

Abstract
Peripheral arterial occlusive disease (PAOD) results from atherosclerosis of large and medium peripheral arteries, as well as the aorta, and has many risk factors, including smoking, diabetes, hypertension, and hyperlipidemia. PAOD often coexists with coronary artery disease and cerebrovascular disease. Cross-matching a population-based list of Icelandic patients with PAOD who had undergone angiography and/or revascularization procedures with a genealogy database of the entire Icelandic nation defined 116 extended families containing 272 patients. A genomewide scan with microsatellite markers revealed significant linkage to chromosome 1p31 with an allele-sharing LOD score of 3.93 (P=1.04×10-5). We designate this locus as “PAOD1.” Subtracting 35 patients with a history of stroke increased the LOD score to 4.93. This suggests that, although PAOD and other vascular diseases share risk factors, genetic factors specific to subtypes of vascular disease may exist.
Introduction
Atherosclerosis is the pathology underlying several of humankind's most lethal diseases, such as myocardial infarction (MI) and stroke. PAOD shares the risk factors of those atherosclerotic diseases, especially smoking, diabetes, hypertension, and hyperlipidemia (Dormandy et al. 1999; Hooi et al. 1999). However, PAOD represents atherosclerosis of the large and medium arteries of the limbs, particularly to the lower extremities, and includes the aorta and iliac arteries. It often coexists with coronary artery disease and cerebrovascular disease. Clinically significant lesions may gradually narrow the peripheral arteries, leading to pain, on walking, that is usually relieved by rest (claudication); ischemic ulcers; gangrene; and, sometimes, limb amputation. Medical therapy is generally ineffective, but operations bypassing or replacing the lesion with artificial or venous grafts improve blood flow distally, at least until the grafts become restenosed (Haustein 1997).
Here we present data on the mapping of a locus for PAOD. This is the first example of linkage to a specific chromosomal location reported for this disease. For this study, we recruited patients with PAOD whose diagnosis had been angiographically and/or surgically confirmed. This phenotype represents the 20% of the patients with PAOD who have the most severe disease based on epidemiological studies in other populations (Dormandy et al. 1999).
Subjects and Methods
Population and Genealogy
This study was approved by the Data Protection Commission of Iceland and the National Bioethics Committee of Iceland. Informed consent was obtained from all patients and their relatives whose DNA samples were used in the linkage scan (see the deCODE Genetics Web site for an example of the informed consent form). The original population-based lists of patients with PAOD covering the years 1980–1995 were derived from the two major hospitals in Iceland. All patients had undergone angiography, and 85% had angioplasty and/or vascular surgery. Those undergoing vascular procedures for trauma were excluded.
A comprehensive genealogy database established at deCODE Genetics was used to cluster the patients in pedigrees (Gulcher and Stefansson 1998). Each version of the computerized genealogy database is reversibly encrypted by the Data Protection Commission of Iceland before arriving at the laboratory (Gulcher et al. 2000). The database uses a patient list, with encrypted personal identifiers, as input and uses recursive algorithms to find all ancestors in the database who are related to any member on the input list, within a given number of generations. The cluster function then searches for ancestors who are common to any two or more members of the input list.
Microsatellite Markers and Genotyping
The marker order and positions for the framework mapping set were obtained from the Marshfield genetic map, except for a three-marker putative inversion on chromosome 8 (Jonsdottir et al. 2000; Yu et al. 2000; Giglio et al. 2001). The DNA samples were genotyped using nearly 1,000 fluorescently labeled primers. We have developed a microsatellite screening set based in part on the ABI Linkage Marker (version 2) screening set and the ABI Linkage Marker (version 2) intercalating set in combination with >500 custom-made markers. All markers were extensively tested for robustness, ease of scoring, and efficiency in 4× multiplex PCRs. The PCR amplifications were set up, run, and pooled on Perkin Elmer/Applied Biosystems 877 Integrated Catalyst Thermocyclers, with a similar protocol for each marker. The reaction volume used was 5 μl, and, for each PCR, 20 ng of genomic DNA was amplified in the presence of 2 pmol of each primer, 0.25 U AmpliTaq Gold, 0.2 mM dNTPs, and 2.5 mM MgCl2 (buffer was supplied by manufacturer). The PCR conditions used were 95°C for 10 min, then 37 cycles of 94°C for 15 s, 55°C for 30 s, and 72°C for 1 min. The PCR products were supplemented with the internal size standard, and the pools were separated and detected on Applied Biosystems model 377 Sequencer, by use of Genescan version 3.0 peak-calling software. Alleles were called automatically with the TrueAllele program (see Cybergenetics Web site), and the program DecodeGT was used to fractionate according to quality and to edit the called genotypes (Palsson et al. 1999). At least 180 Icelandic control subjects were genotyped to derive allelic frequencies.
Statistical Methods for Linkage Analysis
In our analyses, we used multipoint, affected-only allele-sharing methods to assess the evidence for linkage. All relatives who did not have an angiogram or surgery for PAOD were considered to have “unknown” status. All results were obtained using the program Allegro (Gudbjartsson et al. 2000). We used the Spairs scoring function (Whittemore and Halpern 1994; Kruglyak et al. 1996) and the exponential allele-sharing model (Kong and Cox 1997) to generate the relevant statistics with 1 df. When combining the family scores to obtain an overall score, instead of weighting the families equally, which is the default method of Genehunter (Kruglyak et al. 1996), or weighting the affected pairs equally, we used a weighting scheme that is halfway between the two on the log scale; our family weights are the geometric means of the weights of the two schemes. Although not identical, this weighting scheme tends to give results similar to those of the scheme proposed by Weeks and Lange (1988) as an extension of a weighting scheme of Hodge (1984) designed for sibships. We computed the P value two different ways and report the less significant one. The first P value was computed based on large-sample theory; 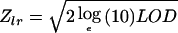 is approximately distributed as a standard normal random variable under the null hypothesis of no linkage (Kong and Cox 1997). Because of the concern with small-sample behavior, we computed a second P value by comparing the observed LOD score to its complete data-sampling distribution under the null hypothesis (Gudbjartsson et al. 2000). When a data set consists of more than a few families, as is the case here, these two P values tend to be very similar. To ensure that the result was a true reflection of the information contained in the material, for us to consider a linkage result significant, it was required not only that the P value be <2×10-5 (Lander and Kruglyak 1995) but also that the information content in the region was ⩾85%. For the families in this study, an information content of 85% corresponded to a marker density of approximately one marker per centimorgan. The information measure we used has been defined elsewhere (Nicolae 1999) and has been implemented in Allegro. This measure is closely related to a classical measure of information (Dempster et al. 1977), having the property that it is between 0, if the marker genotypes are completely uninformative, and 1, if the genotypes determine the exact amount of allele sharing by descent among the affected relatives.
is approximately distributed as a standard normal random variable under the null hypothesis of no linkage (Kong and Cox 1997). Because of the concern with small-sample behavior, we computed a second P value by comparing the observed LOD score to its complete data-sampling distribution under the null hypothesis (Gudbjartsson et al. 2000). When a data set consists of more than a few families, as is the case here, these two P values tend to be very similar. To ensure that the result was a true reflection of the information contained in the material, for us to consider a linkage result significant, it was required not only that the P value be <2×10-5 (Lander and Kruglyak 1995) but also that the information content in the region was ⩾85%. For the families in this study, an information content of 85% corresponded to a marker density of approximately one marker per centimorgan. The information measure we used has been defined elsewhere (Nicolae 1999) and has been implemented in Allegro. This measure is closely related to a classical measure of information (Dempster et al. 1977), having the property that it is between 0, if the marker genotypes are completely uninformative, and 1, if the genotypes determine the exact amount of allele sharing by descent among the affected relatives.
After obtaining a significant allele-sharing LOD score, in an attempt to understand the contribution of this susceptibility locus, we fitted a range of parametric models to the data. Even when fitting parametric models, we performed affected-only analyses, in the sense that an individual is classified either as affected or as having unknown disease status. As a consequence, only ratios of penetrances are relevant. We fitted a range of single-locus dominant, additive, and multiplicative models (Risch 1990). With a complex disease such as PAOD, none of these simple models are likely to be exactly true and the effect of a gene and its variants can only be reliably determined after the at-risk variant, or variants, are identified. However, by calculating the corresponding contribution to the sibling recurrence-risk ratio, the fitted parametric models do provide some rough idea as to how much the gene is contributing to the familial clustering of the disease.
To assess whether the increase in LOD score resulting from subtracting the 35 patients with PAOD who also had stroke would be likely to occur by chance, we selected 1,000 random sets of 35 patients whose status we then changed to “unknown” in an analysis. The P value we present is the fraction of the 1,000 simulations that produced a LOD score increase at the peak locus equal to or greater than that which we observed by changing the affection status of the stroke patients to “unknown.” We used the same method to assess subtraction of the 38 patients with PAOD who also had MI. There, however, we assess the significance of the decrease in LOD score that results by looking at the fraction of the simulations that produced a LOD-score decrease greater or equal to that which we observed by changing the affection status of the MI patients to “unknown.”
Physical and Genetic Mapping
For the locus region, a combination of data from coincident hybridizations of BAC membranes by use of a high density of STSs, and the Finger Printing Contig (FPC) database was used to build large contigs of BACs from the RPCI-11 library provided by Pieter deJong (Children's Hospital Oakland Research Institute). BAC contigs were generated by a method that combines coincident hybridization, versus the RPCI-11 BAC library, with data mining and the FPC program (see Wellcome Trust Sanger Institute Web site). Hybridizations were performed using primers from markers in the region of interest according to their location in the Weizmann Institute Unified Database. To close the gaps, new markers were generated from BAC end sequences.
High-resolution genetic mapping was used (1) to anchor and place in order contigs found by physical mapping and (2) to obtain accurate intermarker distances for the correctly ordered markers (see fig. 1 for marker orders and distances used). Data from 112 Icelandic nuclear families (sibships with their parents, containing two to seven siblings) were analyzed together with the nuclear families available within the PAOD pedigrees. For the purpose of genetic mapping, the 112 nuclear families alone provided 588 meioses, and the total number of meioses available for mapping was >1,000. By comparison, the Marshfield genetic map was constructed on the basis of 182 meioses. The large number of meiotic events within our families provides the ability to map markers to a resolution of 1.0 cM or better. Using a modification of the Allegro program, we then applied the expectation-maximization algorithm (Dempster et al. 1977) to our data to estimate the genetic distances between markers. Combining this information with the physical map resulted in a highly reliable order of markers and better estimates of intermarker distances within the peak region, both of which are important for an accurate linkage analysis (Halpern and Whittemore 1999; Daw et al. 2000).
Figure 1.

Comparison of marker order and genetic distances. Arrows indicate flips in marker order between the two maps. Asterisks (*) denote two markers flipped in the UCSC December 2000 freeze (corrected in the UCSC April 2001 freeze).
Results
We used our genealogy database together with a population-based list of 1,745 patients who had undergone angiography and/or surgery for PAOD to define families of patients with PAOD. Our study was focused on patients who were related to other patients by six or fewer meiotic events. This article reports the results of our linkage scan on a total of 272 patients and 612 relatives within 116 families. Figure 2 exhibits two of the larger families used in the analysis, as well as an example of a family in which a patient with PAOD is related to another patient with PAOD who also had stroke. Our genome-scan results are displayed in figure 3. The most prominent linkage was found to chromosome 1p with a LOD score of 2.95, although other LOD scores >1.5 also occur on 13q and 18q. Additional microsatellite markers were genotyped and mapped for the chromosome 1p peak region. When our map was used, the multipoint analysis increased the LOD score on chromosome 1 to 3.93 (P=1.04×10-5) (fig. 4). In contrast, when the Marshfield map was used, the LOD score was 2.83. The peak is centered on marker D1S2895, with markers D1S411 and D1S2855, telomeric and centromeric, respectively, defining a drop of 1 in LOD score from the peak. We designate this locus as “PAOD1.”
Figure 2.

Three families used in the linkage study of PAOD. Two of the families, A and B, have positive LOD scores at the chromosome 1p31 locus, whereas the cousin pair in family C contributes negatively. Sex indicators have been shuffled for some individuals in the top two generations, and unaffected siblings of patients are not shown, to protect privacy. The blackened squares and circles represent men and women, respectively, who are affected with PAOD. The checkered square represents a patient with PAOD who also had stroke. The slashed symbols represent deceased individuals.
Figure 3.

Full genome scan of all affected family members, by use of nearly 1,000 microsatellite markers. The multipoint LOD score is on the Y-axis, and distance (in centimorgans) from the p-terminus of the chromosome is on the X-axis.
Figure 4.

Multipoint allele-sharing LOD score of chromosome 1 with extra microsatellite markers within PAOD1. The solid line represents the results of all 272 patients with PAOD. The dashed line represents the results of defining affecteds as PAOD without stroke.
Our genetic map for 19 markers in the peak region, along with the publicly available Marshfield genetic map, is displayed in figure 1. The Marshfield map differs from our map both in the order of a number of markers, for which Marshfield has no resolution, and in the genetic distance between markers. At the time of our submission, the December 2000 freeze of the University of California Santa Cruz (UCSC) draft assembly of the human DNA sequence was available. Our order of markers agreed with the public sequence, except for the two markers that are indicated in figure 1. The April 2001 freeze (released June 2001) corrected the order of these two markers but changed the orders of two other pairs (not shown in figure 1). The August 2001 freeze agrees with the order we have.
Some demographic information and the fraction of patients with several risk factors are given in table 1, for the entire patient set. For the risk factors, we have also displayed the fractions for the set of patients in families with nonparametric linkage (NPL) scores ⩾1 (the patients in these families show an excess of sharing genetic material identical by descent over what would be expected simply because of their relationship). There does not appear to be any substantial shifting in the pattern of risk factors for the patients from these families. This suggests that this PAOD locus is not a locus for these risk factors.
Table 1.
Risk Factors and Demographics of Patients in the Study
|
% of Patient Group |
||
| Risk Factora | All (n=272)b | with NPL >1 (n=75) |
| Hyperlipidemia | 33.8 | 39.2 |
| Hypertension | 51.8 | 48.6 |
| Diabetes | 9.9 | 9.5 |
| Smokingc | 77.6 | 85 |
Risk factors were self reported.
Of the 272 patients, 169 (62%) were male and 103 (38%) were female. Mean age was 70.8 years.
Including both current and past smoking.
PAOD is often associated with other forms of atherosclerosis, such as cerebrovascular disease and coronary artery disease, and many of the patients we studied also had a history of stroke and/or MI. When we reanalyzed our data—after defining those 35 patients with PAOD (13% of the total) who also had history of stroke as having unknown disease status, instead of affected status—the LOD score on chromosome 1 increased to 4.93. This increase in the LOD score due to subtraction of the stroke patients is statistically significant (P=.019) (fig. 4). When we subtracted the 38 patients with PAOD who also had MI (14%; designating their affection status as “unknown”), the LOD score decreased to 2.95. However, this decrease was not statistically significant (P=.577) and may simply reflect the reduction of the material.
In an attempt to understand the contribution of this locus to the population, we tried fitting a variety of parametric models, dominant, additive, or multiplicative. For each of these models, we were able to achieve a LOD score >4.0 at this locus. For example, a dominant model assuming that the at-risk allele has a frequency of 14% and the penetrance of a noncarrier is 0 gives a LOD score of 4.26. For this dominant model, 47 of the 116 families give a positive LOD score; of these, 38 families have LOD scores >0.1, and three families have LOD scores between 0.4 and 0.7. The three families, A, B, and C, in figure 2 gave LOD scores of 0.68, 0.62, and −0.12 respectively, for this model. Note that, even for a family with a negative LOD score, some or all of the patients can carry the at-risk allele since they could have inherited it from different sources. A multiplicative model assuming that the at-risk allele has a frequency of 20% and that there is a seven-fold increase in risk for every at-risk allele carried gives a LOD score of 4.23. These dominant and multiplicative models correspond to sibling recurrence-risk ratios of 2.37 and 2.54.
Discussion
In this study of PAOD, we defined the phenotype as surgically corrected and/or angiographically documented PAOD, a definition that has many advantages. This decreases the subjectivity sometimes encountered when defining PAOD based on the patient's history of intermittent claudication. This also eliminates the need of relying on the ankle-brachial blood-pressure ratio, which may vary from observer to observer (Jeelani et al. 2000). Since only ∼20%–25% of patients with PAOD ever go on to have angiography and surgery, this group represents those who are more severely affected by the disease.
The lack of prominent linkage to PAOD1 in our stroke linkage study (in this issue) suggests that a gene at this PAOD locus may confer a specific predisposition to PAOD, rather than to stroke or to PAOD in those patients with stroke. This increase in LOD score with the removal of stroke patients suggests that there are other strong genetic and/or environmental factors that contribute to both PAOD and stroke. A more accurate understanding of the contribution of PAOD1 to PAOD will have to await its isolation.
Although some have predicted that examining the consequences of atherosclerosis, as we did in this study, rather than its underlying risk factors, would not yield sufficient power to detect genes predisposing individuals to this multifactorial common disease, we have shown that the genealogic approach can indeed yield significant linkage. This study suggests that there is both overlap and independence between genetic factors for the major manifestations of atherosclerosis, specifically PAOD and stroke. It further suggests that, at least in Iceland, there exist genes with major influence on one's risk of PAOD. Because no correlation was observed between patients contributing to the LOD score and vascular risk factors such as hyperlipidemia, hypertension, and diabetes, PAOD1 is unlikely to be simply a hypertension, diabetes, or hyperlipidemia locus. This suggests that additional genetic factors may affect the pathogenesis of atherosclerosis—either directly, in the arterial wall where it takes place, or indirectly, through mediators in the blood, such as mononuclear cells, platelets, or coagulation factors.
Acknowledgments
We thank the patients and their families whose contributions have made this study possible. We thank the nurses of Noatun Genetic Research Service Center and the deCODE genotyping facility for their excellent assistance.
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- Center for Medical Genetics, http://research.marshfieldclinic.org/genetics/ (for marker order and positions for the framework mapping set)
- Cybergenetics, http://www.cybgen.com/ (for TrueAllele program)
- deCODE Genetics, http://www.decode.com/ (for sample of informed consent form)
- Weizmann Institute Unified Database for Human Genome Mapping, http://bioinformatics.weizmann.ac.il/udb/ (for marker locations)
- Wellcome Trust Sanger Institute, http://www.sanger.ac.uk/ (for FPC program)
References
- Daw EW, Thompson EA, Wijsman EM (2000) Bias in multipoint linkage analysis arising from map misspecification. Genet Epidemiol 19:366–380 [DOI] [PubMed] [Google Scholar]
- Dempster AP, Laird NM, Rubin DB (1977) Maximum likelihood from incomplete data via the EM algorithm (with discussion). J R Stat Soc B 39:1–38 [Google Scholar]
- Dormandy J, Heeck L, Vig S (1999) The natural history of claudication: risk to life and limb. Semin Vasc Surg 12:123–137 [PubMed] [Google Scholar]
- Giglio S, Broman KW, Matsumoto N, Calvari V, Gimelli G, Neumann T, Ohashi H, Voullaire L, Larizza D, Giorda R, Weber JL, Ledbetter DH, Zuffardi O (2001) Olfactory receptor-gene clusters, genomic-inversion polymorphisms, and common chromosome rearrangements. Am J Hum Genet 68:874–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudbjartsson DF, Jonasson K, Frigge ML, Kong A (2000) Allegro, a new computer program for multipoint linkage analysis. Nat Genet 25:12–13 [DOI] [PubMed] [Google Scholar]
- Gulcher JR, Kristjansson K, Gudbjartsson H, Stefansson K (2000) Protection of privacy by third-party encryption in genetic research in Iceland. Eur J Hum Genet 8:739–742 [DOI] [PubMed] [Google Scholar]
- Gulcher J, Stefansson K (1998) Population genomics: laying the groundwork for genetic disease modeling and targeting. Clin Chem Lab Med 36:523–527 [DOI] [PubMed] [Google Scholar]
- Halpern J, Whittemore AS (1999) Multipoint linkage analysis: a cautionary note. Hum Hered 49:194–196 [DOI] [PubMed] [Google Scholar]
- Haustein KO (1997) State of the art treatment of peripheral occlusive arterial disease (POAD) with drugs vs. vascular reconstruction or amputation. Int J Clin Pharmacol Ther 35:266–274 [PubMed] [Google Scholar]
- Hodge SE (1984) The information contained in multiple sibling pairs. Genet Epidemiol 1:109–122 [DOI] [PubMed] [Google Scholar]
- Hooi JD, Stoffers HE, Knottnerus JA, van Ree JW (1999) The prognosis of non-critical limb ischaemia: a systematic review of population-based evidence. Br J Gen Pract 49:49–55 [PMC free article] [PubMed] [Google Scholar]
- Jeelani NU, Braithwaite BD, Tomlin C, MacSweeney ST (2000) Variation of method for measurement of brachial artery pressure significantly affects ankle-brachial pressure index values. Eur J Vasc Endovasc Surg 20:25–28 [DOI] [PubMed] [Google Scholar]
- Jonsdottir GM, Jonasson K, Gudbjartsson DF, Kong A (2000) Construction of a genetic marker map using genotypes from over 7500 Icelanders. Am J Hum Genet Suppl 67:332 [Google Scholar]
- Kong A, Cox NJ (1997) Allele-sharing models: LOD scores and accurate linkage tests. Am J Hum Genet 61:1179–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES (1996) Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet 58:1347–1363 [PMC free article] [PubMed] [Google Scholar]
- Lander E, Kruglyak L (1995) Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 11:241–247 [DOI] [PubMed] [Google Scholar]
- Nicolae DL (1999) Allele sharing models in gene mapping: a likelihood approach. PhD thesis, University of Chicago, Chicago [Google Scholar]
- Palsson B, Palsson F, Perlin M, Gudbjartsson H, Stefansson K, Gulcher J (1999) Using quality measures to facilitate allele calling in high-throughput genotyping. Genome Res 9:1002–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risch N (1990) Linkage strategies for genetically complex traits. II. The power of affected relative pairs. Am J Hum Genet 46:229–241 [PMC free article] [PubMed] [Google Scholar]
- Weeks DE, Lange K (1988) The affected-pedigree-member method of linkage analysis. Am J Hum Genet 42:315–326 [PMC free article] [PubMed] [Google Scholar]
- Whittemore AS, Halpern J (1994) A class of tests for linkage using affected pedigree members. Biometrics 50:118–127 [PubMed] [Google Scholar]
- Yu A, Zhao C, Broman KW, Jang W, Mungall AJ, Dunham I, Weber JL (2000) Genome-wide comparison of human genetic and physical maps. Am J Hum Genet Suppl 67:10 [Google Scholar]