

Abstract
Alzheimer disease is an age-related neurodegenerative condition. AD is histopathologically characterized by the presence of three main hallmarks: senile plaque (SP, rich in amyloid-beta peptide), neuronal fibrillary tangles (NFT, rich in phosphorylated tau protein), and synapse loss. However, definitive biomarkers for this devastating disease in living people are still lacking. In the present study, we showed that levels of oxidative stress markers are significantly increased in the mitochondria isolated from lymphocytes of subjects with mild cognitive impairment (MCI) compared to cognitively normal individuals. Further, increase in mitochondrial oxidative stress in MCI is associated with the MMSE scores, vitamin E components, and beta-carotene. Further, proteomics approach showed that the alterations in the levels of thioredoxin-dependent peroxide reductase, myosin light polypeptide 6, and ATP synthase subunit beta might be important in the progression and pathogenesis of AD. Increased understanding of oxidative stress and protein alterations in easily obtainable peripheral tissues will be helpful in developing biomarkers to combat this devastating disorder.
Keywords: Proteomics, Oxidative stress, Alzheimer disease, Mild cognitive impairment, lymphocytes, mitochondria, peripheral biomarker
Introduction
Biomarkers will be helpful for diagnosis and monitoring the progression of a disease in addition to serving as a tool for testing the therapeutic efficacy of drugs [1]. Since its first description in 1906, the underlying mechanism(s) for Alzheimer disease (AD) so far is (are) not clearly understood nor has a therapeutic approach been developed that could be useful in dealing with this devastating pathology. Currently AD is the sixth leading cause of death in the USA and is the most common form of dementia. This disorder is currently estimated to affect 5.5 million people aged 65 and older in the USA. With approximately 80 million people in the “Baby Boomer” population, it is expected that the number of AD patients will increase to as many as 16–20 million by 2050 unless means to delay the onset or progression of the disease are developed [2]. Hence, there is an urgent need to develop biomarkers for AD, which if not achieved will lead to a significant public health crisis worldwide.
Presently AD is diagnosed by the presence of memory and cognitive impairment, early brain atrophy in several brain regions detected by structural MRI, a characteristic pattern of decreased glucose metabolism in parietal-temporal association cortices shown by FDG-PET analysis [3, 4], and abnormal cerebrospinal fluid (CSF) markers such as elevated tau and phospho-tau levels and reduced level of amyloid β-peptide (Aβ). Up to now definite diagnosis of AD is achieved histopathologically at autopsy and is characterized by the presence of abnormal protein deposits, including senile plaques [the main component of which is amyloid β-peptide (Aβ)] and neurofibrillary tangles [the main component of which is hyperphosphorylated tau protein], and synaptic loss. Aβ[5], generated by the cleavage of amyloid precursor protein (APP) by beta- and gamma-secretases, has been shown to induce oxidative stress in a number of in vitro and in vivo studies [6–9] and, conversely, oxidative stress can increase production of Aβ [10]. A large number of studies have shown increased levels of markers of oxidative modification in different biomolecules (protein, lipids, carbohydrates and nucleic acids) in AD brain and brain from subjects with arguable its earlies form, mild cognitive impairment (MCI), as well as in peripheral systems [11–20]. Further, increasing evidence implicates Aβ accumulation in mitochondria in early stages of AD [21–24]. Mitochondria are the main source of energy and impairment of mitochondrial function leads to decreased energetics and to increased production of free radicals that can in turn impact cellular functions with consequent neuronal loss.
Previously, we demonstrated that elevated levels of indices of oxidation and lipid peroxidation were elevated in mitochondria isolated from lymphocytes obtained from AD patients [25]. To test the hypothesis that such changes occur early in the progression of AD, in the present study we investigated the levels of oxidative stress markers in the mitochondria isolated from lymphocytes of MCI individuals, and showed an inverse correlation between elevated indices of protein oxidation and lipid peroxidation with MMSE scores, vitamin E components, and beta-carotene. Further, proteomics analysis identified thioredoxin-dependent peroxide reductase, myosin light polypeptide 6, and ATP synthase subunit beta might be important in the progression and pathogenesis of AD.
Materials and Methods
Subjects
Subjects were enrolled at the Memory Clinic of the Institute of Gerontology and Geritatrics, University of Perugia. Demographic information of the MCI and Control patients used for the measurement of oxidative stress parameter is presented in Table I. Demographic information of the AD, MCI and control patients used for the identification of proteins with altered protein levels is presented in Table II. All subjects were evaluated according to a standard protocol including a detailed anamnesis, clinical, and neuropsychological evaluation. Subjects with a history of having a smoking habit and/or alcohol abuse, major organ failure, dyslipidemia, or metabolic alterations were not included. After giving informed consent, patients and controls underwent a 20ml blood sample withdrawal. Samples were immediately processed for mitochondria isolation as described previously [25, 26].
Table I.
Demographic information of control and MCI subjects used for measurement of oxidative stress marker
| Controls | MCI | |
|---|---|---|
| n. | 10 | 12 |
| M/F | 1/9 | 5/7 |
| age | 77.9 ± 5.6 | 80 ± 5.2 |
| MMSE | 28.1 ± 1.5 * | 24.0 ± 1.9 |
| ACE-R | 84.9 ± 6.1 * | 70.2 ± 9.6 |
| GDS | 3.5 ± 1.8 | 4.6 ± 1.0 |
| HIS | 1.7 ± 1.3 | 2.4 ± 1.9 |
| ADL | 5.4 ± 0.5 | 5.3 ± 0.6 |
| IADL | 6.5 ± 1.6 | 5.4 ± 1.5 |
p< 0.001 Mann-Whitney test
MMSE Mini Mental State Examination (range 0–30)
ACE-R Addenbrooke’s Cognitive Examination-Revised (range 0–100)
GDS Geriatric Depression Scale (range 0–15)
HIS Hachinski Ischemic Score (range 0–18)
ADL Activity of Daily Living (range 0–6)
IADL Instrumental Activity of Daily Living (range 0–8)
Table II.
Demographic information of control, MCI, and AD subjects used for the identification of proteins with altered protein levels.
| Controls | MCI | AD | |
|---|---|---|---|
| n. | 5 | 5 | 5 |
| M/F | 1/4 | 5/0 | 0/5 |
| age | 76.6 ± 7.4 | 79.6 ± 7.5 | 82.6 ± 4.6 |
| MMSE | 29.0 ± 1.4 | 24.8 ± 1.1 | 18.8 ± 2.4 |
| ACE-R | 87.6 ± 7.6 | 71.4 ± 9.9 | 44.2 ± 9.9 |
| GDS | 3.8 ± 1.3 | 4.4 ± 1.1 | 5.2 ± 2.3 |
| HIS | 2.0 ± 1.4 | 3.0 ± 2.9 | 2.6 ± 1.1 |
| ADL | 5.2 ± 0.4 | 5.4 ± 0.5 | 4.8 ± 1.3 |
| IADL | 6.0 ± 1.9 | 4.4 ± 0.5 | 2.0 ± 1.2 |
p< 0.001 for MMSE, ACE-R and IADL among groups (Kruskal Wallis test)
MMSE Mini Mental State Examination (range 0–30)
ACE-R Addenbrooke’s Cognitive Examination-Revised (range 0–100)
GDS Geriatric Depression Scale (range 0–15)
HIS Hachinski Ischemic Score (range 0–18)
ADL Activity of Daily Living (range 0–6)
IADL Instrumental Activity of Daily Living (range 0–8)
Materials
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise stated. The Oxyblot oxidized protein kit was obtained from Intergen, Inc. (Purchase, NY). Primary antibodies for 4-hydroxynonenal (HNE) and 3-nitrotyrosine (3-NT) were obtained from Chemicon (Temecula, CA).
Freshly obtained blood was layered on Lymphoprep (Gibco, BRL, Bethesda MD), centrifuged and washed twice. The pellet was resuspended in 400 μl of ice-cold PBS. Eight μl of 2.5% digitonin were added and kept on ice for 5 min inverting gently every 30 s. Each sample was sonicated for 10 s at 20% power for 1 min and then centrifuged at 600g for 10 min at 4°C to eliminate nuclei and unbroken cells. The supernatant was centrifuged at 14,000g for 10 min at 4°C, the pellet resuspended in 400μl ice cold PBS and centrifuged at 7,000g for 10 min at 4°C. The pellet was resuspended in 400μl ice cold PBS and centrifuged at 3,500g for 10 min at 4°C. The pelleted purified mitochondria were immediately frozen and kept at −80°C until analyses. Mitochondria purity was determined spectrophotometrically following ferrocytochrome c oxidation at 550 nm at 25°C [27].
Sample Preparation
To the frozen mitochondria 100μl of Media I buffer [0.32 M sucrose, 0.10 mM Tris HCl (pH 8.0), 0.10 mM MgCl2, 0.08 mM EDTA, 10 μg/ml leupeptin, 0.5 μg/ml pepstatin, and 11.5 μg/ml aprotinin; pH 8.0] was added and samples were vortexed followed by sonication for 10 s at 20% power with a Fisher 550 Sonic Dismembrator (Pittsburgh, PA). Protein concentrations were determined according to the Pierce BCA method (Rockford, IL).
Measurements of Oxidation-derived protein and Lipid Damage Markers in Control and MCI samples
1. Protein Carbonyl Measurement
Protein carbonyl content in the samples was determined according to the manufacturer’s protocol (Oxyblot; Chemicon) and as described previously [28]. Briefly, 5 μl of control and MCI mitochondria samples (4 mg/ml) were incubated for 20 min with 10 mM DNPH and 5 μl of 12% sodium dodecyl sulfate (SDS). Samples were neutralized with 7.5 μl of neutralization solution (2 M Tris in 30% glycerol). The derivatized proteins (250 ng) were transferred onto nitrocellulose membrane by the slot blot technique. Membranes were incubated with blocking buffer for 60 min at 27°C and incubated with rabbit antibodies to DNPH (diluted 1:150) for 90 min, then by anti-rabbit IgG coupled to alkaline phosphatase (1:10,000) for 1 hr at 27°C. After being washed and developed with SigmaFast chromogen (Sigma), blots were scanned into Adobe Photoshop (Adobe Systems, Inc., Mountain View, CA) and quantitated with Scion Image (PC version of Macintosh-compatible NIH Image). To validate the protein carbonyl detection approach, protein carbonyls were reduced by sodium borohydride or incubation with secondary antibody and showed no staining on the blots (data not shown) as described previously [29].
2. 3-Nitrotyrosine Levels (3-NT)
To determine protein-bound 3-NT levels, 5 μl (10ug) of control and MCI mitochondria samples were incubated with Laemmli buffer (10 μl ) (0.125 M Trizma base, pH 6.8, 4% SDS, 20% glycerol) for 20 min. Samples (250 ng of protein) were blotted onto nitrocellulose membranes, and immunochemical methods were performed as described previously [28]. The rabbit anti-3-nitrotyrosine (3-NT) primary antibody was incubated 1:200 in blocking buffer [bovine serum albumin (BSA) 3% in TBS-T] for 2 hr. The membranes were washed three times with tris buffered saline-Tween (TBS-T) and incubated with alkaline phosphatase-conjugated goat anti-rabbit secondary antibody (1:10,000). Densitometric analysis of bands in images of the blots was used to calculate levels of 3-NT. To validate the protein nitration detection approach the nitrotyrosine were reduced by dithionite or use of secondary antibody alone showed no staining on the blot (data not shown) as described previously [30].
3. Protein bound 4-hydroxy-2-nonenal (HNE)
Protein bound 4-hydroxy-2-nonenal (HNE) levels were measured as a marker of lipid peroxidation [18, 28]. The control and MCI mitochondrial samples (5 μl) were incubated with 10 μl Laemmli buffer for 20 min at room temperature, and 250 ng of protein samples were loaded into each well on nitrocellulose membrane in a slot blot apparatus under vacuum. The membranes were incubated with anti-HNE rabbit polyclonal antibody (1:5,000) for 2 h, washed three times with TBS-T, and then incubated with an anti-rabbit IgG alkaline phosphatase-conjugated secondary antibody (1:10,000). Blots were developed with SigmaFast tablets (BCIP/NBT), dried, and quantified in Scion Image.
Protein Alterations: Proteomics Approach
To identify proteins with differential levels between control (n=5), MCI (n=5), and AD (n=5) we used a proteomics approach (See Table II for demographic information) [31].
1. Isoelectric Focusing (IEF)
Fifty micrograms of mitochondrial protein from control, AD and MCI samples were precipitated separately by addition of 15% ice-cold trichloroacetic acid (TCA) for 10 min, followed by centrifugation at 14,000 rpm (23,700 × g) for 5 min at 4 °C. Pellets were washed in Wash Buffer [1:1 (v/v) ethanol:ethyl acetate] a total of four times to remove excess salts. Following the final wash, 200 μl of rehydration buffer [8 M urea, 2 M thiourea, 50 mM DTT, 2.0% (w/v) CHAPS, 0.2% Biolytes, bromophenol blue], was added to the samples and incubated for 2h at RT, and then sonicated for 10 s at 20% power. Samples (200 μl) were applied to 11 cm pH 3–10 ReadyStrip™ IPG strips and actively rehydrated at 20 °C for 18 h at 50 V, followed by isoelectrofocusing at a constant temperature of 20 °C beginning at 300 V for 2 h, 500 V for 2 h, 1,000 V for 2 h, 8,000 V for 8 h, and finishing at 8,000 V for 10 h rapidly. IEF strips were stored at −80 °C until the second dimension of analysis was performed.
2. 2D-PAGE
2D-PAGE was performed to further separate proteins previously separated on IEF strips. IEF strips were thawed and equilibrated for 10 min in equilibration buffer A [50 mM Tris-HCl pH 6.8, 6 M urea, 1% (w/v) SDS, 30% (v/v) glycerol, and 0.5% DTT] and then re-equilibrated for 10 min in equilibration buffer B [50 mM Tris-HCl pH 6.8, 6 M urea, 1% (w/v) SDS, 30% (v/v) glycerol, and 4.5% IA]. All strips were rinsed in a 1× dilution of Tris glycine saline (TGS) running buffer before being placed into Criterion precast linear gradient (8–16%) Tris-HCl polyacrylamide gels. Precision Plus Protein™ Standards and samples were run at a constant voltage of 200 V for 65 min in a 1× dilution of TGS running buffer.
3. SYPRO Ruby® Staining
Following 2D-PAGE, gels were incubated in a Fixing solution [7% (v/v) acetic acid, 10% (v/v) methanol] for 20 min at RTSypro Ruby® Protein Gel Stain (~50 ml) was added to gels and allowed to stain overnight at RT on a gently rocking platform, followed by scanning of the gels with a Molecular Dynamics STORM Phosphoimager (λex/λem: 470/618 nm).
4. Image Analysis
Spot intensities from SYPRO Ruby®-stained 2D-gel images of cognitively normal, MCI and AD samples were quantified densitometrically according to the total spot density using PDQuest analysis software from Bio-Rad (Hercules, CA). Intensities were normalized to total gel densities and/or densities of all valid spots on the gels. For the determination of spots with increased or decreased levels, we normalized spot density in AD/MCI samples compared to MCI/Control samples. Only protein spots with a statistically significant difference based on a Student’s t-test at 95% confidence (i.e., P<0.05) were considered for MS analysis.
In-Gel Trypsin Digestion
Protein spots identified as significantly altered were excised from 2D-gels with a clean, sterilized blade and transferred to Eppendorf microcentrifuge tubes. Gel plugs were then washed with 0.1 M ammonium bicarbonate (NH4HCO3) at RT for 15 min, followed by incubation with 100% acetonitrile at RT for 15 min. After solvent removal, gel plugs were dried in their respective tubes under a flow hood at RT. Plugs were incubated for 45 min in 20 μl of 20 mM DTT in 0.1 M NH4HCO3 at 56 °C. The DTT/NH4HCO3 solution was then removed and replaced with 20 μl of 55 mM IA in 0.1 M NH4HCO3 and incubated with gentle agitation at RT in the dark for 30 min. Excess IA solution was removed and plugs incubated for 15 min with 200 μl of 50 mM NH4HCO3 at RT. A volume of 200 μL of 100% acetonitrile was added to this solution and incubated for 15 min at RT. Solvent was removed and gel plugs were allowed to dry for 30 min at RT under a flow hood. Plugs were rehydrated with 20 ng/μl of modified trypsin (Promega, Madison, WI) in 50 mM NH4HCO3 in a shaking incubator overnight at 37 °C. Enough trypsin solution was added to completely submerge the gel plugs.
Mass Spectrometry
Salts and contaminants were removed from tryptic peptide solutions using C18 ZipTips (Sigma-Aldrich, St. Louis, MO), reconstituted to a volume of ~15 μL in a 50:50 (water:acetonitrile) solution containing 0.1% formic acid. Tryptic peptides were analyzed by MS/MS with an automated Nanomate electrospray (ESI) [Advion Biosciences, Ithaca, NY] Orbitrap XL MS (ThermoScientific, Waltham, MA) platform. The Orbitrap MS was operated in a data-dependent mode whereby the 8 most intense parent ions measured in the Fourier Transform (FT) at 60,000 resolution were selected for ion trap fragmentation with the following conditions: injection time 50 ms, 35% collision energy, MS/MS spectra were measured in the FT at 7500 resolution, and dynamic exclusion was set for 120 sec. Each sample was acquired for a total of ~2.5 min. MS/MS spectra were searched against the International Protein Index (IPI) Database (downloaded 03/05/09) using SEQUEST with the following specifications: 2 trypsin miscleavages, fixed carbamidomethyl modification, variable Methionine oxidation, parent tolerance 10 ppm, and fragment tolerance of 25 mmu or 0.01 Da. Results were filtered with the following criteria: Xcorr > 1.5, 2.0, 2.5, 3.0 for +1, +2, +3, and +4 charge states, respectively, Delta CN >0.1, and P-value (protein and peptide) <0.01. Accession numbers from IPI were cross-correlated with SwissProt accession numbers for final protein identification. It should be noted that proteins identified with a single peptide were kept for further analyses if multiple spectral counts (SC, number of observed MS/MS spectra) were observed in a single analysis or if the peptide was identified in a separate analysis and workup of the same protein spot.
Vitamin E and beta-carotene measurement methods
Analyses were performed as described previously [26]. Blood samples were taken from subjects after a minimum of 2 hours fasting; serum and plasma samples were aliquoted and frozen at −80 °C. Plasma tocopherols, tocotrienols, alpha-tochopherylquinone, 5-NO2-gamma-tocopherol, and beta-carotene were measured with reverse phase high performance liquid chromatography (HPLC) using electrochemical-coularray system (ESA, Chelmsford, MA, USA). Aliquots of 200 μL were mixed and extracted 3 times with a 1:2 ratio of ethanol to hexane, concentrated to dryness with high-purity nitrogen gas, and reconstituted in 300 μL mobile phase. Beta-tocopherol (Superchrome, Milan, Italy); alpha-, gamma-, and delta-tocopherol, beta-carotene (Sigma-Aldrich, Milan, Italy), alpha-, gamma-, and delta-tocotrienol (LGC-Promochem, Milan, Italy), beta-tocotrienol (Matreya-DBA, Pleasant-Gap, PA, USA), alpha-tochopherylquinone (Research Organics, Rome, Italy), and 5-NO2-gamma-tocopherol (gift from Prof. K. Hensley, University of Toledo Health Sciences Center, Toledo, OH, USA) were used as standards. After filtration, analyte separation was conducted at room temperature on a Discovery-C18-column (Sigma-Aldrich). Mobile phase (30 mmol lithium acetate/L, 83% HPLC grade acetonitrile, 12% HPLC grade methanol, and 0.2% HPLC grade acetic acid, pH 6.5) was delivered at 1 mL/minute.
2D Western blot
Proteins were first separated by 2D gel electrophoresis followed by protein transfer to nitrocellulose membrane. The membrane was blocked with 3% bovine serumlbumin (BSA) in PBST for 1 h at 4°C. The membrane was incubated with anti-peroxiredoxin polyclonal antibody (Abcam, MA) (1:1000) for 2 h in 1% BSA for 2 h at room temperature, and then the membrane was washed three times in PBST for 5 min each. An anti-rabbit IgG alkaline phosphatase secondary antibody was diluted 1:3000 in PBST and incubated with the membranes for 1 h at room temperature. The membranes were washed in PBST three times for five minutes and developed using Sigmafast Tablets (BCIP/NBT substrate).
Statistical Analysis
Two-tailed, Student’s t-tests were used to analyze differences in oxidative stress markers between MCI and age-matched controls samples. A p-value of less than 0.05 was considered statistically significant. Correlation analyses were assessed for significance using GraphPad Prism 5.
Results
Oxidative stress markers
In our laboratory we measured the amount of protein carbonyls and 3-nitrotyrosine (3-NT) levels in biological samples to assess the oxidation status of protein [32]. The levels of protein carbonyls (p<0.05) (Fig 1A) are significantly increased in MCI lymphocyte mitochondria compared to controls. Figure 1B shows levels of HNE-bound protein, a product of lipid peroxidation, are significantly increased in mitochondria isolated from MCI lymphocytes compared to control (p<0.05). Figure 1C shows that levels of 3-NT (p<0.05) (Fig 1C) are significantly increased in MCI lymphocyte mitochondria compared to controls.
Figure 1.
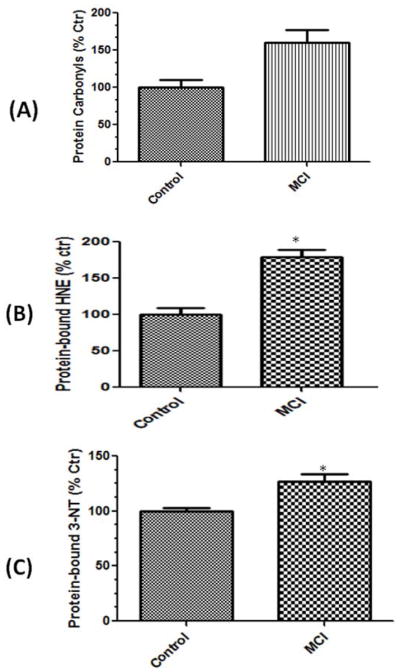
Protein carbonyls (A), protein-bound HNE (B), and protein-resident 3-nitrotyrosine levels (3-NT) were found to be significantly increased in mitochondria isolated from lymphocytes from mild cognitive impairment (MCI) patients compared to those of respective controls. Data are shown as percent control. N= 10 for controls, 12 for MCI. p*<0.05.
Correlation analysis of oxidative stress markers and MMSE
The levels of protein carbonyls showed an inverse correlation (r2=−0.22) with MMSE score (Fig 2A) of MCI patients; however, this correlation is not statistically significant (p=0.3). In contrast, protein-bound HNE showed a significant (p=0.02) negative correlation (r2=−0.58) with MMSE score (Fig 2B). The levels of protein-bound 3NT did not show any significant (p=0.07) correlation with MMSE score (Fig 2C), although we observed a trend towards a negative correlation with MMSE score (r2=−0.45). The correlation analysis of oxidative stress with MMSE suggests that the increased protein-bound HNE is related to the decrease in cognitive functions in MCI, and further support the hypothesis that lipid peroxidation is an early event in the pathogenesis of AD [33].
Figure 2.

A correlative analysis between markers of oxidative stress and MMSE. Linear correlation analysis between protein carbonyls/ protein-bound HNE, protein-bound 3-NT and MMSE showed a significant negative correlation between the oxidative stress markers and MMSE scores (see text).
Correlation analysis of Vitamin E components and beta-carotene with oxidative stress markers
The Vitamin E and beta-carotene data used in this study were a part of recently published paper by Meccoci’s laboratory, and showed that alterations in the levels of vitamin E have an effect on MMSE scores [26]. In the current study, we carried out correlation analysis between Vitamin E components, and beta-carotene with the markers of oxidative stress, and only those components that showed a significant correlation are reported here. Figure 3 shows that the amount of tocopheryl quinone (r2=−0.48, p=0.02), delta-tocopherol (r2=−0.52, p=0.01), and beta-carotene (r2=−0.47, p=0.03) showed significant inverse correlations with protein carbonyls. Further, the levels of gamma-tocotrienol (r2=−0.55, p=0.02), alpha-tocotrienol (r2=−0.65, p=0.01), delta-tocopherol (r2=−0.64, p=0.008), gamma-tocopherol (r2=−0.68, p=0.004), and beta-carotene (r2=−0.62, p=0.01) showed a significant inverse correlation with protein-bound HNE. A significant negative correlation is observed between protein-bound 3-NT and gamma-tocotrienol (r2=−0.52, p=0.04), delta-tocopherol (r2=−0.52, p=0.03), tocopheryl quinone (r2=−0.51, p=0.04), gamma-tocopherol (r2=−0.69, p=0.003), and beta-carotene (r2=−0.64, p=0.007) (Figure 4). The levels of 5-nitro-γ-tocopherol (5-NO2-γ-tocopherol), a product of reaction between gamma-tocopherol and RNS [34], showed a significant positive correlation with protein carbonyls (r2=0.54, p=0.01), protein-bound HNE (r2=0.79, p=0.0003), and protein-bound 3-NT (r2=−0.75, p=0.001) (Figure 5).
Figure 3.

A correlative analysis between protein carbonyls and tocopheryl quinine/delta-tocopherol/, and beta-carotene showed a significant negative correlation, whereas 5-NO2-gamma-tocopherol showed a significant positive correlation with protein carbonyls (see text).
Figure 4.

A correlative analysis between protein-bound HNE and gamma-tocotrienol/alpha-tocotrienol/ delta-tocopherol/ gamma-tocopherol/ and beta-carotene showed a significant negative correlation, whereas 5-NO2-gamma-tocopherol showed a significant positive correlation with protein-bound HNE (see text).
Figure 5.

A correlative analysis between protein-bound 3NT and gamma-tocotrienol/delta-tocopherol/tocopheryl quinine/gamma-tocopherol/ and beta-carotene showed a significant negative correlation, whereas 5-NO2-gamma-tocopherol showed a significant positive correlation with protein-bound 3-NT (see text).
Alterations of protein levels in mitochondria
Two-dimensional gel-based proteomic analyses were used to identify mitochondrial proteins that showed altered levels in MCI and AD compared to age-matched controls. From these experiments we present results from three comparisons: control vs. MCI, control vs. AD, and MCI vs. AD. The comparison of mitochondrial proteins between control and MCI provide insight into which specific protein(s) alteration might be critical in the transition from MCI to AD. The proteins with altered levels between control and MCI are shown in Figure 6. Table III list the characteristics of the proteins including the number of peptide sequences, scores, molecular weight (MW), isoelectric point (pI), and p-values and fold-change levels. All the proteins from lymphocyte mitochondria that were identified as altered between controls and MCI show increased levels and are grouped into 4 categories: cellular energetics that include -glyceraldehyde 3-phosphate deydrogenase (GADPH), lactate dehydrogenase B chain (LDH), and ATP synthase subunit beta; structural proteins: annexin, beta-centractin, and myosin light polypeptide 6; cell signaling-Rho GDP-dissociation inhibitor 2 (RhoGDI); and cellular defense: thioredoxin-dependent peroxide reducase/ peroxiredoxin III (PDXIII).
Figure 6. Identification of the proteins that are of differential levels in mitochondria isolated from the between control and MCI lymphocytes.

Representative 2D-gel images of the proteins isolated from the mitochondria of (A) Control and (B) MCI lymphocytes. Protein showing altered levels was indicated by spot ID, and their identification was from nanospray ESI-MS/MS analyses. N=5 for control and MCI.
Table III.
Characteristic of proteins showing altered levels between Controls vs. MCI
| Spot | Protein Identified | Accession # | Coverage | Number of peptides | Score | MW (kDa) | pI | P value | Fold |
|---|---|---|---|---|---|---|---|---|---|
| 1409 | Glycerald ehyde-3-phosphate dehydrogenase | P04406 | 17.31 | 5 | 39.67 | 66 | 8.12 | 0.05 | 3.5 (i) |
| 4306 | Annexin | E7EMC6 | 21.52 | 6 | 71.36 | 36.9 | 5.91 | 0.02 | 2.0 (i) |
| 5202 | Thioredox independent peroxide reductase | E9PH29 | 14.71 | 3 | 17.69 | 25.8 | 7.46 | 0.02 | 3.3 (i) |
| 5304 | L-lactate dehydrogenase B chain | P07195 | 28.74 | 8 | 49.59 | 36.6 | 6.05 | 0.05 | 3.3 (i) |
| 7203 | Rho GDP-dissociation inhibitor 2 | P52566 | 51.74 | 8 | 75.29 | 23 | 5.21 | 0.01 | 3.5 (i) |
| 7501 | ATP synthase subunit beta, | P06576 | 55.01 | 21 | 188.34 | 56.5 | 5.4 | 0.06 | 1.8 (i) |
| 8103 | Myosin light polypeptide 6 | F8VZV5 | 62.96 | 9 | 49.05 | 15 | 5.00 | 0.02 | 5.4 (i) |
‘i’ indicates increase and ‘d’ indicate decrease
Figure 7 shows a representative 2D-gel image of spots observed to statistically change in density in control vs AD. The characteristics of the identified proteins are listed in Table IV. The levels of most of the proteins are significantly decreased in AD compared to controls, except for ATP synthase subunit beta, and Ras suppressor protein 1. The proteins with differential levels between control and AD are grouped into 4 categories: cellular energetics :ATP synthase subunit beta; structural proteins: vimentin, actin related protein 2/3 complex subunit 2; cell signaling: Ras spressor protein 1; and cellular defense: heat shock protein 70, thioredoxin.
Figure 7. Identification of the proteins that are of differential levels in mitochondria isolated from control and AD lymphocytes.

Representative 2D-gel images of the proteins isolated from the mitochondria of (A) Control and (B) AD lymphocytes. Protein showing altered levels was indicated by spot ID, and their identification was from nanospray ESI-MS/MS analyses. N=5 for control and AD
Table IV.
Characteristic of proteins showing altered levels between Controls vs. AD
| Spot | Protein Identified | Accession # | Coverage | Number of peptides | Score | MW (kDa) | pI | P value | Fold |
|---|---|---|---|---|---|---|---|---|---|
| 2110 | Profilin-1 | P07737 | 27.5 | 3 | 26.12 | 15 | 8.27 | 0.02 | 0.01 (d) |
| 2302 | Ras suppressor protein 1 | Q15404 | 31.41 | 6 | 60.36 | 31.5 | 8.65 | 0.03 | 2.4 (i) |
| 3305 | Actin-related protein 2/3 complex subunit 2 | O15144 | 42.33 | 13 | 68.13 | 34.3 | 7.36 | 0.01 | 0.06 (d) |
| 4403 | Heat shock cognate 71 kDa protein | E9PN89 | 8.33 | 2 | 9.46 | 34.8 | 7.53 | 0.001 | 0.01 (d) |
| 7003 | Thioredoxin | B1ALW1 | 38.82 | 4 | 15.00 | 9.4 | 6.04 | 0.04 | 0.02 (d) |
| 7501 | ATP synthase subunit beta, | P06576 | 55.01 | 21 | 188.34 | 56.5 | 5.4 | 0.03 | 1.5 (i) |
| 8101 | Vimentin | F5H288 | 19.9 | 11 | 62.47 | 47 | 5.00 | 0.02 | 0.02 (d) |
‘i’ indicates increase and ‘d’ indicate decrease
Figures 8 shows a representative 2D-gel image of mitochondrial protein spots observed to statistically change in density in MCI vs AD. The characteristics of the identified proteins are listed in Table V. The level of all the identified proteins is significantly decreased in AD compared to MCI. The proteins with differential levels between MCI and AD are grouped into 2 categories: structural proteins: actin cytoplasmic 1, myosin light polypeptide 6; and cellular defense: thioredoxin-dependent peroxide reductase, thioredoxin.
Figure 8. Identification of the proteins that are of differential levels in mitochondria isolated from MCI and AD lymphocytes.

Representative 2D-gel images of the proteins isolated from the mitochondria of (A) MCI and (B) AD lymphocytes. Protein showing altered levels was indicated by spot ID, and their identification was from nanospray ESIMS/MS analyses. N=6 for MCI and AD
Table V.
Characteristic of proteins showing altered levels between MCI vs. AD
| Spot | Protein Identified | Accession # | Cover age | Number of peptides | Score | MW (kDa) | pI | p value | Fold |
|---|---|---|---|---|---|---|---|---|---|
| 4403 | Heat shock cognate 71 kDa protein | E9PN89 | 8.33 | 2 | 9.46 | 34.8 | 7.53 | 0.0001 | 0.02 (d) |
| 5202 | Thioredoxin dependent peroxide reductase | E9PH29 | 14.71 | 3 | 17.69 | 25.8 | 7.46 | 8.5×10−7 | 0.02 (d) |
| 5503 | Protein disulfide isomerase A3 | B3KQT9 | 40.42 | 19 | 103.68 | 54.1 | 7.21 | 0.01 | 0.14 (d) |
| 6301 | Actin, cytoplasmic 1, N terminally processed | B4DW52 | 49.28 | 12 | 184.08 | 38.6 | 5.35 | 0.03 | 0.29 (d) |
| 7003 | Thioredoxin | B1ALW1 | 38.82 | 4 | 15.00 | 9.4 | 6.04 | 0.0002 | 0.01 (d) |
| 8103 | Myosin light polypeptide 6 | F8VZV5 | 62.96 | 9 | 49.05 | 15 | 5.00 | 0.02 | 0.32 (d) |
To validate the proteomics results, 2D Western blot analysis was performed for peroxiredoxin III (Figure 9). The spot corresponding to peroxiredoxin III appeared at the same location on the gel from where we excised and submitted spot for MS identification of the protein, thereby validating the correct identification of this protein and providing confidence on the identification of all proteins.
Figure 9. A representative blot validating the location of peroxiredoxin III on blot using 2D Western blot.

Spot number 5202 on gel (A) corresponding to peroxiredoxin III was validated by 2D Western blot (B) that was probed with anti-peroxiredoxin III antibody, consistent with the molecular weight, pI and location of peroxiredoxin III, thereby confirming its identity following proteomics analysis.
Discussion
Mitochondria are one of the main sources for production of reactive oxygen species. Oligomeric Aβ has been shown to appear in the mitochondria during the early stage of MCI [23, 35]. Oligomeric Aβ has been suggested to be highly reactive and is capable of inserting itself in the lipid bilayer consequently inducing free radical production because of the presence of methionine at the 35th amino acid residue [9]. Met in Aβ is capable of forming a sulfuranyl radical and subsequently abstracting an allylic hydrogen atom from acyl chains of unsaturated lipids present in the lipid bilayer [36]. As evidenced by increased markers of protein oxidation, peripheral mitochondria are highly vulnerable to oxidative stress demonstrating early oxidative stress in the progression of AD, similar to what is observed in brain.
Previous studies from our laboratory showed that the mitochondria isolated from AD lymphocytes showed increased markers of oxidative stress, and the oxidative stress inversely correlated with MMSE scores [25]. MCI samples in this study showed a significant relationship between an increased protein-bound HNE and MMSE scores. This observation is consistent with lipid peroxidation as a process that is an early event in the progression of disease and the reported cognitive decline in the patients suffering from this devastating disorder [37].
Beta-carotene is a precursor of vitamin A, and has been reported to have ability to scavenge peroxyl radicals. Further, in vitro studies showed that beta-carotene can inhibit Aβ aggregation in a dose-dependent manner. The levels of beta-carotene were reported to be decreased in brain, serum and plasma of AD and MCI subjects [26, 38, 39]. Recently published data on levels of beta-carotene using the same sample used in the current study showed a significant decrease in MCI plasma compared to control and it correlated inversely to MMSE score [26]. In the present study, we found that the levels of beta-carotene are inversely proportional to the markers of oxidative stress. Decreased levels of beta-carotene might lead to increased aggregation of Aβ and consequently to oxidative stress-mediated damage and be important in AD pathogenesis.
Vitamin E is a major lipid-soluble vitamin known to be good chain-breaking antioxidant [40, 41]. Vitamin E includes a group of 8 compounds referred to as tocopherols and tocotrienols [40]. Previous studies have reported reduced levels of alpha-tocopherol in the CSF, plasma of subjects with AD, and mild cognitive impairment (MCI) [26, 42–45]. The exact role of vitamin E in AD pathogenesis is still controversial. A number of clinical trials using alpha-tocopherol failed to show any protective effect as therapeutics in clinical studies involving late-stage AD or MCI patients [42, 46, 47]. In vitro and in vivo studies showed that other forms of Vitamin E, i.e., gamma-tocopherol and tocotrienols, were more effective than alpha-tocopherol in reducing the oxidative stress [26, 48]. In the present study, we showed that with the increase oxidative stress in mitochondria isolated from lymphocytes in patients with MCI the levels of delta-tocopherol decreased, and suggest that the alteration in the levels of delta-tocopherol might be an early event in the progression of AD.
Two markers of Vitamin E damage, i.e, 5-NO2-gamma-tocopherol and alpha-tocopherylquinone showed a significant difference with respect to oxidative stress markers. Gamma-tocopherol has been shown to protect against peroxynitrite-induced lipid peroxidation [49]. A previous study showed increased levels of 5-NO2-gamma-tocopherol in AD brain [34]. In this study, we found a direct correlation between the levels of 5-NO2-gamma-tocopherol and the oxidative stress markers. The increased nitration of gamma-tocopherol could be due to increased nitration observed in the MCI samples which could potentially serve as a marker for monitoring the nitrosative stress in AD pathogenesis. Alpha-tocopherylquinone is a metabolite of alpha-tocopherol, which upon reduction to tocopherylhydroquinone has been shown to have antioxidant activity [50]. The decrease in the levels of alpha-tocopherylquinone with concomitant increased in oxidative stress markers reported here suggest one factor important in the elevated oxidative stress.
In addition to changes in oxidative stress, analysis of the mitochondrial proteome showed altered protein levels in MCI and AD mitochondria compared to controls. A large number of proteins were found to have altered expression during the progression of AD which can be functionally categorized as energetics, structural, antioxidant, and cell signaling. Interestingly, we found that proteins involved in energetics, i.e., GAPDH, lactate dehydrogenase B chain, ATP synthase subunit beta are significantly increased in the MCI samples compared to control, indicating that cell might be experiencing stress and there is an energy demand to overcome this stress. Previous studies from our laboratory and others showed alteration of these proteins in AD and MCI brain [51–53, 54, 55, 56]. It is widely accepted that early in the progression of AD glucose metabolism is affected as shown by PET analysis [3]. We earlier identified a number of proteins in the glycolytic pathway as oxidatively modified [56]. GAPDH is not just a glycolytic enzyme, but has multifunctional roles and is found in different subcellular locations in brain [57]. One role of GAPDH is to capture nitric oxide. Hence, the increase in the GAPDH we found here also could be due to increased nitric oxide formation in MCI lymphocytes as indexed by the observed increased protein-bound 3-NT levels.
Further, levels of ATP synthase beta subunits were found to significantly increased in mitochondria isolated from lymphocytes of MCI/AD patients compared to cognitively normal individuals. A recent study using a transgenic model of AD showed that the levels of ATP synthase beta subunit increases and is linked to both Aβ oligomers and amyloid deposition [58]. Previous studies from our laboratory showed that increased levels of ROS/RNS lead to modification of biomolecules including proteins, which in most cases make them dysfunctional [56]. The increased oxidation of mitochondrial proteins reported here might affect the function of mitochondria leading to increased lactate dehydrogenase (LDH) levels to meet the cellular energy demands.
Rho GDP-dissociation inhibitor 2 level is significantly increased in MCI samples compared to controls. RhoGDIs play a negative regulatory role for RhoGTPases, which regulate cytoskeleton and other cellular functions including proliferation, differentiation, and apoptosis. Hence, an increase in the levels of RhoGDI could lead to decrease in RhoGTPases activity and thereby modulating apoptotic cell death following increased oxidation.
The decreased levels of heat shock cognate 71 protein and PDI were found in mitochondria isolated from the lymphocytes during the transition of the disease from MCI to AD. Heat shock cognate 71 is also decreased in AD mitochondria compared to controls. Both PDI and heat shock cognate protein have Cys residues at positions Cys5 and Cys267, respectively that are sensitive to oxidation. The observation of increased oxidative stress together with decreased levels of these proteins is consistent with increased accumulation of damaged protein reported in AD.
Vimentin is an intermediate filament that supports the morphology, organization and function of mitochondria. The protein ARPC2/3 is important in regulation of actin filament polymerization. Both vimentin and ARPC2/3 levels were found to be decreased in AD compared to controls, which are consistent with cytoskeletal reorganization and may contribute to fragmentation of mitochondria [59]; such changes would consequently increase ROS/RNS production, and decrease cellular energetics.
PRDX3 and myosin light polypeptide 6 were found to be significantly increased in levels in MCI mitochondria compared to control; however, during the transition of MCI to AD the levels of both these proteins were significantly decreased in the mitochondria, suggesting that the altered levels of these protein might be important in the progression of AD.
PRDX3 is found exclusively in the mitochondrion and comprises 5% of the total mitochondrial matrix. PRDX3 is a 2-cysteine peroxidase of the mammalian peroxiredoxin (Prx), a family of six isoforms that is capable of catalyzing H2O2 reduction [60, 61]. During the reduction of H2O2 the active NH2-terminal Cys of one subunit in the PRDX3 homodimer is oxidized to cystine sulfenic acid (Cys-SOH), which forms disulfide linkage with the C-terminal Cys-SH of the other homodimer subunit. Thioredoxin (Trx) then reduces the thiolate form of the active cysteine that lowers the active site pKa and preserves the anionic charged state of NH2-terminal Cys-SH. Recent studies suggested that the mammalian Trx/Prx redox system is critical for the antioxidant regeneration and regulation of intracellular ROS level generated by respiration and metabolism of dopamine [62, 63].
As discussed earlier Aβ is found in the mitochondria early during the progression of the disease, which can initiate the process of lipid peroxidation in the mitochondrial membrane leading to the alterations in the function and composition of mitochondrial [22, 23], eventually leading to impairment of the electron transport chain (ETC), consequently increased formation of ROS and oxidative stress. The increased levels of PRDX3 during the early stage of the disease suggest that oxidative stress occurs during the early stages of disease progression. Hence, increased levels of PRDX3 at the MCI stage of AD suggests a compensatory mechanism to protect against elevated oxidative stress as observed by the increased markers of oxidative stress in the MCI samples. Elevated levels of PRDX3 were induced by hypoxia which protects the cells against excitotoxic cell death and apoptosis. Lubec et al., 2001 showed decreased levels of PRDX 3 in AD and Down syndrome brain [64].
Interestingly, both the levels of PRDX3 and thioredoxin are decreased in mitochondria isolated from AD lymphocytes. The reduced levels of PRDX3 leads to increased formation of ROS including H2O2 that could accelerate apoptosis via increased production of oxidative stress by-products like HNE, with increased rates of mitochondrial membrane potential collapse, release of cytochrome c, and activation of caspase [65]. A previous study showed increased levels of oxidative stress markers, altered antioxidant enzymes levels and activities, and decreased plasma levels of GSH and increased levels of GSSG [66] in the lymphocytes of AD patients, consistent with an elevated oxidative environment in AD. Further, others showed that the levels of plasma antioxidants such as α-carotene and β-carotene etc., were significantly lower in patients with AD compared with controls [13]. The imbalance in the levels of antioxidants and increased levels of oxidative insult conceivably could be due to Aβ in the plasma of AD subjects. However, studies related with plasma Aβ levels showed contradictory results.
Myosin light polypeptide 6 levels is increased in MCI mitochondria from lymphocytes compared to the control. However, during the transition from MCI to AD the level of this protein is significantly decreased. Myosin light polypeptide is a regulatory light chain of myosin, and hence important for the function of myosin. Decreased levels of myosin during the progression of AD may compromise mitochondrial structure and consequently the function leading to loss of cellular energetics, increased production of free radicals, and eventually to cell death.
In conclusion, the mitochondria isolated from MCI lymphocytes showed increased oxidative stress that correlated with altered levels of a number of vitamin E components suggesting the increased oxidative stress markers in the peripheral system may potentially reflect the brain damage and could potentially serve as a biomarker for progression, diagnosis, or treatment of AD. Further, identification of affected mitochondrial proteins in common during the progression of AD suggests these proteins might be instrumental in progression, diagnosis or treatment early in the disease. Further, identification of proteins that are targets of oxidation could be helpful in understanding better the mechanism of disease progression and pathogenesis. Such studies are currently underway in our laboratory.
Elevated oxidative stress was found in mitochondria isolated from lymphocytes in MCI.
Proteomics analyses of mitochondria from lymphocytes in MCI and AD were performed.
Key proteins were identified as differentially expressed in these mitochondria.
Oxidative stress in peripheral mitochondria is an early event in progression of AD.
Potential biomarkers for pathogenesis and progression of AD were found.
Acknowledgments
This work was supported in part by a grant from the NIH to D.A.B [AG-05119], and by a grant from Italian Ministry of Research (MIUR) PRIN 20078TC4E5 to PM
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gustaw-Rothenberg K, Lerner A, Bonda DJ, Lee HG, Zhu X, Perry G, Smith MA. Biomarkers in Alzheimer’s disease: past, present and future. Biomark Med. 2010;4:15–26. doi: 10.2217/bmm.09.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol. 2003;60:1119–22. doi: 10.1001/archneur.60.8.1119. [DOI] [PubMed] [Google Scholar]
- 3.Rapoport SI. In vivo PET imaging and postmortem studies suggest potentially reversible and irreversible stages of brain metabolic failure in Alzheimer’s disease. Eur Arch Psychiatry Clin Neurosci. 1999;249(Suppl 3):46–55. doi: 10.1007/pl00014174. [DOI] [PubMed] [Google Scholar]
- 4.Fox NC, Crum WR, Scahill RI, Stevens JM, Janssen JC, Rossor MN. Imaging of onset and progression of Alzheimer’s disease with voxel-compression mapping of serial magnetic resonance images. Lancet. 2001;358:201–5. doi: 10.1016/S0140-6736(01)05408-3. [DOI] [PubMed] [Google Scholar]
- 5.Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–66. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 6.Boyd-Kimball D, Mohmmad Abdul H, Reed T, Sultana R, Butterfield DA. Role of phenylalanine 20 in Alzheimer’s amyloid beta-peptide (1–42)-induced oxidative stress and neurotoxicity. Chem Res Toxicol. 2004;17:1743–9. doi: 10.1021/tx049796w. [DOI] [PubMed] [Google Scholar]
- 7.Boyd-Kimball D, Sultana R, Poon HF, Lynn BC, Casamenti F, Pepeu G, Klein JB, Butterfield DA. Proteomic identification of proteins specifically oxidized by intracerebral injection of amyloid beta-peptide (1–42) into rat brain: implications for Alzheimer’s disease. Neuroscience. 2005;132:313–24. doi: 10.1016/j.neuroscience.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 8.Butterfield DA, Boyd-Kimball D. The critical role of methionine 35 in Alzheimer’s amyloid beta-peptide (1–42)-induced oxidative stress and neurotoxicity. Biochim Biophys Acta. 2005;1703:149–56. doi: 10.1016/j.bbapap.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 9.Butterfield DA, Galvan V, Lange MB, Tang H, Sowell RA, Spilman P, Fombonne J, Gorostiza O, Zhang J, Sultana R, Bredesen DE. In vivo oxidative stress in brain of Alzheimer disease transgenic mice: Requirement for methionine 35 in amyloid beta-peptide of APP. Free Radic Biol Med. 2010 doi: 10.1016/j.freeradbiomed.2009.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Markesbery WR. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic Biol Med. 1997;23:134–47. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- 11.Fukuda M, Kanou F, Shimada N, Sawabe M, Saito Y, Murayama S, Hashimoto M, Maruyama N, Ishigami A. Elevated levels of 4-hydroxynonenal-histidine Michael adduct in the hippocampi of patients with Alzheimer’s disease. Biomed Res. 2009;30:227–33. doi: 10.2220/biomedres.30.227. [DOI] [PubMed] [Google Scholar]
- 12.Markesbery WR, Lovell MA. 4-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer’s disease. Neurobiol Aging. 1998;19:33–6. doi: 10.1016/s0197-4580(98)00009-8. [DOI] [PubMed] [Google Scholar]
- 13.Mecocci P, Polidori MC, Cherubini A, Ingegni T, Mattioli P, Catani M, Rinaldi P, Cecchetti R, Stahl W, Senin U, Beal MF. Lymphocyte oxidative DNA damage and plasma antioxidants in Alzheimer disease. Arch Neurol. 2002;59:794–8. doi: 10.1001/archneur.59.5.794. [DOI] [PubMed] [Google Scholar]
- 14.Montine KS, Olson SJ, Amarnath V, Whetsell WO, Jr, Graham DG, Montine TJ. Immunohistochemical detection of 4-hydroxy-2-nonenal adducts in Alzheimer’s disease is associated with inheritance of APOE4. Am J Pathol. 1997;150:437–43. [PMC free article] [PubMed] [Google Scholar]
- 15.Pratico D, Sung S. Lipid peroxidation and oxidative imbalance: early functional events in Alzheimer’s disease. J Alzheimers Dis. 2004;6:171–5. doi: 10.3233/jad-2004-6209. [DOI] [PubMed] [Google Scholar]
- 16.Volkel W, Sicilia T, Pahler A, Gsell W, Tatschner T, Jellinger K, Leblhuber F, Riederer P, Lutz WK, Gotz ME. Increased brain levels of 4-hydroxy-2-nonenal glutathione conjugates in severe Alzheimer’s disease. Neurochem Int. 2006;48:679–86. doi: 10.1016/j.neuint.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 17.Hensley K, Maidt ML, Yu Z, Sang H, Markesbery WR, Floyd RA. Electrochemical analysis of protein nitrotyrosine and dityrosine in the Alzheimer brain indicates region-specific accumulation. J Neurosci. 1998;18:8126–32. doi: 10.1523/JNEUROSCI.18-20-08126.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lauderback CM, Hackett JM, Huang FF, Keller JN, Szweda LI, Markesbery WR, Butterfield DA. The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer’s disease brain: the role of Abeta1–42. J Neurochem. 2001;78:413–6. doi: 10.1046/j.1471-4159.2001.00451.x. [DOI] [PubMed] [Google Scholar]
- 19.Moreira PI, Nunomura A, Nakamura M, Takeda A, Shenk JC, Aliev G, Smith MA, Perry G. Nucleic acid oxidation in Alzheimer disease. Free Radic Biol Med. 2008;44:1493–505. doi: 10.1016/j.freeradbiomed.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 20.Nunomura A, Tamaoki T, Tanaka K, Motohashi N, Nakamura M, Hayashi T, Yamaguchi H, Shimohama S, Lee HG, Zhu X, Smith MA, Perry G. Intraneuronal amyloid beta accumulation and oxidative damage to nucleic acids in Alzheimer disease. Neurobiol Dis. 2010;37:731–7. doi: 10.1016/j.nbd.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, Xu HW, Stern D, McKhann G, Yan SD. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. Faseb J. 2005;19:2040–1. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- 22.Reddy PH. Amyloid beta, mitochondrial structural and functional dynamics in Alzheimer’s disease. Exp Neurol. 2009;218:286–92. doi: 10.1016/j.expneurol.2009.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sultana R, Butterfield DA. Oxidatively modified, mitochondria-relevant brain proteins in subjects with Alzheimer disease and mild cognitive impairment. J Bioenerg Biomembr. 2009;41:441–6. doi: 10.1007/s10863-009-9241-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ankarcrona M, Mangialasche F, Winblad B. Rethinking Alzheimer’s disease therapy: are mitochondria the key? J Alzheimers Dis. 2010;20(Suppl 2):S579–90. doi: 10.3233/JAD-2010-100327. [DOI] [PubMed] [Google Scholar]
- 25.Sultana R, Mecocci P, Mangialasche F, Cecchetti R, Baglioni M, Butterfield DA. Increased protein and lipid oxidative damage in mitochondria isolated from lymphocytes from patients with Alzheimer’s disease: insights into the role of oxidative stress in Alzheimer’s disease and initial investigations into a potential biomarker for this dementing disorder. J Alzheimers Dis. 2011;24:77–84. doi: 10.3233/JAD-2011-101425. [DOI] [PubMed] [Google Scholar]
- 26.Mangialasche F, Xu W, Kivipelto M, Costanzi E, Ercolani S, Pigliautile M, Cecchetti R, Baglioni M, Simmons A, Soininen H, Tsolaki M, Kloszewska I, Vellas B, Lovestone S, Mecocci P. Tocopherols and tocotrienols plasma levels are associated with cognitive impairment. Neurobiol Aging. 2012;33:2282–90. doi: 10.1016/j.neurobiolaging.2011.11.019. [DOI] [PubMed] [Google Scholar]
- 27.Pallotti F, Lenaz G. Isolation and subfractionation of mitochondria from animal cells and tissue culture lines. Methods Cell Biol. 2007;80:3–44. doi: 10.1016/S0091-679X(06)80001-4. [DOI] [PubMed] [Google Scholar]
- 28.Sultana R, Ravagna A, Mohmmad-Abdul H, Calabrese V, Butterfield DA. Ferulic acid ethyl ester protects neurons against amyloid beta-peptide(1–42)-induced oxidative stress and neurotoxicity: relationship to antioxidant activity. J Neurochem. 2005;92:749–58. doi: 10.1111/j.1471-4159.2004.02899.x. [DOI] [PubMed] [Google Scholar]
- 29.Aksenov MY, Aksenova MV, Butterfield DA, Geddes JW, Markesbery WR. Protein oxidation in the brain in Alzheimer’s disease. Neuroscience. 2001;103:373–83. doi: 10.1016/s0306-4522(00)00580-7. [DOI] [PubMed] [Google Scholar]
- 30.Butterfield DA, Reed TT, Perluigi M, De Marco C, Coccia R, Keller JN, Markesbery WR, Sultana R. Elevated levels of 3-nitrotyrosine in brain from subjects with amnestic mild cognitive impairment: implications for the role of nitration in the progression of Alzheimer’s disease. Brain Res. 2007;1148:243–8. doi: 10.1016/j.brainres.2007.02.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sultana R, Boyd-Kimball D, Cai J, Pierce WM, Klein JB, Merchant M, Butterfield DA. Proteomics analysis of the Alzheimer’s disease hippocampal proteome. J Alzheimers Dis. 2007;11:153–64. doi: 10.3233/jad-2007-11203. [DOI] [PubMed] [Google Scholar]
- 32.Butterfield DA, Stadtman ER. Protein Oxidation processes in aging brain. Adv Cell Aging Gerontol. 1997;2:161–91. [Google Scholar]
- 33.Butterfield DA, Reed T, Perluigi M, De Marco C, Coccia R, Cini C, Sultana R. Elevated protein-bound levels of the lipid peroxidation product, 4-hydroxy-2-nonenal, in brain from persons with mild cognitive impairment. Neurosci Lett. 2006;397:170–3. doi: 10.1016/j.neulet.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 34.Williamson KS, Gabbita SP, Mou S, West M, Pye QN, Markesbery WR, Cooney RV, Grammas P, Reimann-Philipp U, Floyd RA, Hensley K. The nitration product 5-nitro-gamma-tocopherol is increased in the Alzheimer brain. Nitric Oxide. 2002;6:221–7. doi: 10.1006/niox.2001.0399. [DOI] [PubMed] [Google Scholar]
- 35.Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006;15:1437–49. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- 36.Butterfield DA, Sultana R. Methionine-35 of abeta(1–42): importance for oxidative stress in Alzheimer disease. J Amino Acids. 2011;2011:198430. doi: 10.4061/2011/198430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Butterfield DA, Bader Lange ML, Sultana R. Involvements of the lipid peroxidation product, HNE, in the pathogenesis and progression of Alzheimer’s disease. Biochim Biophys Acta. 2010;1801:924–9. doi: 10.1016/j.bbalip.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jimenez-Jimenez FJ, Molina JA, de Bustos F, Orti-Pareja M, Benito-Leon J, Tallon-Barranco A, Gasalla T, Porta J, Arenas J. Serum levels of beta-carotene, alpha-carotene and vitamin A in patients with Alzheimer’s disease. Eur J Neurol. 1999;6:495–7. doi: 10.1046/j.1468-1331.1999.640495.x. [DOI] [PubMed] [Google Scholar]
- 39.Zaman Z, Roche S, Fielden P, Frost PG, Niriella DC, Cayley AC. Plasma concentrations of vitamins A and E and carotenoids in Alzheimer’s disease. Age Ageing. 1992;21:91–4. doi: 10.1093/ageing/21.2.91. [DOI] [PubMed] [Google Scholar]
- 40.Parks E, Traber MG. Mechanisms of vitamin E regulation: research over the past decade and focus on the future. Antioxid Redox Signal. 2000;2:405–12. doi: 10.1089/15230860050192189. [DOI] [PubMed] [Google Scholar]
- 41.Sung S, Yao Y, Uryu K, Yang H, Lee VM, Trojanowski JQ, Pratico D. Early vitamin E supplementation in young but not aged mice reduces Abeta levels and amyloid deposition in a transgenic model of Alzheimer’s disease. Faseb J. 2004;18:323–5. doi: 10.1096/fj.03-0961fje. [DOI] [PubMed] [Google Scholar]
- 42.Farina N, Isaac MG, Clark AR, Rusted J, Tabet N. Vitamin E for Alzheimer’s dementia and mild cognitive impairment. Cochrane Database Syst Rev. 2012;11:CD002854. doi: 10.1002/14651858.CD002854.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hensley K, Barnes LL, Christov A, Tangney C, Honer WG, Schneider JA, Bennett DA, Morris MC. Analysis of postmortem ventricular cerebrospinal fluid from patients with and without dementia indicates association of vitamin E with neuritic plaques and specific measures of cognitive performance. J Alzheimers Dis. 2011;24:767–74. doi: 10.3233/JAD-2011-101995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Perkins AJ, Hendrie HC, Callahan CM, Gao S, Unverzagt FW, Xu Y, Hall KS, Hui SL. Association of antioxidants with memory in a multiethnic elderly sample using the Third National Health and Nutrition Examination Survey. Am J Epidemiol. 1999;150:37–44. doi: 10.1093/oxfordjournals.aje.a009915. [DOI] [PubMed] [Google Scholar]
- 45.Rinaldi P, Polidori MC, Metastasio A, Mariani E, Mattioli P, Cherubini A, Catani M, Cecchetti R, Senin U, Mecocci P. Plasma antioxidants are similarly depleted in mild cognitive impairment and in Alzheimer’s disease. Neurobiol Aging. 2003;24:915–9. doi: 10.1016/s0197-4580(03)00031-9. [DOI] [PubMed] [Google Scholar]
- 46.Sano M, Ernesto C, Thomas RG, Klauber MR, Schafer K, Grundman M, Woodbury P, Growdon J, Cotman CW, Pfeiffer E, Schneider LS, Thal LJ. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. The Alzheimer’s Disease Cooperative Study. N Engl J Med. 1997;336:1216–22. doi: 10.1056/NEJM199704243361704. [DOI] [PubMed] [Google Scholar]
- 47.Galasko DR, Peskind E, Clark CM, Quinn JF, Ringman JM, Jicha GA, Cotman C, Cottrell B, Montine TJ, Thomas RG, Aisen P. Antioxidants for Alzheimer disease: a randomized clinical trial with cerebrospinal fluid biomarker measures. Arch Neurol. 2012;69:836–41. doi: 10.1001/archneurol.2012.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Suzuki YJ, Tsuchiya M, Wassall SR, Choo YM, Govil G, Kagan VE, Packer L. Structural and dynamic membrane properties of alpha-tocopherol and alpha-tocotrienol: implication to the molecular mechanism of their antioxidant potency. Biochemistry. 1993;32:10692–9. doi: 10.1021/bi00091a020. [DOI] [PubMed] [Google Scholar]
- 49.Christen S, Woodall AA, Shigenaga MK, Southwell-Keely PT, Duncan MW, Ames BN. gamma-tocopherol traps mutagenic electrophiles such as NO(X) and complements alpha-tocopherol: physiological implications. Proc Natl Acad Sci U S A. 1997;94:3217–22. doi: 10.1073/pnas.94.7.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Niki E. Tocopherylquinone and tocopherylhydroquinone. Redox Rep. 2007;12:204–10. doi: 10.1179/135100007X200353. [DOI] [PubMed] [Google Scholar]
- 51.Castegna A, Thongboonkerd V, Klein JB, Lynn B, Markesbery WR, Butterfield DA. Proteomic identification of nitrated proteins in Alzheimer’s disease brain. J Neurochem. 2003;85:1394–401. doi: 10.1046/j.1471-4159.2003.01786.x. [DOI] [PubMed] [Google Scholar]
- 52.Castegna A, Aksenov M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part II: dihydropyrimidinase-related protein 2, alpha-enolase and heat shock cognate 71. J Neurochem. 2002;82:1524–32. doi: 10.1046/j.1471-4159.2002.01103.x. [DOI] [PubMed] [Google Scholar]
- 53.Castegna A, Aksenov M, Aksenova M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part I: creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radic Biol Med. 2002;33:562–71. doi: 10.1016/s0891-5849(02)00914-0. [DOI] [PubMed] [Google Scholar]
- 54.Choi J, Levey AI, Weintraub ST, Rees HD, Gearing M, Chin LS, Li L. Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson’s and Alzheimer’s diseases. J Biol Chem. 2004;279:13256–64. doi: 10.1074/jbc.M314124200. [DOI] [PubMed] [Google Scholar]
- 55.Sultana R, Poon HF, Cai J, Pierce WM, Merchant M, Klein JB, Markesbery WR, Butterfield DA. Identification of nitrated proteins in Alzheimer’s disease brain using a redox proteomics approach. Neurobiol Dis. 2006;22:76–87. doi: 10.1016/j.nbd.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 56.Sultana R, Butterfield DA. Role of oxidative stress in the progression of Alzheimer’s disease. J Alzheimers Dis. 2010;19:341–53. doi: 10.3233/JAD-2010-1222. [DOI] [PubMed] [Google Scholar]
- 57.Butterfield DA, Hardas SS, Lange ML. Oxidatively modified glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and Alzheimer’s disease: many pathways to neurodegeneration. J Alzheimers Dis. 2010;20:369–93. doi: 10.3233/JAD-2010-1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Takano M, Yamashita T, Nagano K, Otani M, Maekura K, Kamada H, Tsunoda S, Tsutsumi Y, Tomiyama T, Mori H, Matsuura K, Matsuyama S. Proteomic analysis of the hippocampus in Alzheimer’s disease model mice by using two-dimensional fluorescence difference in gel electrophoresis. Neurosci Lett. 2013;534:85–9. doi: 10.1016/j.neulet.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 59.Tang HL, Lung HL, Wu KC, Le AH, Tang HM, Fung MC. Vimentin supports mitochondrial morphology and organization. Biochem J. 2008;410:141–6. doi: 10.1042/BJ20071072. [DOI] [PubMed] [Google Scholar]
- 60.Fujii J, Ikeda Y. Advances in our understanding of peroxiredoxin, a multifunctional, mammalian redox protein. Redox Rep. 2002;7:123–30. doi: 10.1179/135100002125000352. [DOI] [PubMed] [Google Scholar]
- 61.Kil IS, Lee SK, Ryu KW, Woo HA, Hu MC, Bae SH, Rhee SG. Feedback control of adrenal steroidogenesis via H2O2-dependent, reversible inactivation of peroxiredoxin III in mitochondria. Mol Cell. 2012;46:584–94. doi: 10.1016/j.molcel.2012.05.030. [DOI] [PubMed] [Google Scholar]
- 62.Drechsel DA, Patel M. Respiration-dependent H2O2 removal in brain mitochondria via the thioredoxin/peroxiredoxin system. J Biol Chem. 2010;285:27850–8. doi: 10.1074/jbc.M110.101196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lopert P, Day BJ, Patel M. Thioredoxin reductase deficiency potentiates oxidative stress, mitochondrial dysfunction and cell death in dopaminergic cells. PLoS One. 2012;7:e50683. doi: 10.1371/journal.pone.0050683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim SH, Fountoulakis M, Cairns N, Lubec G. Protein levels of human peroxiredoxin subtypes in brains of patients with Alzheimer’s disease and Down syndrome. J Neural Transm Suppl. 2001:223–35. doi: 10.1007/978-3-7091-6262-0_18. [DOI] [PubMed] [Google Scholar]
- 65.Chang TS, Cho CS, Park S, Yu S, Kang SW, Rhee SG. Peroxiredoxin III, a mitochondrion-specific peroxidase, regulates apoptotic signaling by mitochondria. J Biol Chem. 2004;279:41975–84. doi: 10.1074/jbc.M407707200. [DOI] [PubMed] [Google Scholar]
- 66.Calabrese V, Sultana R, Scapagnini G, Guagliano E, Sapienza M, Bella R, Kanski J, Pennisi G, Mancuso C, Stella AM, Butterfield DA. Nitrosative stress, cellular stress response, and thiol homeostasis in patients with Alzheimer’s disease. Antioxid Redox Signal. 2006;8:1975–86. doi: 10.1089/ars.2006.8.1975. [DOI] [PubMed] [Google Scholar]