholar]
- 38.Salvatore G, Nappi TC, Salerno P, Jiang Y, Garbi C, Ugolini C, et al. A cell proliferation and chromosomal instability signature in anaplastic thyroid carcinoma. Cancer Res. 2007;67:10148–58. doi: 10.1158/0008-5472.CAN-07-1887. [DOI] [PubMed] [Google Scholar]
- 39.Schmit TL, Zhong W, Setaluri V, Spiegelman VS, Ahmad N. Targeted depletion of Polo-like kinase (Plk) 1 through lentiviral shRNA or a small-molecule inhibitor causes mitotic catastrophe and induction of apoptosis in human melanoma cells. J Invest Dermatol. 2009;129:2843–53. doi: 10.1038/jid.2009.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takahashi T, Sano B, Nagata T, Kato H, Sugiyama Y, Kunieda K, et al. Polo-like kinase 1 (PLK1) is overexpressed in primary colorectal cancers. Cancer Sci. 2003;94:148–52. doi: 10.1111/j.1349-7006.2003.tb01411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weichert W, Denkert C, Schmidt M, Gekeler V, Wolf G, Kobel M, et al. Polo-like kinase isoform expression is a prognostic factor in ovarian carcinoma. Br J Cancer. 2004;90:815–21. doi: 10.1038/sj.bjc.6601610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wolf G, Elez R, Doermer A, Holtrich U, Ackermann H, Stutte HJ, et al. Prognostic significance of polo-like kinase (PLK) expression in non-small cell lung cancer. Oncogene. 1997;14:543–9. doi: 10.1038/sj.onc.1200862. [DOI] [PubMed] [Google Scholar]
- 43.Wolf G, Hildenbrand R, Schwar C, Grobholz R, Kaufmann M, Stutte HJ, et al. Polo-like kinase: a novel marker of proliferation: correlation with estrogen-receptor expression in human breast cancer. Pathol Res Pract. 2000;196:753–9. doi: 10.1016/S0344-0338(00)80107-7. [DOI] [PubMed] [Google Scholar]
- 44.Li H, Wang Y, Liu X. Plk1-dependent phosphorylation regulates functions of DNA topoisomerase IIalpha in cell cycle progression. J Biol Chem. 2008;283:6209–21. doi: 10.1074/jbc.M709007200. [DOI] [PubMed] [Google Scholar]
- 45.Song B, Liu XS, Davis K, Liu X. Plk1 phosphorylation of Orc2 promotes DNA replication under conditions of stress. Mol Cell Biol. 2011;31:4844–56. doi: 10.1128/MCB.06110-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu ZQ, Liu X. Role for Plk1 phosphorylation of Hbo1 in regulation of replication licensing. Proc Natl Acad Sci U S A. 2008;105:1919–24. doi: 10.1073/pnas.0712063105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang Y, Bai J, Shen R, Brown SA, Komissarova E, Huang Y, et al. Polo-like kinase 3 functions as a tumor suppressor and is a negative regulator of hypoxia-inducible factor-1 alpha under hypoxic conditions. Cancer Res. 2008;68:4077–85. doi: 10.1158/0008-5472.CAN-07-6182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim JK, Diehl JA. Nuclear cyclin D1: an oncogenic driver in human cancer. J Cell Physiol. 2009;220:292–6. doi: 10.1002/jcp.21791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Motokura T, Bloom T, Kim HG, Juppner H, Ruderman JV, Kronenberg HM, et al. A novel cyclin encoded by a bcl1-linked candidate oncogene. Nature. 1991;350:512–5. doi: 10.1038/350512a0. [DOI] [PubMed] [Google Scholar]
- 50.Xiong Y, Connolly T, Futcher B, Beach D. Human D-type cyclin. Cell. 1991;65:691–9. doi: 10.1016/0092-8674(91)90100-d. [DOI] [PubMed] [Google Scholar]
- 51.Diehl JA, Sherr CJ. A dominant-negative cyclin D1 mutant prevents nuclear import of cyclin-dependent kinase 4 (CDK4) and its phosphorylation by CDK-activating kinase. Mol Cell Biol. 1997;17:7362–74. doi: 10.1128/mcb.17.12.7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee MH, Reynisdottir I, Massague J. Cloning of p57KIP2, a cyclin-dependent kinase inhibitor with unique domain structure and tissue distribution. Genes Dev. 1995;9:639–49. doi: 10.1101/gad.9.6.639. [DOI] [PubMed] [Google Scholar]
- 53.Polyak K, Lee MH, Erdjument-Bromage H, Koff A, Roberts JM, Tempst P, et al. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell. 1994;78:59–66. doi: 10.1016/0092-8674(94)90572-x. [DOI] [PubMed] [Google Scholar]
- 54.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 55.Kamb A, Shattuck-Eidens D, Eeles R, Liu Q, Gruis NA, Ding W, et al. Analysis of the p16 gene (CDKN2) as a candidate for the chromosome 9p melanoma susceptibility locus. Nat Genet. 1994;8:23–6. doi: 10.1038/ng0994-22. [DOI] [PubMed] [Google Scholar]
- 56.Ruas M, Peters G. The p16INK4a/CDKN2A tumor suppressor and its relatives. Biochim Biophys Acta. 1998;1378:F115–77. doi: 10.1016/s0304-419x(98)00017-1. [DOI] [PubMed] [Google Scholar]
- 57.Wolfel T, Hauer M, Schneider J, Serrano M, Wolfel C, Klehmann-Hieb E, et al. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science. 1995;269:1281–4. doi: 10.1126/science.7652577. [DOI] [PubMed] [Google Scholar]
- 58.Sharpless NE, Bardeesy N, Lee KH, Carrasco D, Castrillon DH, Aguirre AJ, et al. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature. 2001;413:86–91. doi: 10.1038/35092592. [DOI] [PubMed] [Google Scholar]
- 59.Monahan KB, Rozenberg GI, Krishnamurthy J, Johnson SM, Liu W, Bradford MK, et al. Somatic p16(INK4a) loss accelerates melanomagenesis. Oncogene. 2010;29:5809–17. doi: 10.1038/onc.2010.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim HJ, Cho JH, Kim JR. Downregulation of Polo-Like Kinase 1 Induces Cellular Senescence in Human Primary Cells Through a p53-Dependent Pathway. J Gerontol A Biol Sci Med Sci. 2013 doi: 10.1093/gerona/glt017. [DOI] [PubMed] [Google Scholar]
- 61.Kato J, Matsushime H, Hiebert SW, Ewen ME, Sherr CJ. Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes Dev. 1993;7:331–42. doi: 10.1101/gad.7.3.331. [DOI] [PubMed] [Google Scholar]
- 62.Horowitz JM, Park SH, Bogenmann E, Cheng JC, Yandell DW, Kaye FJ, et al. Frequent inactivation of the retinoblastoma anti-oncogene is restricted to a subset of human tumor cells. Proc Natl Acad Sci U S A. 1990;87:2775–9. doi: 10.1073/pnas.87.7.2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Classon M, Dyson N. p107 and p130: versatile proteins with interesting pockets. Exp Cell Res. 2001;264:135–47. doi: 10.1006/excr.2000.5135. [DOI] [PubMed] [Google Scholar]
- 64.Dyson N, Howley PM, Munger K, Harlow E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989;243:934–7. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- 65.Whyte P, Buchkovich KJ, Horowitz JM, Friend SH, Raybuck M, Weinberg RA, et al. Association between an oncogene and an anti-oncogene: the adenovirus E1A proteins bind to the retinoblastoma gene product. Nature. 1988;334:124–9. doi: 10.1038/334124a0. [DOI] [PubMed] [Google Scholar]
- 66.Liu X, Marmorstein R. Structure of the retinoblastoma protein bound to adenovirus E1A reveals the molecular basis for viral oncoprotein inactivation of a tumor suppressor. Genes Dev. 2007;21:2711–6. doi: 10.1101/gad.1590607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Henley SA, Dick FA. The retinoblastoma family of proteins and their regulatory functions in the mammalian cell division cycle. Cell Div. 2012;7:10. doi: 10.1186/1747-1028-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bracken AP, Ciro M, Cocito A, Helin K. E2F target genes: unraveling the biology. Trends Biochem Sci. 2004;29:409–17. doi: 10.1016/j.tibs.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 69.Chen HZ, Tsai SY, Leone G. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer. 2009;9:785–97. doi: 10.1038/nrc2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhu H, Chang BD, Uchiumi T, Roninson IB. Identification of promoter elements responsible for transcriptional inhibition of polo-like kinase 1 and topoisomerase IIalpha genes by p21(WAF1/CIP1/SDI1) Cell Cycle. 2002;1:59–66. [PubMed] [Google Scholar]
- 71.Olsson AY, Feber A, Edwards S, Te Poele R, Giddings I, Merson S, et al. Role of E2F3 expression in modulating cellular proliferation rate in human bladder and prostate cancer cells. Oncogene. 2007;26:1028–37. doi: 10.1038/sj.onc.1209854. [DOI] [PubMed] [Google Scholar]
- 72.Tategu M, Nakagawa H, Sasaki K, Yamauchi R, Sekimachi S, Suita Y, et al. Transcriptional regulation of human polo-like kinases and early mitotic inhibitor. J Genet Genomics. 2008;35:215–24. doi: 10.1016/S1673-8527(08)60030-2. [DOI] [PubMed] [Google Scholar]
- 73.Ahn J, Murphy M, Kratowicz S, Wang A, Levine AJ, George DL. Down-regulation of the stathmin/Op18 and FKBP25 genes following p53 induction. Oncogene. 1999;18:5954–8. doi: 10.1038/sj.onc.1202986. [DOI] [PubMed] [Google Scholar]
- 74.el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. Definition of a consensus binding site for p53. Nat Genet. 1992;1:45–9. doi: 10.1038/ng0492-45. [DOI] [PubMed] [Google Scholar]
- 75.Murphy M, Ahn J, Walker KK, Hoffman WH, Evans RM, Levine AJ, et al. Transcriptional repression by wild-type p53 utilizes histone deacetylases, mediated by interaction with mSin3a. Genes Dev. 1999;13:2490–501. doi: 10.1101/gad.13.19.2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Menendez D, Inga A, Resnick MA. The expanding universe of p53 targets. Nat Rev Cancer. 2009;9:724–37. doi: 10.1038/nrc2730. [DOI] [PubMed] [Google Scholar]
- 77.Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137:413–31. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 78.Uchiumi T, Longo DL, Ferris DK. Cell cycle regulation of the human polo-like kinase (PLK) promoter. J Biol Chem. 1997;272:9166–74. doi: 10.1074/jbc.272.14.9166. [DOI] [PubMed] [Google Scholar]
- 79.McKenzie L, King S, Marcar L, Nicol S, Dias SS, Schumm K, et al. p53-dependent repression of polo-like kinase-1 (PLK1) Cell Cycle. 2010;9:4200–12. doi: 10.4161/cc.9.20.13532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lucibello FC, Truss M, Zwicker J, Ehlert F, Beato M, Muller R. Periodic cdc25C transcription is mediated by a novel cell cycle-regulated repressor element (CDE) Embo J. 1995;14:132–42. doi: 10.1002/j.1460-2075.1995.tb06983.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Muller GA, Engeland K. The central role of CDE/CHR promoter elements in the regulation of cell cycle-dependent gene transcription. Febs J. 2010;277:877–93. doi: 10.1111/j.1742-4658.2009.07508.x. [DOI] [PubMed] [Google Scholar]
- 82.Zwicker J, Lucibello FC, Wolfraim LA, Gross C, Truss M, Engeland K, et al. Cell cycle regulation of the cyclin A, cdc25C and cdc2 genes is based on a common mechanism of transcriptional repression. Embo J. 1995;14:4514–22. doi: 10.1002/j.1460-2075.1995.tb00130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–25. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 84.Quaas M, Muller GA, Engeland K. p53 can repress transcription of cell cycle genes through a p21(WAF1/CIP1)-dependent switch from MMB to DREAM protein complex binding at CHR promoter elements. Cell Cycle. 2012;11:4661–72. doi: 10.4161/cc.22917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.de Carne Trecesson S, Guillemin Y, Belanger A, Bernard AC, Preisser L, Ravon E, et al. Escape from p21-mediated oncogene-induced senescence leads to cell dedifferentiation and dependence on anti-apoptotic Bcl-xL and MCL1 proteins. J Biol Chem. 2011;286:12825–38. doi: 10.1074/jbc.M110.186437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kreis NN, Sommer K, Sanhaji M, Kramer A, Matthess Y, Kaufmann M, et al. Long-term downregulation of Polo-like kinase 1 increases the cyclin-dependent kinase inhibitor p21(WAF1/CIP1) Cell Cycle. 2009;8:460–72. doi: 10.4161/cc.8.3.7651. [DOI] [PubMed] [Google Scholar]
- 87.Lei M, Erikson RL. Plk1 depletion in nontransformed diploid cells activates the DNA-damage checkpoint. Oncogene. 2008;27:3935–43. doi: 10.1038/onc.2008.36. [DOI] [PubMed] [Google Scholar]
- 88.Ando K, Ozaki T, Yamamoto H, Furuya K, Hosoda M, Hayashi S, et al. Polo-like kinase 1 (Plk1) inhibits p53 function by physical interaction and phosphorylation. J Biol Chem. 2004;279:25549–61. doi: 10.1074/jbc.M314182200. [DOI] [PubMed] [Google Scholar]
- 89.Yang X, Li H, Zhou Z, Wang WH, Deng A, Andrisani O, et al. Plk1-mediated phosphorylation of Topors regulates p53 stability. J Biol Chem. 2009;284:18588–92. doi: 10.1074/jbc.C109.001560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liu XS, Li H, Song B, Liu X. Polo-like kinase 1 phosphorylation of G2 and S-phase-expressed 1 protein is essential for p53 inactivation during G2 checkpoint recovery. EMBO Rep. 2010;11:626–32. doi: 10.1038/embor.2010.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lin YC, Sun SH, Wang FF. Suppression of Polo like kinase 1 (PLK1) by p21(Waf1) mediates the p53-dependent prevention of caspase-independent mitotic death. Cell Signal. 2011;23:1816–23. doi: 10.1016/j.cellsig.2011.06.016. [DOI] [PubMed] [Google Scholar]
- 92.Chiyoda T, Sugiyama N, Shimizu T, Naoe H, Kobayashi Y, Ishizawa J, et al. LATS1/WARTS phosphorylates MYPT1 to counteract PLK1 and regulate mammalian mitotic progression. J Cell Biol. 2012;197:625–41. doi: 10.1083/jcb.201110110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jang YJ, Ji JH, Choi YC, Ryu CJ, Ko SY. Regulation of Polo-like kinase 1 by DNA damage in mitosis. Inhibition of mitotic PLK-1 by protein phosphatase 2A. J Biol Chem. 2007;282:2473–82. doi: 10.1074/jbc.M605480200. [DOI] [PubMed] [Google Scholar]
- 94.Sun X, Shimizu H, Yamamoto K. Identification of a novel p53 promoter element involved in genotoxic stress-inducible p53 gene expression. Mol Cell Biol. 1995;15:4489–96. doi: 10.1128/mcb.15.8.4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yoo HY, Kumagai A, Shevchenko A, Dunphy WG. Adaptation of a DNA replication checkpoint response depends upon inactivation of Claspin by the Polo-like kinase. Cell. 2004;117:575–88. doi: 10.1016/s0092-8674(04)00417-9. [DOI] [PubMed] [Google Scholar]
- 96.Bates S, Phillips AC, Clark PA, Stott F, Peters G, Ludwig RL, et al. p14ARF links the tumour suppressors RB and p53. Nature. 1998;395:124–5. doi: 10.1038/25867. [DOI] [PubMed] [Google Scholar]
- 97.Stott FJ, Bates S, James MC, McConnell BB, Starborg M, Brookes S, et al. The alternative product from the human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. Embo J. 1998;17:5001–14. doi: 10.1093/emboj/17.17.5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Uchida C, Miwa S, Kitagawa K, Hattori T, Isobe T, Otani S, et al. Enhanced Mdm2 activity inhibits pRB function via ubiquitin-dependent degradation. Embo J. 2005;24:160–9. doi: 10.1038/sj.emboj.7600486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ruan K, Ye F, Li C, Liou YC, Lin SC, Lin SY. PLK1 interacts and phosphorylates Axin that is essential for proper centrosome formation. PLoS One. 2012;7:e49184. doi: 10.1371/journal.pone.0049184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tsvetkov LM, Tsekova RT, Xu X, Stern DF. The Plk1 Polo box domain mediates a cell cycle and DNA damage regulated interaction with Chk2. Cell Cycle. 2005;4:609–17. [PubMed] [Google Scholar]
- 101.Schmit TL, Nihal M, Ndiaye M, Setaluri V, Spiegelman VS, Ahmad N. Numb Regulates Stability and Localization of the Mitotic Kinase PLK1 and Is Required for Transit through Mitosis. Cancer Res. 2012;72:3864–72. doi: 10.1158/0008-5472.CAN-12-0714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fu Z, Malureanu L, Huang J, Wang W, Li H, van Deursen JM, et al. Plk1-dependent phosphorylation of FoxM1 regulates a transcriptional programme required for mitotic progression. Nat Cell Biol. 2008;10:1076–82. doi: 10.1038/ncb1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tang J, Yang X, Liu X. Phosphorylation of Plk1 at Ser326 regulates its functions during mitotic progression. Oncogene. 2008;27:6635–45. doi: 10.1038/onc.2008.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang H, Shi X, Paddon H, Hampong M, Dai W, Pelech S. B23/nucleophosmin serine 4 phosphorylation mediates mitotic functions of polo-like kinase 1. J Biol Chem. 2004;279:35726–34. doi: 10.1074/jbc.M403264200. [DOI] [PubMed] [Google Scholar]