

Abstract
Francisella tularensis is an infectious, gram-negative, intracellular microorganism, and the cause of tularemia. Invasion of host cells by intracellular pathogens like Francisella is initiated by their interaction with different host cell membrane receptors and the rapid phosphorylation of different downstream signaling molecules. PI3K and Syk have been shown to be involved in F. tularensis host cell entry, and both of these signaling molecules are associated with the master regulator serine/threonine kinase mTOR; yet the involvement of mTOR in F. tularensis invasion of host cells has not been assessed. Here, we report that infection of macrophages with F. tularensis triggers the phosphorylation of mTOR downstream effector molecules, and that signaling via TLR2 is necessary for these events. Inhibition of mTOR or of PI3K, ERK, or p38, but not Akt signaling, downregulates the levels of phosphorylation of mTOR downstream targets, and significantly reduces the number of F. tularensis cells invading macrophages. Moreover, while phosphorylation of mTOR downstream effectors occurs via the PI3K pathway, it also involves PLCγ1 and Ca2+ signaling. Furthermore, abrogation of PLC or Ca2+ signaling revealed their important role in the ability of F. tularensis to invade host cells. Together, these findings suggest that F. tularensis invasion of primary macrophages utilize a myriad of host signaling pathways to ensure effective cell entry.
Introduction
Francisella tularensis subspecies tularensis (Type A) and subspecies holartica (Type B) are highly infectious, Gram-negative, intracellular pathogens that cause tularemia, a disease with significant morbidity and mortality in humans and other mammals. Due to its ease of infection and means of dissemination, these F. tularensis subspecies are classified as select agents [1,2]. F. tularensis can infect a variety of host cells, but macrophages seem to be a very effective cell type for the replication and survival of this bacterium [3,4]. The F. tularensis Live Vaccine Strain (LVS) derived from subspecies holartica causes an attenuated form of infection in humans and has been used as a vaccine, although it is not licensed. Conversely, F. tularensis LVS infection of mice does cause a pathology that resembles that observed in humans infected with virulent Francisella strains. Since the intracellular life cycle of F. tularensis LVS is similar to that of type A Francisella, its use in research has been of great advantage [5].
Intracellular bacteria like F. tularensis have devised sophisticated mechanisms that exploit, trigger, and activate host signal transduction pathways for their internalization into mammalian cells. Central to the internalization of bacteria, including that of F. tularensis, is the rearrangement of the actin cytoskeleton [6]. Actin remodeling during bacterial infection can occur through the participation of various host cell receptors linked to downstream signaling molecules necessary for bacterial host cell entry that ultimately will be associated with the regulation of actin proteins. For instance, the phosphoinositide kinase-3 (PI3K), the tyrosine kinase Syk, and the extracellular regulated kinase (ERK) have been implicated in the internalization of F. tularensis [3,7], and these molecules directly interact with actin [8] or participate in actin regulation [9,10]. Downstream of the PI3K/Akt pathway is the master regulator serine/threonine kinase mammalian target of rapamycin (mTOR), which has been shown to be involved in the modulation of actin via downstream effectors of mTOR complex 1 (mTORC1) [11] and mTORC2 [12,13]. Yet, the importance of the mTOR pathway in F. tularensis invasion has not been assessed. Evidence suggest that phospholipases play a role in phagocytosis, e.g., phospholipase C (PLC), which is activated downstream of PI3K, has been shown to be important for FcγR-mediated phagocytosis [14] and for host cell uptake of Escherichia coli [15,16]. Moreover, PLCγ1 was shown to be involved in the modulation of mTOR in an Akt-independent manner [17]; however, whether the PLC pathway is associated with the regulation of mTOR downstream effector molecules and with F. tularensis infection is not known.
The kinase mTOR is found in all eukaryotes [18,19] and plays a major role in key aspects of cell biology, including membrane trafficking, cell growth and survival [20-22]. Studies on the involvement of mTOR in actin regulation have shown that knockdown of rictor resulted in defective actin cytoskeleton rearrangement [12,13]. Moreover, ERK and Akt signaling molecules are regulated by mTORC2, and these molecules seem to be implicated in actin regulation, as exemplified by the necessity for ERK in the cell entry of Francisella novicida [7] and Chlamydia pneumoniae [23], and for Akt in the internalization of Pseudomonas aeruginosa [24]. Downstream effectors of mTORC1, such as the 70 kDa ribosomal S6 kinase (p70S6K), known to be important in cell growth, have been recently reported to also regulate the actin cytoskeleton [11]. In addition, phosphorylation of its downstream effector ribosomal protein S6 was enhanced during phagocytosis in macrophages [25]. Rapamycin, a powerful and specific inhibitor of mTOR downstream signaling [18,26,27], can abrogate the mTORC1 pathway through its binding to FK506-binding protein 12 (FKBP12). The region of mTOR that binds the FKBP12-rapamycin complex is known as the FRB domain. mTOR forms a scaffold complex with other proteins that regulate different arms of the mTOR signaling cascade. The association of raptor with mTOR (mTORC1) is indispensable for mTOR signaling, as shown by RNA interference in cultured mammalian cells [28]. However, raptor does not affect the catalytic function of mTOR, but serves as a scaffold for the juxtaposition of mTOR with its substrates p70S6K and eukaryotic initiation factor 4E-binding protein 1 (4E-BP1) [29,30]. Hence, binding of the FKBP12-rapamycin complex to the mTOR FRB promotes a substantial dissociation of raptor from mTOR, thereby separating mTOR from its substrates and abrogating mTOR signaling, but not its intrinsic autophosphorylating catalytic activity [18]. Albeit rapamycin was known to affect only the mTORC1 pathway, recent studies demonstrated that although mTORC2 does not interact with FKBP12-rapamycin, the assembly of mTORC2 is inhibited by rapamycin following long-term treatment [31]. Since mTORC2 was reported to phosphorylate and activate AktSer473, rapamycin cell treatment consequently affected AktSer473 phosphorylation [31]. Akt activation occurs following extracellular agonist-induced PI3K stimulation, and phosphorylation at Thr308 and at Ser473 residues of Akt allows its full activation. Akt is critical in cellular processes like growth, differentiation and proliferation, and is believed to bridge the PI3K pathway to mTORC1 signaling [22]. Thus, while Akt is an upstream activator of mTORC1, Akt is also the target of mTOR via mTORC2.
PLC is important in mediating signal transduction from extracellular and intracellular stimuli and in its involvement in phagocytic signaling [32-34]. PLCγ signaling can be activated downstream of PI3K through interactions between their SH2 and/or PH domains with phosphatidyl-inositol-3,4,5-triphosphate, thus linking the PI3K and the PLCγ/Ca2+ signaling pathways [35]. Furthermore, PLCγ1 has been shown to control the activation of the mTORC1 downstream effector molecule p70S6K in an Akt-independent manner, acting in parallel with the classical PI3K/Akt pathway [17].
The PI3K/Akt/mTOR cascade can respond to a myriad of stimuli through specific receptors such as Toll-like receptors. Activation of mammalian host cell transduction pathways is an essential step for the invasion of intracellular pathogens, including F. tularensis. F. tularensis is a TLR2 agonist that induces activation of the mitogen-activated protein kinases (MAPK) pathway, shown to play a role in the regulation of mTOR downstream targets [36-40]. Yet, the involvement of TLR2 signaling in F. tularensis cell entry in the context of the mTOR signaling cascade has not been studied.
The aims of this study were to (i) determine the phosphorylation of specific signaling molecules associated with the mTOR signaling cascade in response to F. tularensis LVS infection, and (ii) to identify specific signaling molecules/pathways that play a significant role in F. tularensis LVS invasion. Our results suggest that the mTOR signaling cascade plays a prominent role in F. tularensis infection of primary macrophages, thus, augmenting our understanding of the signaling events involved in the invasion process by this bacterium. Furthermore, inhibition of mTOR and PI3K signaling affected the architecture of the actin cytoskeleton, inducing numerous thick, short filaments and small patches distributed throughout the cell, which significantly affected bacterial cell entry. Moreover, inhibition of MEK/ERK and p38 MAPK, but not Akt signaling resulted in a decrease in the phosphorylation of mTOR downstream effector molecules and of internalized F. tularensis in primary macrophages. Our results further show that PLC and Ca2+ signaling are also important for F. tularensis infection, since their inhibition significantly decreased the ability of the bacteria to invade the host cells, and abrogation of these signaling molecules affected the phosphorylation of mTOR downstream effectors. Overall, our findings suggest that phosphorylation of the mTOR signaling cascade via various cell signaling pathways is a critical strategy used by F. tularensis to ensure host cell invasion.
Materials and Methods
Ethics Statement
All studies were done in accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals of the National Institute of Health. All protocols involving animal research were approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham (UAB; Protocol number 09112 under Institutional Animal Assurance Number A-3255-01).
Bacteria
F. tularensis LVS (ATCC 29684; American Type Culture Collection, Rockville, MD), a gift from Karen Elkins (Food and Drug Administration, Rockville, MD), was grown and maintained as described previously [41,42].
Mice and peritoneal macrophage cultures
C57BL/6 wild type (WT), TLR2 knockout (KO), TLR1KO, TLR6KO and TLR4KO mice were bred and maintained within an environmentally controlled, pathogen-free animal facility at the University of Alabama at Birmingham (UAB). The original WT mice were obtained from NCI/Frederick. The TLRKO breeding pairs, backcrossed >8 times onto the C57BL/6 background, were originally obtained under a material transfer agreement from Shizuo Akira (Osaka University, Osaka, Japan). All studies used 8-10 week old female mice, and all protocols were approved by the UAB Institutional Animal Care and Use Committee.
Peritoneal macrophages from C57BL/6 WT or from the corresponding TLRKO mice were induced by i.p. injection of 1 ml of sterile 3% BBL Brewer modified thioglycollate medium (BD Biosciences), and were isolated as previously described [43]. Cells were washed and cultured in 24-well plates (1 x 106 cells) in RPMI 1640 medium supplemented with 5% fetal calf serum, 2 mM L-glutamine, 50 μM 2-mercaptoethanol, 1 mM sodium pyruvate, 1.5 mg/ml of sodium bicarbonate, and 25 mM HEPES (complete medium). The cells were allowed to adhere overnight at 37°C in a humidified 5% CO2 incubator. Non-adherent cells were then removed by washing several times with complete medium.
Immunoassay and Western analysis
Macrophages from C57BL/6 WT mice were pre-incubated with medium only, rapamycin (3 h) or with selective inhibitors (1 h) followed by stimulation with F. tularensis LVS [multiplicity of infection (MOI)=20] for the indicated time periods. Macrophages derived from KO mice were stimulated with F. tularensis LVS for the indicated times. Cells were then washed with PBS and lysed as previously described [43,44]. Protein concentrations in whole cell extracts were assessed using the Micro BCATM Protein Assay Kit (Pierce, Rockford, IL). Equivalent amounts of protein from cell lysates were separated by SDS-PAGE on a 7.5 or 10% Tris–HCl gel (Bio-Rad Laboratories, Hercules, CA). Protein was electrotransferred to immobilon-P transfer membranes (Millipore, Bedford, MA) and Western analysis was carried out with specific antibodies against the total and/or phosphorylated forms of extracellular signal-regulated kinase (ERK) (Thr202/Tyr204), p38 mitogen-activated protein kinase (Thr180/Tyr182), Akt (Ser473), PI3K p85 (Tyr458/p55), p70S6K (Thr389) and (Thr421/Ser424), 4E-BP1 (Ser65), eI-F4E (Ser209), S6 (Ser235/236) and (Ser240/244) and rictor (Cell Signaling Technology, Inc. Danvers, MA). Detection of antibodies was carried out using horseradish peroxidase-linked rabbit or mouse anti-IgG antibody, followed by ECL Western blot detection reagents (GE Healthcare UK, Buckinghamshire, England). Densitometer scans of blots were done using the AlphaImager 2000 documentation and analysis system (Alpha Innotech, San Leandro, CA). In some experiments, cells were pre-incubated with the selective p38 MAPK inhibitor SB203580 (10 µM), the MEK1/2/ERK1/2 inhibitor UO126 (10 µM), the Akt inhibitor VIII (isoenzyme selective Akt1/2; 500 nM), the Raf1 inhibitor InSolution™ Raf1 Kinase Inhibitor I (1 µM), or the PLC inhibitor U73122 (3 µM) (EMD Milipore; Rockland, MA); or the Ca2+ chelator BAPTA-AM™ (10 µM), the PI3K inhibitor wortmannin (100 nM), or the mTOR inhibitor rapamycin (50 µg/ml) (Sigma-Aldrich; St. Louis, MO). The concentration of the inhibitors used was determined to be optimal in preliminary studies that tested different concentrations of each inhibitor. The effect of the inhibitors on macrophage viability was assessed by trypan blue exclusion, and on bacterial viability by microbiologic analysis as previously described [41]. The concentration of each inhibitor used in our study had no affect on macrophage or bacterial viablility. All experiments were repeated 3 to 5 times unless otherwise stated.
Transfection assay
Murine macrophage-like RAW 264.7 cells were cultured in 96- or 24-well plates in complete medium until 50-70% confluency was reached. Cells cultured in 96-well plates were transfected with nonspecific siRNA or with siRNA (100 nM) targeted to the Akt1/2 or PLCγ1 genes (Santa Cruz, Santa Cruz, CA) using Lipofectamine™ RNAiMAX (0.3 µl), whereas cells cultured in 24-well plates were transfected with non-specific siRNA or siRNA targeted to the mTOR gene (Santa Cruz, Santa Cuz, CA) using Lipofectamine™ RNAiMax (1.5 µl) transfection reagent (Life Technologies, Grand Island, NY), according to the manufacturer’s instructions. Transfected cells were incubated at 37°C for 5 days and rested for 4 h prior to the addition of F. tularensis LVS. Cells were then washed, lysed and assessed by Western analysis.
Immunoprecipitation assay
Macrophages from C57BL/6 WT mice were cultured in 60-mm cell culture plates overnight. Cells were pretreated with rapamycin (50 µg/ml; 3 h) or media only, followed by infection with F. tularensis LVS (MOI=20). Cells were washed twice with PBS and lysed with 500 µl of lysis buffer. Lysates were collected and assessed for protein content. Equal amounts of protein were transferred to microfuge tubes and 5 µl of anti-rictor antibody (0.1 mg/0.4 ml; Bethyl Labs, Montgomery, TX) or 5 µl of IgG control antibody (0.1 mg/ml; Santa Cruz) were added to the correspondent samples. Samples were rotated overnight at 4°C. Afterwards, 50 µl of protein A sepharose CL-4B (GE Heathcare UK, Buckinghamshire, England), prepared in a 50% slurry, was added to each sample and incubated for 2 h. Samples were then washed 3 times in lysis buffer and 45 µl of Laemmli buffer with 5% mercaptoethanol was added to each sample and assessed by Western analysis.
7-Methyl GTP Pull-down assay
Macrophages from C57BL/6 WT mice were cultured in 60-mm cell culture plates overnight. Cells were pretreated with rapamycin (50 µg/ml; 3 h) or media only, followed by infection with F. tularensis LVS (MOI=20). Cells were washed twice with PBS and lysed with 750 µl of TritonX buffer. Lysates were collected and assessed for protein content. Equal amounts of protein were transferred to microfuge tubes and 30 µl of 7-Methyl GTP sepharose (GE Heathcare UK, Buckinghamshire, England) was added to each sample. The samples were rotated for 2 h at 4°C. Samples were then washed 3 times in TritonX buffer, and 45 µl of Laemmli buffer with 5% mercaptoethanol was added to each sample and assessed by Western analysis.
Bacterial invasion
Thioglycollate-induced peritoneal macrophages from WT mice were harvested, washed and cultured in complete medium at 5 x 105 cells/well overnight in 24-well plates. Cells derived from WT mice were incubated or not with rapamycin (50 µg/ml; 3 h), U73122 (3 µM; 1 h), UO126 (10 µM; 1 h), SB203580 (10 µM; 1 h), Akt VIII (500 nM; 1 h), wortmannin (100 nM; 1 h), BAPTA (10 µM; 1 h) or cytochalasin D (2.5 µg/ml; 1 h) prior to incubation with bacteria. The indicated concentration of each inhibitor was shown to be optimal in preliminary studies. RAW cells transfected with control siRNA or with siRNA directed to the mTOR gene were cultured in complete medium in 24-well plates (see above). Freshly harvested F. tularensis LVS (MOI=20) were added to WT derived cells and to a portion of the transfected RAW cells for 90 min to assess bacterial invasion. Following incubation, cells were washed (5x) with PBS+ at room temperature and complete media containing gentamycin (50 µg/ml) was then added to the cultures for no more than 45 min to kill extracellular bacteria. Afterwards, cells were washed as described above and lysed for 5 min with ice-cold distilled water (150 µl). Lysates were serially diluted, plated on Mueller-Hinton II agar plates, and plates were incubated at 37°C in a 5% CO2 atmosphere [45]. Colonies were counted after 72-96 h. Non-transfected RAW cells treated or not with rapamycin were incubated or not with bacteria and used as controls. Each condition was set up in triplicate, and the experiment was repeated a minimum of three times. None of the inhibitors had an effect on macrophage viability as determined by trypan blue exclusion. In addition, preliminary studies demonstrated that the inhibitors had no effect on bacterial viability as determined by microbiologic analysis of bacterial suspensions incubated with or without each inhibitor. The number of internalized bacteria was expressed as a percent of the control infected cells [23]. Additionally, transfected RAW cells were washed following incubation with bacteria, as described above, and then the cells were lysed and lysates were assessed for the phosphorylation of mTOR downstream effectors by Western analysis.
Fluorescence confocal microscopy
Cells were plated in 6-well plates with coverslips in each well and either left untreated or treated with rapamycin or wortmannin, as described above. One set of wells (control, +rapamycin or +wortmannin) was supplemented with buffer alone, while the other received F. tularensis (MOI= 20), and then all sets were incubated for 90 min. Cells were then quickly washed with ice-cold phosphate-buffered saline (PBS) and fixed with 3% paraformaldehyde in PBS for 10 min at room temperature followed by permeabilization with 0.1% Triton X-100 in PBS for 7 min at room temperature. The coverslips were then incubated for 45 min at room temperature with Phalloidin conjugated to Alexa-Fluor 594 (Molecular Probes, Eugene, OR) to stain actin and mouse monoclonal F. tularensis anti-lipopolysaccharide (LPS) antibody (FB11; 1:2000 dilution; Advanced Immunochemical Inc., Long Beach, CA) to visualize F. tularensis bacteria. Cells were washed with 0.2% Tween 20 in PBS and incubated for 30 min at room temperature with goat anti-mouse Alexa-Flour 488 secondary antibody (Molecular Probes, Eugene, OR). Cells were washed with 0.2% Tween 20 in PBS, nuclei were labeled with Hoescht stain and coverslips were mounted in 9:1 glycerol/PBS with 0.1% p-phenylenediamine (Sigma Aldrich, St. Louis, MO) on glass slides. Z-stack confocal images were acquired using a Nikon Eclipse TE 2000-U and Velocity 3D Image Analysis software (Perkin Elmer, Waltham, MA).
Statistics
Statistical significance between infected control cells and infected cells pretreated with inhibitors was evaluated by Student’s two-tailed t test using the InStat program (Graphpad Software, San Diego, CA).
Results
mTOR signaling is involved in F. tularensis LVS invasion of primary macrophages.
To assess the involvement of mTOR signaling in the invasion of host cells by F. tularensis LVS, freshly isolated peritoneal macrophages were pretreated or not with the mTOR inhibitor rapamycin or with media containing DMSO (control). Cells were then co-cultured for 90 min with freshly harvested F. tularensis LVS and invasion was assessed. Rapamycin treatment of macrophages significantly reduced F. tularensis invasion to 19% compared to F. tularensis infected, untreated control cells (Figure 1A). Since mTOR is downstream of the PI3K/Akt pathway, we likewise assessed if signaling via PI3K was required for F. tularensis LVS invasion. Pretreatment of cells with the PI3K inhibitor wortmannin significantly reduced bacterial entry to 37% (Figure 1A), whereas inhibition of the PI3K downstream target Akt with the inhibitor Akt VIII (inhibitor of Akt1/2) did not significantly reduce the number of bacteria invading macrophages compared to infected, untreated controls (Figure 1A). No statistical difference was observed in bacterial invasion of host cells following treatment of cells with wortmannin or rapamycin. These results indicate that PI3K and mTOR signaling play important roles in F. tularensis LVS invasion of primary murine macrophages. Recent studies have demonstrated differences between F. tularensis LVS grown in Brain Heart Infusion broth (BHI) and that grown in Mueller-Hinton broth (MHB) [46,47]. Since the bacteria used in our studies were grown in MHB, we sought to verify that infection of cells with F. tularensis grown in BHI compared to those grown in MHB resulted in a similar phosphorylation pattern of mTOR downstream effector molecules, and a similar ability to invade macrophages. Therefore, macrophages were incubated with freshly harvested bacteria grown in BHI [46] or in MHB [41,42] for 90 min and invasion was assessed. No significant differences were seen in the number of bacteria internalized by macrophages using F. tularensis grown in BHI or that grown in MHB (Figure S1A).
Figure 1. Effect of mTOR, PI3K and Akt inhibitors on the internalization of F. tularensis LVS.

(A) Peritoneal macrophages derived from WT mice were pretreated or not with rapamycin (50 µg/ml; 3 h), wortmannin (100 nM; 1 h) or Akt VIII (500 nM; 1 h) and infected with freshly harvested F. tularensis LVS (MOI=20) for 90 min to assess bacterial invasion. Values are the mean ± SEM of 5 independent experiments, each done in triplicate; **p < 0.001; *p < 0.05 compared with infected control cells treated with DMSO. Peritoneal macrophages derived from WT mice were pretreated or not with the inhibitors as described above, exposed to F. tularensis LVS for 0-120 min and then lysed. (B, D, E) Total p70S6K, S6, 4E-BP1, eI-F4E and Akt, and phosphorylated p70S6K (Thr389 and Thr421/Ser424), 4E-BP1 (Ser65), S6 (Ser235/236 and Ser240/244), eI-F4E (Ser209) and Akt (Ser473) were assessed by Western analysis. Samples analyzed contained an equal amount of protein. Unstimulated control cells (time 0) were incubated with the respective inhibitors for the correspondent pre-incubation period. Prior to the addition of bacteria, cells were not washed including unstimulated controls. Unstimulated cells served as negative controls. (C) Peritoneal macrophages were pretreated with rapamycin (50 µg/ml) and exposed to F. tularensis LVS for 0-120 min. An equal amount of protein from each lysate was pulled-down using 7-methyl GTP sepharose beads. Pull-down products were assessed for eI-F4G by Western analysis. All gels are representative of three to five independent experiments.
To delineate the phosphorylation events of the downstream mTOR signaling cascade that take place as a consequence of F. tularensis infection, peritoneal macrophages were pre-treated or not with rapamycin and then exposed to Francisella for various times. The phosphorylation of mTORC1 downstream effector proteins was then assessed as previously described [21,22,48,49]. Phosphorylation of p70S6KThr389 induced by F. tularensis LVS infection was seen at 30 min, and was increased at 60 and 120 min, whereas p70S6KThr421/Ser424 phosphorylation was detected at 10 min and peaked at 30 min (Figure 1B). However, when cell cultures were treated with rapamycin prior to the addition of bacteria, phosphorylation of p70S6K was suppressed (Figure 1B). Phosphorylation of S6 ribosomal protein, a downstream target of p70S6K, was also observed in cells infected with Francisella (Figure 1B); however, treatment of cells with rapamycin prior to the addition of bacteria abrogated phosphorylation of S6Ser235/236, whereas the level of phosphorylated S6Ser240/244 (pS6Ser240/244) detected was notably less than that observed in non-rapamycin treated, infected macrophages (Figure 1B). Similarly, phosphorylation of 4E-BP1, which involves the other arm of the mTORC1 downstream signaling cascade [29,30], was also suppressed in rapamycin-treated cells (Figure 1B). Following exposure of cell cultures not treated with rapamycin to bacteria, a peak in the level of 4E-BP1 phosphorylation was seen at 60 min, as evidenced by an upward shift in electrophoretic mobility (Figure 1B), consistent with previous studies establishishing that phosphorylation of specific sites on 4E-BP1 retards its electrophoretic mobility [50,51]. 4E-BP1 positively or negatively regulates the function of the 4F-translation initiation complex (eI-F4E) by reversibly associating with it. Specifically, hypo- or dephosphorylated 4E-BP1 avidly binds eI-F4E, thereby inhibiting its phosphorylation and translational activity, whereas when 4E-BP1 is phosphorylated, this interaction is disrupted, leading to eI-F4E activation [50,52]. Western analysis of phosphorylated eI-F4E (peI-F4E) in macrophages treated with rapamycin and infected with F. tularensis LVS revealed less phosphorylation at 60 and 120 min, compared to that seen with lysates of cells exposed to bacteria only (Figure 1B). Importantly, the resulting phosphorylation of the mTORC1 downstream proteins was not due to differences in total protein, except in the case of 4E-BP1 where rapamycin affects total 4E-BP1 (Figure 1B). It is noteworthy that phosphorylation of these downstream proteins was also blocked when rapamycin was removed from the cultures by washing prior to the addition of bacteria (not shown). Finally, the phosphorylation patterns of mTOR downstream effectors induced by F. tularensis LVS grown in BHI were similar to that seen with F. tularensis LVS grown in MHB (Figure S1B).
To determine if the downregulatory effect on eI-F4E phosphorylation by rapamycin resulted in the dampening of the translation initiation machinery, we next carried out a 7-methyl GTP pulldown assay and assessed binding to eI-F4G by Western analysis. No eI-F4G was detected in lysates from cultures treated with rapamycin, suggesting that the absence of mTOR signaling with F. tularensis LVS infection does not allow the formation of the eI-F4F in spite of the low level of peI-F4E observed in the presence of rapamycin (Figure 1C).
To confirm that phosphorylation of mTOR downstream signaling molecules is critical for the internalization of Francisella into primary macrophages, RAW cells were transfected with siRNA control or siRNA to mTOR and exposed to Francisella for 90 min for the assessment of invasion and phosphorylation of mTOR downstream effectors. Transfection of cells with siRNA to mTOR resulted in a significant decrease (77%) in the number of invading bacteria, compared to that seen with siRNA control transfected RAW cells (Figure S2A), which was similar to that seen in primary macrophages (Figure 1A) and RAW (not shown) cells treated with rapamycin prior to the addition of bacteria. Moreover, the phosphorylation pattern of mTOR’s downstream effector proteins of cells transfected with siRNA to mTOR, but not with siRNA control, was similar to that observed in peritoneal macrophages treated with rapamycin prior to the addition of bacteria (Figure S2B).
Next, we determined the role of PI3K signaling on the phosphorylation of mTOR downstream effectors in macrophages exposed to Francisella in the presence or absence of wortmannin. Our findings revealed that inhibition of PI3K signaling in F. tularensis LVS-infected macrophages by pretreatment with wortmannin abrogated phosphorylation of p70S6K at Thr389 and at Thr421/Ser424 (Figure 1D), as well as phosphorylation of 4E-BP1, but not phosphorylation of eI-F4E (Figure 1D). Furthermore, phosphorylation of S6 at Ser235/236 was also inhibited. A low level of pS6 at Ser240/244 was apparent at 30 and 60 min (Figure 1D), suggesting that a PI3K-independent pathway is also involved in the regulation of S6 phosphorylation at Ser240/244. Lastly, inhibition of PI3K also abrogated AktSer473 phosphorylation (Figure 1D). The above data shows, as previously reported [19,53-55], that inhibition of PI3K affected the phosphorylation of downstream targets of mTORC1; however, it further suggests that PI3K inhibition also affected mTORC2, since mTORC2 has been shown to regulate AktSer473 phosphorylation [31,56]. The observed phosphorylation of the respective proteins derived from cultures that were infected only or treated with wortmannin and then infected were not due to differences in total protein (Figure 1D). To further establish the direct involvement of Akt in the regulation of mTOR’s downstream molecules, we evaluated if chemical inhibition of Akt rendered similar results to those obtained with wortmannin. Downregulation of phosphorylated AktSer473 (pAktSer473) was achieved by use of the Akt inhibitor VIII in macrophages exposed to Francisella; however, essentially no change was seen in p70S6K phosphorylation at Thr389 or at Thr421/Ser424 or in the phosphorylation of 4E-BP1, eI-F4E or S6 at Ser235/236 or at Ser240/244 (Figure 1E). The resulting phosphorylation of the respective proteins derived from cultures infected only or treated with Akt VIII and then infected, was not due to differences in total protein (Figure 1E). To demonstrate that these results were indeed due to the specific inhibition of Akt, we next transfected RAW cells with siRNA to Akt and cultured them or not with F. tularensis (Figure S3). While complete abrogation of AktSer473 phosphorylation was seen (Figure S3), phosphorylation of p70S6KThr389 was not suppressed as seen with wortmannin (Figure 1D). These studies suggest the participation of a PI3K-dependent, Akt-independent regulation of the downstream targets of mTORC1 following Francisella infection of primary macrophages. Taken together, the phosphorylation data of the mTORC1 downstream effector molecules in infected macrophages (Figures 1B, 1D, 1E and Figure S2B) and that of Francisella invasion in the presence or absence of inhibitors (Figures 1A and Figure S2A) indicate that mTORC1 downstream effector molecules are central for the infection of peritoneal macrophages by F. tularensis LVS.
It has been reported that infection of brain endothelial cells by Cronobacter sakazakii disassembled actin fibers, but that cytochalasin D and PI3K inhibitors effectively blocked the bacterial effect on actin and cell invasion [57]. Furthermore, inhibition of mTOR signaling using rapamycin or stable inhibition of raptor (mTORC1) and rictor (mTORC2), decreased actin cytoskeleton remodeling [58]. Thus, we next assessed by fluorescence microscopy the effect of rapamycin and wortmannin, on the actin cytoskeleton. Our findings revealed that wortmannin and rapamycin significantly alter the architecture of the actin cytoskeleton by inducing numerous thick, short filaments and small patches distributed throughout the cell (Figure 2A). Moreover, rapamycin-treated and wortmannin-treated cells contained significantly fewer F. tularensis than control cells (Figure 2B).
Figure 2. Effects of wortmannin and rapamycin on actin cytoskeleton and F. tularensis entry into cells.
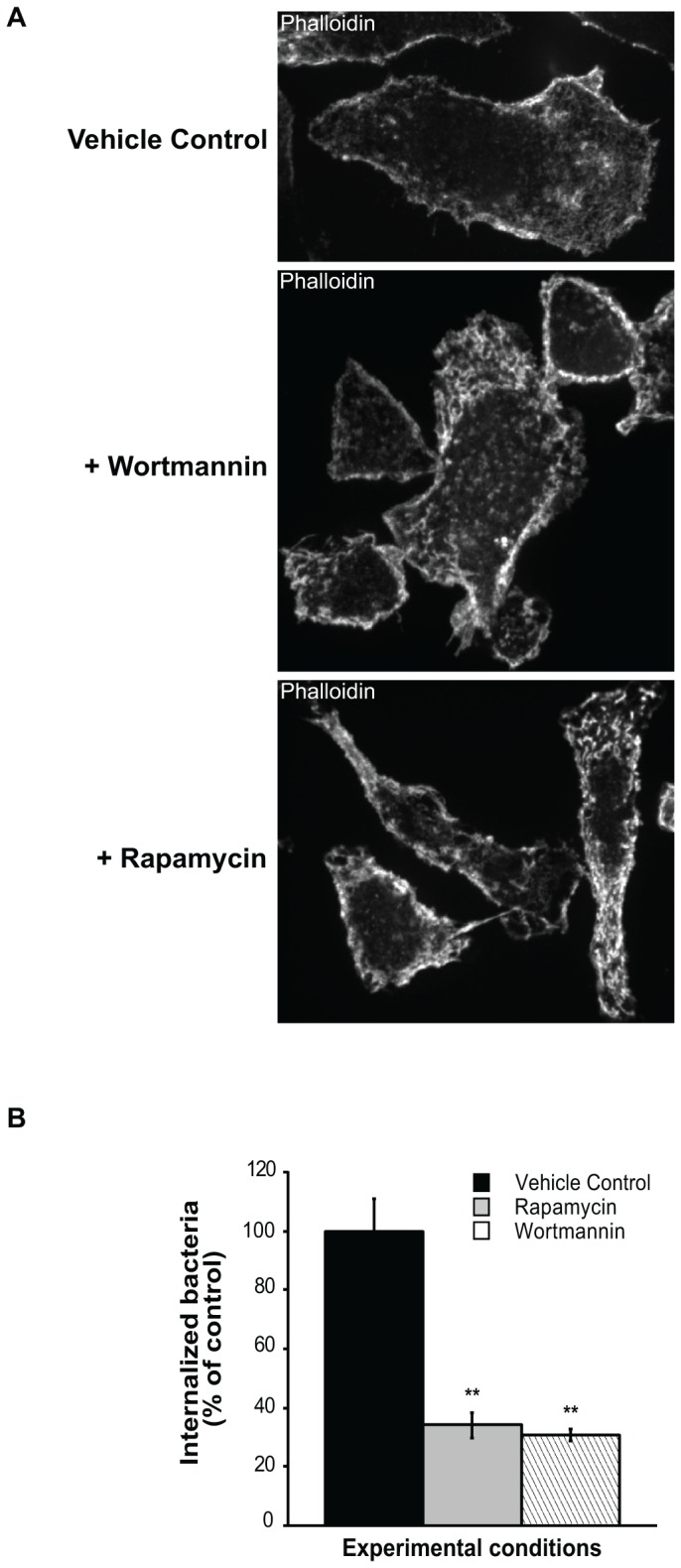
(A) Peritoneal macrophages were treated with vehicle control, wortmannin (1 h) or rapamycin (3 h) and stained with fluorescent β-phalloidin to detect F-actin. Wortmannin and rapamycin significantly alter the architecture of the actin cytoskeleton and induce numerous thick, short filaments and small patches distributed throughout the cell. Representative cells are shown. (B) Isolated macrophages were treated with vehicle control, wortmannin (for 1 h) or rapamycin (for 3 h), and then F. tularensis was added for 90 min. Cells were processed by immunofluorescence with an F. tularensis anti-LPS antibody to detect internalized bacteria. One hundred (100) cells for each treatment were counted in randomly selected fields. The number of bacteris in treated cells is expressed as percent normalized to the control (100%). The p values of rapamycin- and wortmannin-treated cells are <0.001 relative to control.
PLC and Ca2+ signaling play a role in the invasion of macrophages by F. tularensis LVS and in the regulation of downstream effectors of mTORC1 and mTORC2.
Markova et al. [17] reported a novel Akt-independent signaling pathway regulating p70S6K activation via phospholipase Cγ1 (PLCγ1), Ca2+ signaling and protein kinase C in leukemic cells. Since our results support the notion of an Akt-independent phosphorylation of the mTORC1 cascade in peritoneal macrophages infected with Francisella, we questioned if PLC and Ca2+ signaling could play a role in the phosphorylation of the mTORC1 downstream effectors in our system. Cells were exposed to Francisella for various times after pre-incubation or not with the PLC inhibitor U73122 [59] or with the intracellular Ca2+ chelator 1,2-bis (o-aminophenoxy)ethane-N,N,N’N’-tetraacetic acid, sodium (BAPTA) (Calbiochem Biosciences Inc. Gibbstown, NJ), and then lysed and subjected to Western analysis. Treatment of cells with U73122 or BAPTA prior to their incubation with F. tularensis LVS decreased the level of phosphorylated p70S6K (pp70S6K) at Thr389 and at Thr421/Ser424, as well as that of 4E-BP1 (p4E-BP1) (Figure 3A). Downregulation of S6Ser240/244 phosphorylation was more evident in the presence of U73122 than of BAPTA (Figure 3A), whereas S6Ser235/236 phosphorylation was affected more by Ca2+ than by PLC signal inhibition, suggesting a differential regulation of these S6 phosphorylation sites by PLC and Ca2+ signaling in peritoneal macrophages exposed to Francisella. The resulting phosphorylation of the respective proteins derived from cultures infected only or treated with the inhibitors and then infected, was not due to differences in total protein, except in the case of 4E-BP1 when BAPTA was used since it seems to affect total 4E-BP1 (Figure 3A). The relative fold changes in phosphorylated proteins observed by densitometry analysis were normalized against the total protein level of the appropriate target protein in a given lysate (Figure 3A).
Figure 3. Effect of PLCγ1/Ca2+ signaling on mTOR downstream targets and F. tularensis host cell internalization.

Peritoneal macrophages derived from WT mice were pretreated or not with U73122 (3 µM; 1 h) or BAPTA (10 µM; 1 h), infected with F. tularensis LVS (MOI=20) for 0-120 min and lysed. (A) Total p70S6K, S6, 4E-BP1 and Akt, and phosphorylated p70S6K (Thr389 and Thr421/Ser424), 4E-BP1 (Ser65), S6 (Ser235/236 and Ser240/244) and Akt (Ser473) were assessed by Western analysis. Samples analyzed contained equal amount of protein. (A) The band densities determined by densitometry for pAktSer473 were normalized to the total protein levels in a given lysate. Unstimulated control cells (time 0) were incubated with the respective inhibitors for the correspondent pre-incubation period. Prior to the addition of bacteria, cells were not washed including unstimulated controls. Unstimulated cells served as negative controls. (B) RAW cells were transfected with PLCγ1 siRNA (100 nM), and after 5 days, the cells were washed, rested and infected with F. tularensis LVS (MOI=20) for 0-120 min. Total PLCγ1 and PLCγ2, and phosphorylated p70S6K (Thr389 and Thr421/Ser424), 4E-BP1 (Ser65) and Akt (Ser473) were assessed by Western analysis. Samples analyzed contained equal amount of protein. Unstimulated cells served as negative controls. All gels are representative of three to five independent experiments. (C) Peritoneal macrophages derived from WT mice were pre-treated or not with U73122 or BAPTA, as described above, and exposed to freshly harvested F. tularensis LVS (MOI=20) for 90 min to assess bacterial invasion. Values are the mean ± SEM of 5 independent experiments, each done in triplicate; **p < 0.001; *p < 0.05 compared with infected control cells treated with DMSO.
PLC can be subdivided into β, γ, δ and ε isotypes [60]. PLCγ signaling can be activated downstream from PI3K through interactions between their SH2 and/or PH domains with phosphatidyl-inositol-3,4,5-triphosphate, thus linking the PI3K and the PLCγ/Ca2+ signaling pathways [34,35,61]. Considering that PLCγ1 is ubiquitously expressed [60] and is involved in the internalization of bacteria, e.g., E. coli [15], we next used siRNA against PLCγ1 to assess if this isotype played a role in the phosphorylation of molecules of the mTOR cascade as shown with the specific chemical inhibitor. Indeed, siRNA inhibition of PLCγ1, but not PLCγ2 in RAW cells infected with Francisella, resulted in downregulation of pp70S6K and p4E-BP1 (Figure 3B), in agreement with the findings obtained with U73122. These findings support a role for PLCγ1 in the activation of mTOR downstream effector molecules. Unlike the findings of Markova et al. [17], inhibition of PLC or Ca2+ signaling slightly downregulated the level of pAktSer473 (Figure 3A), and inhibition of PLCγ1 by siRNA yielded similar results (Figure 3B). The resulting phosphorylation of AktSer473 derived from cultures that were infected only or treated with inhibitor and then infected was not due to differences in total Akt protein. The relative fold changes in pAktSer473, as determined by densitometry analysis, were normalized against the total protein levels (Figure 3A). These findings suggest that PLCγ1 and Ca2+ signaling participate in the regulation of the downstream targets of mTOR in an Akt-dependent and -independent manner in peritoneal macrophages infected with F. tularensis LVS. Moreover, PLC and Ca2+ signaling play important roles in the internalization of F. tularensis into primary macrophages since their specific inhibition significantly reduced bacterial invasion to 11% and 40%, respectively, compared to Francisella infected control cells (Figure 3C).
The p38 and ERK1/2 pathways play critical roles in the regulation of mTOR signaling and their inhibition decreases F. tularensis invasion of primary macrophages.
Studies have demonstrated that ERK1/2 and p38 signaling is associated with actin regulation [8,62], and indeed their participation in host cell invasion by different pathogens has been reported [7,23,63]. Moreover, a role for ERK1/2 and p38 MAPK in the regulation of the mTOR pathway has been demonstrated through the activation of p70S6K, since the auto-inhibitory pseudosubstrate region in the C-terminal domain contains the consensus Ser/Thr-Pro sequence, and therefore, can be phosphorylated by members of the proline-directed protein kinases such as ERK1/2 and p38 [36,64]. Furthermore, the ERK1/2 and p38 pathways have been shown to be indirectly involved in the phosphorylation of the cap-binding protein eI-F4E. To first determine if ERK and p38 play a role in the host cell entry of F. tularensis LVS, peritoneal macrophages were pre-treated or not with inhibitors of MEK/ERK (UO126) or p38 (SB203580) signaling, exposed to bacteria, and then the number of invading Francisella assessed. Inhibition of MEK/ERK significantly decreased F. tularensis LVS host cell invasion to 63% compared to infected control cells (Figure 4A), lending support to the findings by Parsa et al. [7] that ERK plays a role in phagocytosis of F. novicida. In addition, our findings implicate for the first time p38 in the invasion process of F. tularensis LVS into primary macrophages, since inhibition of this MAPK significantly reduced bacterial internalization to 78% compared to F. tularensis-infected, untreated cells (Figure 4A). Moreover, the combined inhibition of MEK/ERK and p38 signaling resulted in a significant decrease (51%) of invading bacteria as compared to invasion by F. tularensis in untreated cells, suggesting a synergistic effect upon inhibition of these pathways (Figure 4A). Next, we determined if ERK and p38 signaling were involved in the phosphorylation events of the downstream targets of mTOR. Pretreatment of macrophages with UO126 or SB203580 prior to bacterial infection resulted in decreased levels of pp70S6KThr389 compared to that seen with infected only cells, whereas pretreatment with both inhibitors completely abrogated p70S6KThr389 phosphorylation (Figure 4B). Conversely, inhibition of either MEK/ERK or p38 resulted in a decrease in the levels of pp70S6KThr421/Ser424, whereas inhibition of both MAPKs resulted in a marked downregulation in pp70S6KThr421/Ser424 compared to Francisella infected control cells (Figure 4B). These findings indicate that while p38 and ERK are critically involved in the phosphorylation of p70S6K at Thr389, phosphorylation at Thr421/Ser424 is partially independent of these MAPKs. Furthermore, inhibition of ERK or p38 resulted in a decrease in the level of pS6Ser235/236 and dual inhibition of these MAPKs almost completely abolished it (Figure 4B). However, pretreatment with either UO126 or SB203580 had essentially no effect on the level of pS6Ser240/244, whereas a downregulatory effect was observed when both inhibitors were used (Figure 4B), suggesting that the p38 and the MEK/ERK pathways in peritoneal macrophages are primarily implicated in the regulation of S6 phosphorylation at Ser235/236, but not at Ser240/244 following Francisella infection. The phosphorylation pattern of the respective proteins derived from cultures infected only or treated with the inhibitors and then infected was not due to differences in total protein (Figure 4B). Densitometry analysis depicting relative fold changes in the indicated protein were normalized to the total protein levels of the respective target protein in the given cell lysate (Figure 4B). Lastly, inhibition of the p38 and MEK/ERK pathways similarly affected the other arm of mTORC1 downstream signaling because the concomitant use of p38 and MEK/ERK inhibitors downregulated further the level of p4E-BP1 and peI-F4E as compared to the effects exerted by each inhibitor alone (Figure 4B). Taken together, the effects exerted on the phosphorylation of mTOR downstream effectors in macrophages exposed to Francisella (Figure 4B), and on F. tularensis invasion (Figure 4A) in the presence or absence of pathway-specific inhibitors, SB203580 and/or UO126, suggest that the role played by p38 and MEK/ERK signaling in the invasion of primary macrophages by Francisella likely involves the mTOR downstream signaling cascade.
Figure 4. MEK/ERK and/or p38 inhibition affects Francisella cell invasion and phosphorylation of mTOR downstream effectors.

(A) Peritoneal macrophages derived from WT mice were pretreated or not with UO126 (10 µM; 1 h), SB203580 (10 µM; 1 h) or with UO126 and SB203580 (1 h) and exposed to freshly harvested F. tularensis LVS (MOI=20) for 90 min to assess bacterial invasion. Values are the mean ± SEM of 5 independent experiments, each done in triplicate; **p < 0.001; *p < 0.05 compared with infected control cells treated with DMSO. Peritoneal macrophages derived from WT mice were pretreated or not with UO126, SB203580 or with a combination of both inhibitors as described above, exposed to F. tularensis LVS (MOI=20) for 0-120 min and then lysed. (B) Total p70S6K, S6, 4E-BP1 and eI-F4E, and phosphorylated p70S6K (Thr389 and Thr421/Ser424), 4E-BP1 (Ser65), S6 (Ser235/236 and Ser240/244) and eI-F4E (Ser209) were assessed by Western analysis. Samples analyzed contained equal amount of protein. Unstimulated control cells (time 0) were incubated with the respective inhibitors for the correspondent pre-incubation period. Prior to the addition of bacteria, cells were not washed including unstimulated controls. Unstimulated cells served as negative controls. (B) Band densities determined by densitometry were normalized to the total protein levels of the appropriate target protein in a given lysate. All gels are representative of three to five independent experiments.
Since Akt is an important component of the mTOR signaling cascade, we next determined if the p38 and/or MEK/ERK pathways are involved in the regulation of AktSer473 phosphorylation. Our findings revealed that while inhibition of MEK/ERK signaling had essentially no effect on AktSer473 phosphorylation (Figure 5A), inhibition of p38 resulted in a decrease in pAktSer473 levels. Although this effect was less pronounced than that observed with rapamycin (Figure 5B), the results suggest that p38 signaling is also involved in the regulation of AktSer473 phosphorylation. Unexpectedly, when SB203580 was used in combination with UO126, increased levels of pAktSer473 were observed compared to SB203580 alone (Figure 5C). The phosphorylation pattern of the respective proteins derived from cultures infected only or treated with the inhibitors and then infected was not due to differences in total protein (Figures 5A, 5B, 5C). Densitometry analysis depicting relative fold changes in the indicated protein were normalized to the total protein levels of the respective target protein in the given cell lysate (Figures 5A, 5B, 5C). Since our findings revealed the importance of p38 and ERK1/2 MAPKs in the modulation of mTOR downstream effector molecules, we next determined if mTOR signaling is involved in the phosphorylation of AktSer473, p38 and ERK in peritoneal macrophages exposed to F. tularensis. Downregulation in the level of pERK1/2 and pAktSer473 was observed in rapamycin treated cells compared to non-treated host cells that were exposed to Francisella for various periods of time (Figure 6A). However, an increase in the level of phosphorylated p38 (pp38) was observed in rapamycin-treated cells compared to that seen in infected only macrophages (Figure 6A). These results suggest that these molecules are downstream of mTOR or that a molecule(s) mediated by mTOR could act back on upstream signaling pathways. Furthermore, the observed downregulation of pERK1/2 and pAktSer473 (Figure 6A) was in line with a report demonstrating that mTORC2 is involved in the modulation of these molecules and that rapamycin downregulated their phosphorylation [56]. The phosphorylation pattern of the respective proteins was not due to differences in total protein (Figure 6A). Immunoprecipitation of rictor in rapamycin-treated, but not in non-treated cells, revealed reduced levels of mTOR (Figure 6B), suggesting, as previously reported, that rapamycin partially affects the mTOR-rictor interaction [31]. Lastly, our findings suggest that mTOR signaling is a negative regulator of p38 phosphorylation as indicated by densitometry analysis (Figure 6A).
Figure 5. Inhibition of MEK and p38 differentially affect Akt (Ser473) phosphorylation.

Peritoneal macrophages derived from WT mice were pretreated with rapamycin (50 µg/ml; 3 h) or UO126 (10 µM; 1 h); or with rapamycin or SB203580 (10 µM; 1 h); or with SB203580 or with UO126 and SB203580 (1 h), exposed to F. tularensis LVS (MOI=20) for 0-120 min and then lysed. (A, B, C) Total protein and phosphorylated Akt (Ser473) were analyzed by Western analysis. Samples analyzed contained equal amount of protein. Unstimulated control cells (time 0) were incubated with the respective inhibitors for the correspondent preincubation period. Prior to the addition of bacteria, cells were not washed including unstimulated controls. Unstimulated cells served as negative controls. (A, B, C) The band densities determined by densitometry were normalized to the total protein levels of the appropriate target protein in a given lysate. All gels are representative of three to five independent experiments.
Figure 6. Rapamycin affects the phosphorylation of Akt (Ser473), ERK1/2 and p38, and the mTOR-rictor interaction.
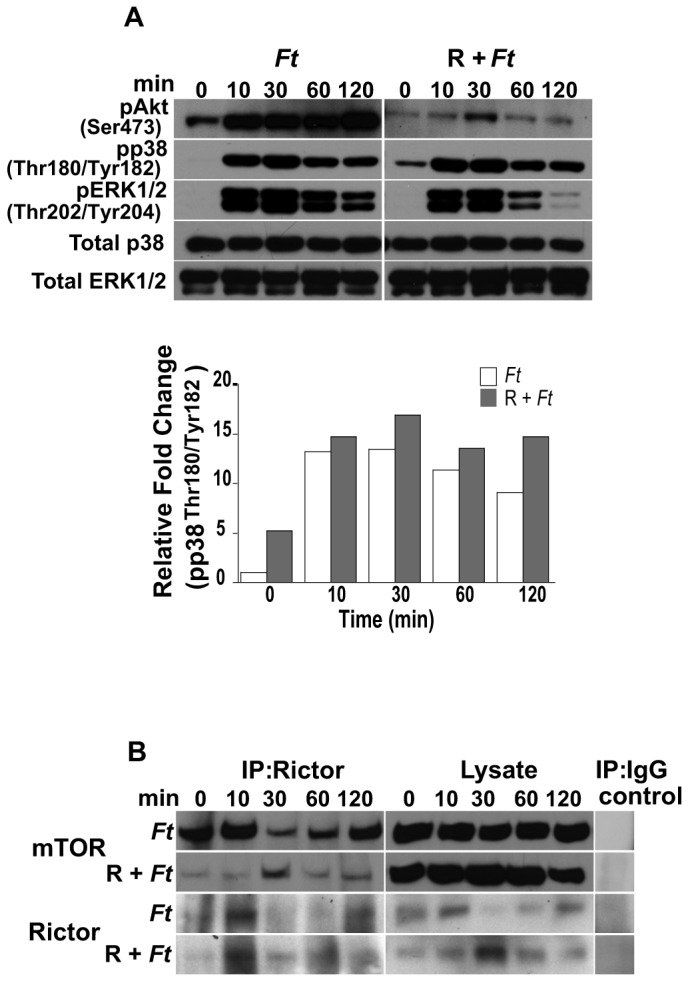
Peritoneal macrophages derived from WT mice were pretreated or not with rapamycin (50 µg/ml; 1 h), exposed to F. tularensis LVS for 0-120 min and then lysed. (A) Total p38 and ERK1/2, and phosphorylated Akt (Ser473), p38 (Thr180/Tyr182) and ERK1/2 (Thr202/Tyr204) were assessed by Western analysis. Samples analyzed contained an equal amount of protein. Unstimulated control cells (time 0) were incubated with rapamycin for the correspondent pre-incubation period. Prior to the addition of bacteria, cells were not washed including unstimulated controls. Unstimulated cells served as negative controls. The band densities determined by densitometry were normalized to the total protein levels of the appropriate target protein in a given lysate. (B) Peritoneal macrophages were pretreated or not with rapamycin and infected with F. tularensis LVS as described above. At the indicated times, cell lysates were prepared and immunoprecipitated with anti-rictor or IgG control antibody. Immunoprecipitates and lysates of mTOR and rictor from macrophages pretreated with rapamycin and infected with F. tularensis LVS or infected with bacteria alone were assessed by Western analysis. All gels are representative of three to five independent experiments.
TLR2 signaling is necessary for the involvement of the mTOR cascade and MAPK in Francisella infection of peritoneal macrophages.
TLRs are pattern recognition receptors that can distinguish molecules broadly shared among microbial pathogens. Recognition of these molecules via TLRs triggers the activation of downstream signaling cascades. Since it had been previously shown that F. tularensis LVS is a TLR2/6 and TLR2/1 agonist [42,65], we next determined the importance of TLR2, TLR1 and TLR6 in the phosphorylation events of mTORC1 downstream effectors. Exposure of TLR2KO macrophages to Francisella resulted in a lack of pp70S6KThr389, pp70S6KThr421/Ser424, p4E-BP1, peI-F4E, pS6Ser235/236 and a reduction in pS6Ser240/244 compared to that seen with WT and TLR4KO cells (Figure 7A). Assessment of the corresponding total protein levels in TLR2KO and WT macrophages revealed no differences, indicating that the lack or reduced phosphorylation was not due to a decrease in total protein (Figure 7A). Interestingly, F. tularensis infection of TLR4KO macrophages resulted in an increase in peI-F4E compared to that seen with WT cells, suggesting that an F. tularensis-induced TLR4 signal possibly limits phosphorylation of eI-F4E in WT macrophages (Figure 7A). Our findings further revealed that TLR1 and TLR6 signaling is not necessary for the phosphorylation events of the mTORC1 downstream signaling cascade, as macrophages derived from these KO mice exposed to bacteria showed levels of pp70S6KThr389, p4E-BP1 and pS6Ser235/236 comparable to the levels detected in WT cells (Figure 7B). These findings indicate that TLR2 is the main pattern recognition receptor involved in the regulation of mTORC1 downstream effector molecules in primary macrophages exposed to F. tularensis LVS. Furthermore, while comparable levels of pAktSer473 were seen in WT and TLR4KO macrophages exposed to Francisella, the level of pAktSer473 in TLR2KO cells was downregulated (Figure 7C), suggesting that TLR2 signaling also plays a role in mTORC2 AktSer473 phosphorylation. Levels of pAktSer473 in TLR1KO and TLR6KO cells were comparable to those observed in WT and TLR4KO macrophages (not shown).
Figure 7. TLR2 is necessary for the phosphorylation of the mTOR downstream signaling cascade upon Francisella infection.

Peritoneal macrophages derived from WT, and TLRKO mice were exposed to F. tularensis LVS (MOI=20) for 0-120 min and then lysed. Total p70S6K, S6, 4E-BP1 and eI-F4E, and phosphorylated (A) p70S6K (Thr389 and Thr421/Ser424), 4E-BP1 (Ser65), S6 (Ser235/236 and Ser240/244) and eI-F4E (Ser209) were assessed in WT, TLR2KO and TLR4KO derived macrophages by Western analysis; (B) Phosphorylated p70S6K (Thr389) 4E-BP1 (Ser65) and S6 (Ser235/236) in WT, TLR1KO and TLR6KO derived macrophages was assessed by Western analysis; (C) Total protein and phosphorylated Akt (Ser473) in WT, TLR2 and TLR4KO derived macrophages were assessed by Western analysis. Samples analyzed contained an equal amount of protein. (A) Phosphorylation of the respective proteins was not influenced by differences in total protein in TLRKO cells. Unstimulated cells served as negative controls. All gels are representative of three to five independent experiments.
Next, we determined if the lack of TLR2 signaling that resulted in 4E-BP1 dephosphorylation and abrogation of p70S6K, S6 and eI-F4E phosphorylation (Figure 7A) was related to a lack of phosphorylation of p38 and/or the MEK/ERK pathway, since these play a critical role in the activation of the mTOR signaling cascade (Figure 4B). Exposure of TLR2KO macrophages to F. tularensis did not induce phosphorylation of p38 and ERK1/2 MAPK, whereas phosphorylation was seen in WT and TLR4KO macrophages at 10, 30 and 60 min following bacterial stimulation (Figure 8A). The lack of phosphorylation of these MAPK in TLR2KO macrophages was not due to a lack or decrease in total protein, as reflected by the comparable levels of total ERK and p38 proteins detected in WT, TLR2KO and TLR4KO macrophages (Figure 8A). These results indicate that in primary macrophages exposed to F. tularensis LVS, TLR2 signaling is necessary for the regulation of mTOR’s downstream effector molecules via the p38 and MEK/ERK pathways. Since downregulation in the phosphorylation of mTOR downstream effector targets was seen in TLR2KO cells, we predicted that reduced bacterial entry would be seen upon exposure of TLR2KO derived macrophages to Francisella. Indeed, the ability of the bacteria to invade TLR2KO compared to WT macrophages was significantly reduced to approximately 40% (Figure 8B). Taken together, the studies suggest that TLR2 signaling by F. tularensis LVS is necessary for the involvement of the mTOR downstream signaling cascade, important for Francisella invasion of primary macrophages.
Figure 8. TLR2 signaling is important in Francisella invasion of primary cells.

(A) Peritoneal macrophages derived from WT, TLR2KO and TLR4KO mice were exposed to F. tularensis LVS (MOI=20) for 0-120 min and then lysed. Total protein and phosphorylated p38 (Thr180/Tyr182) and ERK1/2 (Thr202/Tyr204) were assessed by Western analysis. Samples analyzed contained an equal amount of protein. Unstimulated cells served as negative controls. All gels are representative of three to five independent experiments. (B) Peritoneal macrophages derived from WT and TLR2KO mice were exposed to freshly harvested F. tularensis LVS (MOI=20) for 90 min to assess bacterial invasion. Values are the mean ± SEM of 5 independent experiments, each done in triplicate; **p < 0.001; *p < 0.05 compared with infected control cells treated with DMSO.
Discussion
Intracellular pathogenic bacteria have developed strategies by which they manipulate host cell signaling pathways to facilitate their invasion, intracellular multiplication and survival. The participating downstream signaling events, directly or indirectly, exert a regulatory effect on the host cytoskeleton critical for the internalization of bacteria. In the present study, we have analyzed signaling molecular events in the context of the mTOR downstream signaling cascade in the internalization of F. tularensis LVS into primary macrophages.
Remodeling of the actin cytoskeleton is central to the invasion of host cells by bacteria, including that of F. tularensis, since treatment of peritoneal macrophages with the depolymerizing agent cytochalasin D prior to bacterial exposure essentially abolished Francisella host cell entry (not shown). Furthermore, inhibition of mTOR or of PI3K signaling by rapamycin or wortmannin, respectively, resulted in an alteration of the actin cytoskeleton architecture and inhibited the phosphorylation of mTOR downstream effector molecules. Moreover, in the presence of rapamycin or wortmannin F. tularensis invasion of primary macrophages was significantly reduced. Studies have shown that mTOR and downstream effector molecules are involved in actin cytoskeleton reorganization. For instance, inhibition of mTOR signaling using rapamycin or stable inhibition of raptor (mTORC1) and rictor (mTORC2), decreased actin cytoskeleton remodeling [58]. Moreover, Breven et al. [66] demonstrated that p70S6K, and activated mTOR were enriched at the actin arc of Swiss3T3 fibroblasts suggesting a role in actin cytoskeleton reorganization, and p70S6K was shown to be an important regulator of actin cytoskeleton by decreasing the rate and extent of actin depolymerization [11,67]. Furthermore, enhancement of phagocytosis by the endogenous lipid mediator Resolvin E1 involves the phosphorylation of the ribosomal protein S6 [25], and both p70S6K and 4E-BP1 were involved in the regulation of F-actin reorganization via the mTOR-raptor complex [68]. The mTOR downstream signaling cascade is also regulated by the PI3K pathway, located upstream of mTOR, and inhibition of PI3K by wortmannin abrogates phosphorylation of mTOR downstream effector molecules [9,53], a finding also observed in our studies. Furthermore, inhibition of PI3K significantly inhibited C. sakazakii invasion of brain endothelial cells and prevented actin rearrangement necessary for invasion [57].
While inhibition of rictor (mTORC2) decreased actin cytoskeleton remodeling [58], and Akt was shown to promote phagocytosis via the activation of p70S6K [69], in our studies, chemical inhibition of Akt with the specific inhibitor Akt VIII did not significantly decrease the number of internalized F. tularensis in primary macrophages, and not surprisingly, siRNA to Akt or chemical inhibition of Akt did not abrogate phosphorylation of mTOR downstream effector molecules, including that of p70S6K. These observations suggested an Akt-independent mechanism for the phosphorylation of mTOR downstream signaling molecules. These findings gain support from studies demonstrating that phosphorylation of mTOR downstream effectors was Akt-independent in leukemic cells [17], transformed B lymphocytes [70] and prostaglandin-F2α-treated bovine luteal cells [71]. However, to our knowledge, this is the first report showing that exposure of primary immune cells to F. tularensis LVS results in the phosphorylation of mTOR downstream effectors in an Akt-independent manner.
Analysis of mRNA transcripts in rapamycin-treated cells demonstrated downregulation of several transcripts linked to phagocytosis [72]. It is known that treatment of cells with rapamycin limits the availability of eI-F4E via its sequestration into an inactive complex with the hypophosphorylated 4E-BPs [72]. While such an event can repress global translation rates, the most affected mRNAs containing TOP elements were always downregulated and involved transcripts implicated in phagocytosis [72]. Interestingly, Syk was one of those transcripts [72], and Syk has been found to play a role in the invasion of host cells by Francisella [7]. Moreover, macrophages treated with rapamycin, but not with wortmannin, and exposed to F. tularensis LVS, revealed an apparent downregulation of ERK1/2 phosphorylation. This MAPK is the downstream effector of Syk and was implicated by Parsa et al. [7] and supported by our findings to be involved in F. tularensis host cell infection. Indeed, the MEK/ERK pathway has been implicated in host cell entry by other pathogens like C. pneumoniae [23]. In addition to the involvement of the MEK/ERK pathway in the invasion process of F. tularensis into host cells shown in the present study and that of others [7], our findings revealed that p38 signaling also plays a role in Francisella invasion of peritoneal macrophages. The pathogen Rickettsia rickettsii has been shown to use p38 signaling for host cell entry [63]. Furthermore, it has been reported that activation of p38 can increase the stability of actin microfilaments in the presence of the actin depolymerizing agent cytochalasin D [62]. During the exposure of host cells to stress, p38 signaling may mediate an increased stability of the actin cytoskeleton, thus constituting an important event of the cell response to external stimuli [62]. Along these lines, F. tularensis LVS infection of macrophages induced the phosphorylation of p38, but in the presence of rapamycin, as shown in this study and by others [58,68], alteration of the actin cytoskeleton was observed and the level of pp38 was increased, suggesting that rapamycin contributes to the cell’s stress. Our findings further revealed that while inhibition of p38 and or MEK/ERK significantly decreased the number of F. tularensis invading macrophages, it also resulted in the downregulation of pp70S6K and pS6, and that the simultaneous inhibition of these signaling molecules, had an additive effect. The involvement of the MEK/ERK and p38 pathways in the regulation of the mTOR downstream effector molecules was not unexpected, since various studies favor a role for MAPK in the phosphorylation of the S/T-P (proline-directed serine/threonine) sites in the autoinhibitory C-terminal domain for the modulation of p70S6K [36,64]. Activation of p70S6K initially requires that the interaction between the p70S6K C-terminal domain containing an auto-inhibitory pseudosubstrate region and the N-terminal domain be relieved, since it is thought that the inactive conformation of p70S6K is in place when the acidic N-terminal domain interacts with the basic C-terminal region [64]. This allows for a hierarchical phosphorylation at multiple sites, such as at Thr421/Ser424 in the C-terminus, consequently loosening the conformation and permitting the phosphorylation of Thr389 [73].
Differential requirements for ERK1/2 and p38 on p70S6K phosphorylation have been observed contingent on the stimuli. For instance, UV-irradiation induced phosphorylation of p70S6KThr389 was abrogated by ERK1/2 and p38 inhibitors, while that at Thr421/Ser424 was blocked by ERK1/2, but not p38 inhibition [74]. In addition, upon cell stimulation with amino acids, phosphorylation of p70S6K at Thr421/Ser424, but not at Thr389 was observed, and only the dual use of the inhibitors UO126 and SB203580 suppressed phosphorylation at these sites [75]. These reports, along with our findings using live bacteria, provide support that the nature of the stimulus is a relevant determinant for the differential participation of ERK1/2 and/or p38 in the regulation of p70S6K phosphorylation. Moreover, the involvement of MEK/ERK in the phosphorylation of the 40S ribosomal protein S6 at Ser235/236 seen in our studies is supported by the findings of Roux et al. [76], although these investigators did not show an involvement of p38 at this phosphorylation site, as reported in the present study and by other investigators [75]. Interestingly, p38, but not ERK1/2 signaling exert a regulatory effect on AktSer473 phosphorylation, yet inhibition of the MEK/ERK and p38 pathways in cells exposed to Francisella resulted in an increase in the level of pAktSer473, as compared with that seen by the inhibition of p38 only. While it has been previously reported that p38 regulates AktSer473 phosphorylation via MK2 (MAPKAPK-2), and that inhibition of p38 by SB203580 downregulates this process [77,78], how can we explain the increase in the level of pAktSer473 upon suppression of the MEK/ERK and p38 pathways? Menges et al. [79] showed that in cell arrest Raf via MEK/ERK inhibits Akt phosphorylation. Thus, in the presence of UO126, the ability of Raf to inhibit Akt phosphorylation would be lost. Indeed, inhibition of Raf in the presence of SB203580 resulted in increased levels of pAktSer473 (data not shown), hence supporting this notion. Given the reported involvement of p38 in the induction of G1/S and G2/M cell cycle checkpoints in response to stimuli [80-82], inhibition of p38 signaling could cause cell cycle arrest, and under these circumstances, Raf via MEK/ERK would have exerted a downregulatory effect on AktSer473 phosphorylation, as observed in our studies.
The involvement of phospholipases in phagocytic signaling has been well documented [83]. Phosphorylation of PLCγ1 was shown to be critical for the invasion of endothelial cells by E. coli [15]. The involvement of PLC, and specifically PLCγ1, in the regulation of the downstream targets of mTOR in primary cells in the context of a bacterial infection suggests that such modulation could account, at least partially, for the significant reduction in Francisella host cell entry when cells were pre-treated with the PLC inhibitor U73122. Since the cell permeable molecule BAPTA is enzymatically converted to the non-permeable, calcium chelator BAPTA inside the cell, only intracellular calcium is chelated [84]. Thus, our data suggests that PLC/PLCγ1 downstream signaling involved intracellular calcium, which also plays an important role in the invasion of F. tularensis. Furthermore, this inhibition was associated with a downregulated phosphorylation of mTORC1 downstream effectors as shown in other systems [17].
TLRs are highly conserved pattern recognition receptors and their recognition of pathogen associated molecular patterns induces the activation of host signal transduction pathways that result in an immune response that ultimately will aid in bacterial clearance [85]. However, in recent years, it has been reported that TLR signaling promotes phagocytic activity through the upregulation of genes involved in phagocytosis [86]. Moreover, antibodies to TLR4 inhibit internalization of E. coli by epithelial cells, and no bacteria could be recovered after pre-treatment with cytochalasin D or with antibodies against TLR4, suggesting that TLR4 participates in downstream signaling events that mediate bacterial internalization [87]. Furthermore, TLR2, but not TLR4, has been shown to be essential for efficient internalization of Aspergillus fumigatus conidia, since infection of macrophages deficient in TLR2 rendered reduced conidial cell entry [88]. Results of our studies show that TLR2 signaling is significantly implicated in the internalization of F. tularensis LVS into primary macrophages, and, in the context of our investigations, the involvement of downstream effector molecules of mTOR, as well as signaling molecules associated with their regulation via TLR2 signaling, are important for this process.
Overall, and to the best of our knowledge, our findings reveal for the first time that the integrity of the actin cytoskeleton architecture and the phosphorylation of mTOR downstream effector molecules via mTOR and PI3K signaling, are central for the invasion of F. tularensis LVS into primary murine macrophages through TLR2 signaling. Moreover, the phosphorylation of mTOR downstream proteins via PLC/Ca2+ and MAPKs signaling was also shown to play a role in Francisella invasion of macrophages (Figure S4).
Supporting Information
F. tularensis LVS grown in BHI or in MHB show similar bacterial host cell entry. (A) Peritoneal macrophages derived from WT mice were exposed to freshly harvested F. tularensis LVS (MOI=20) grown in Brain Hear Infusion broth (BHI) or in Muller-Hinton broth (MHB) for 90 min to assess bacterial invasion. (B) Peritoneal macrophages were exposed to F. tularensis LVS grown in BHI or MHB for 0-120 min and then lysed. Total p70S6K and phosphorylated p70S6K (Thr389 and Thr421/Ser424), 4E-BP1 (Ser65), S6 (Ser235/236 and Ser240/244) and eI-F4E (Ser209) were assessed by Western analysis. Samples analyzed contained an equal amount of protein. Unstimulated cells (time 0) served as negative controls. The gel is representative of three to five independent experiments.
(TIF)
mTOR siRNA decreased internalization of Francisella and downregulates phosphorylation of mTOR downstream signaling cascade. (A) RAW cells were transfected with control siRNA or with siRNA to mTOR (100 nM), and after 5 days, cells were washed, rested and infected with F. tularensis LVS for 90 min to assess bacterial invasion. Values are the mean ± SEM of 5 independent experiments, each done in triplicate; **p < 0.001; *p < 0.05 compared with infected control transfected with control siRNA. (B) RAW cells were transfected with control siRNA or with siRNA to mTOR (100 nM) or non-transfected, and after 5 days, cells were washed, rested, infected with F. tularensis LVS (MOI=20) for 0-90 min and then lysed. Total p70S6K and Akt, and phosphorylated p70S6K (Thr389 and Thr421/Ser424), 4E-BP1 (Ser65), S6 (Ser235/236 and Ser240/244), eI-F4E (Ser209) and Akt (Ser473) were assessed by Western analysis. Samples contained equal amount of protein. RAW cells transfected with control siRNA were used as negative controls. Gels are representative of three to five independent experiments.
(TIF)
Phosphorylation of p70S6K in RAW cells transfected with siRNA to Akt1/2. RAW cells were transfected or not with siRNA to Akt1/2 (100 nM), and after 5 days, cells were washed, rested, infected with F. tularensis LVS (MOI=20) for 0 and 120 min and lysed. Total and phosphorylated p70S6K (Thr389) and Akt (Ser473) was assessed by Western analysis. Samples analyzed contained equal amount of protein. Unstimulated cells served as negative controls. Gels are representative of three to five independent experiments.
(TIF)
Proposed model of the signaling pathways involved in the phosphorylation of mTOR downstream effector molecules associated with F. tularensis invasion of primary macrophages via TLR2 signaling.
(TIF)
Acknowledgments
The authors wish to thank Dr. Stefanie Vogel for her helpful suggestions during the course of these studies. The authors also wish to thank Dr. Tatjana Coric a post-doctoral fellow in the laboratory of Dr. Mary Ann Bjornsti for her instrumental guidance in our siRNA studies.
Funding Statement
The work was mainly supported by the United States Public Health Service grant AL-56460 (to SMM) and by National Institutes of Health T32 AI-007051(MWE). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Oyston PCF, Sjostedt A, Titball RW (2004) Tularemia: bioterrorism defence renews interest in Francisella tularensis . Nat Rev Microbiol 2: 967-978. doi: 10.1038/nrmicro1045. PubMed: 15550942. [DOI] [PubMed] [Google Scholar]
- 2. Santic M, Al-Khodor S, Kwaik YA (2010) Cell biology and molecular ecology of Francisella tularensis . Cell Microbiol 12: 129-139. doi: 10.1111/j.1462-5822.2009.01400.x. PubMed: 19863554. [DOI] [PubMed] [Google Scholar]
- 3. Clemens DL, Horwitz MA (2007) Uptake and intracellular fate of Francisella tularensis in human macrophages. Ann N Y Acad Sci 1105: 160-186. doi: 10.1196/annals.1409.001. PubMed: 17435118. [DOI] [PubMed] [Google Scholar]
- 4. Long GW, Oprandy R, Narayanan RB, Fortier AH, Porter KR et al. (1993) Detection of Francisella tularensis in blood by polymerase chain reaction. J Clin Microbiol 31: 152-154. PubMed: 8417022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Elkins KL, Cowley SC, Bosio CM (2003) Innate and adaptive immune responses to an intracellular bacterium Francisella tularensis live vaccine strain. Microbes Infect 5: 135-142. doi: 10.1016/S1286-4579(02)00084-9. PubMed: 12650771. [DOI] [PubMed] [Google Scholar]
- 6. May RC, Machesky LM (2001) Phagocytosis and the actin cytoskeleton. J Cell Sci 114: 1061-1077. PubMed: 11228151. [DOI] [PubMed] [Google Scholar]
- 7. Parsa KVL, Butchar JP, Rajaram MVS, Cremer TJ, Tridandapani S (2008) The tyrosine kinase Syk promotes phagocytosis of Francisella through the activation of Erk. Mol Immunol 45: 3012-3021. doi: 10.1016/j.molimm.2008.01.011. PubMed: 18295889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Leinweber BD, Leavis PC, Grabarek Z, Wang C-LA, Morgan KG (1999) Extracellular regulated kinase (ERK) interaction with actin and the calponin homology (CH) domain of actin-binding proteins. Biochem J 344: 117-123. doi: 10.1042/0264-6021:3440117. PubMed: 10548541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Crowley MT, Costello PS, Fitzer-Attas CJ, Turner M, Meng F et al. (1997) A critical role for Syk in signal transduction and phagocytosis mediated by Fcγ receptors on macrophages. J Exp Med 186: 1027-1039. doi: 10.1084/jem.186.7.1027. PubMed: 9314552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Martin TF (1998) Phosphoinositide lipids as signaling molecules: common themes for signal transduction, cytoskeletal regulation, and membrane trafficking. Annu Rev Cell Dev Biol 14: 231-264. doi: 10.1146/annurev.cellbio.14.1.231. PubMed: 9891784. [DOI] [PubMed] [Google Scholar]
- 11. Ip CKM, Wong AST (2012) p70 S6 kinase and actin dynamics. Spermatogenesis 2: 1-9. doi: 10.4161/spmg.19885. PubMed: 22553484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jacinto E, Loewith R, Schmidt A, Lin S, Rüegg MA et al. (2004) Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol 6: 1122-1128. doi: 10.1038/ncb1183. PubMed: 15467718. [DOI] [PubMed] [Google Scholar]
- 13. Sarbassov DD, Ali SM, Kim D-H, Guertin DA, Latek RR et al. (2004) Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol 14: 1296-1302. doi: 10.1016/j.cub.2004.06.054. PubMed: 15268862. [DOI] [PubMed] [Google Scholar]
- 14. Botelho RJ, Teruel M, Dierckman R, Anderson R, Wells A et al. (2000) Localized biphasic changes in phosphatidylinositol-4,5-biphosphate at sites of phagocytosis. J Cell Biol 151: 1353-1368. doi: 10.1083/jcb.151.7.1353. PubMed: 11134066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sukumaran SK, McNamara G, Prasadarao NV (2003) Escherichia coli K-1 interaction with human brain micro-vascular endothelial cells triggers phospholipase C-γ1 activation downstream of phosphatidylinositol 3-kinase. J Biol Chem 278: 45753-45762. doi: 10.1074/jbc.M307374200. PubMed: 12952950. [DOI] [PubMed] [Google Scholar]
- 16. Sukumaran SK, Prasadarao NV (2002) Regulation of protein kinase C in Escherichia coli K1 invasion of human brain microvascular endothelial cells. J Biol Chem 277: 12253-12262. doi: 10.1074/jbc.M110740200. PubMed: 11805101. [DOI] [PubMed] [Google Scholar]
- 17. Markova B, Albers C, Breitenbuecher F, Melo JV, Brümmendorf TH et al. (2010) Novel pathway in Bcr-Abl signal transduction involves Akt-independent, PLC-γ1-driven activation of mTOR/p70S6-kinase pathway. Oncogene 29: 739-751. doi: 10.1038/onc.2009.374. PubMed: 19881535. [DOI] [PubMed] [Google Scholar]
- 18. Oshiro N, Yoshino K-I, Hidayat S, Tokunaga C, Hara K et al. (2004) Dissociation of raptor from mTOR is a mechanism of rapamycin-induced inhibition of mTOR function. Genes Cells 9: 359-366. doi: 10.1111/j.1356-9597.2004.00727.x. PubMed: 15066126. [DOI] [PubMed] [Google Scholar]
- 19. Raught B, Gingraas AC, Sonenberg N (2001) The target of rapamycin (TOR). Proteins - Proc Natl Acad Sci U_S_A 98: 7037-7044. doi: 10.1073/pnas.121145898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sarbassov DD, Ali SM, Sabatini DM (2005) Growing roles for the mTOR pathway. Curr Opin Cell Biol 17: 596-603. doi: 10.1016/j.ceb.2005.09.009. PubMed: 16226444. [DOI] [PubMed] [Google Scholar]
- 21. Schmelzle T, Hall MN (2000) TOR, a central controller of cell growth. Cell 103: 253-262. doi: 10.1016/S0092-8674(00)00117-3. PubMed: 11057898. [DOI] [PubMed] [Google Scholar]
- 22. Wullschleger S, Loewith R, Hall MN (2006) TOR signaling in growth and metabolism. Cell 124: 471-484. doi: 10.1016/j.cell.2006.01.016. PubMed: 16469695. [DOI] [PubMed] [Google Scholar]
- 23. Coombes BK, Mahony JB (2002) Identification of MEK- and phosphoinositide 3-kinase-dependent signaling as essential events during Chlamydia pneumoniae invasion of HEp2 cells. Cell Microbiol 4: 447-460. doi: 10.1046/j.1462-5822.2002.00203.x. PubMed: 12102690. [DOI] [PubMed] [Google Scholar]
- 24. Kierbel A, Gassama-Diagne A, Mostov K, Engel JN (2005) The phosphoinositol-3-kinase-protein kinase B/Akt pathway is critical for Pseudomonas aeruginosa strain PAK internalization. Mol Biol Cell 16: 2577-2585. doi: 10.1091/mbc.E04-08-0717. PubMed: 15772151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ohira T, Arita M, Omori K, Recchiuti A, Van Dyke TE et al. (2010) Resolvin E1 receptor activation signals phosphorylation and phagocytosis. J Biol Chem 285: 3451-3461. doi: 10.1074/jbc.M109.044131. PubMed: 19906641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen J, Zheng XF, Brown EJ, Schreiber SL (1995) Identification of an 11-kDa FKBP12-rapamycin-binding domain within the 289-kDa FKBP12-rapamycin-associated protein and characterization of a critical serine residue. Proc Natl Acad Sci U S A 92: 4947-4951. doi: 10.1073/pnas.92.11.4947. PubMed: 7539137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huang S, Bjornsti M-A, Houghton PJ (2003) Rapamycins: mechanism of action and cellular resistance. Cancer Biol Ther 2: 222-232. PubMed: 12878853. [DOI] [PubMed] [Google Scholar]
- 28. Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR et al. (2002) mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110: 163-175. doi: 10.1016/S0092-8674(02)00808-5. PubMed: 12150925. [DOI] [PubMed] [Google Scholar]
- 29. Hara K, Maruki Y, Long X, Yoshino K, Oshiro N et al. (2002) Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110: 177-189. doi: 10.1016/S0092-8674(02)00833-4. PubMed: 12150926. [DOI] [PubMed] [Google Scholar]
- 30. Nojima H, Tokunaga C, Eguchi S, Oshiro N, Hidayat S et al. (2003) The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS). Motif - J Biol Chem 278: 15461-15464. doi: 10.1074/jbc.C200665200. [DOI] [PubMed] [Google Scholar]
- 31. Sarbassov DD, Ali SM, Sengupta S, Sheen J-H, Hsu PP et al. (2006) Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 22: 159-168. doi: 10.1016/j.molcel.2006.03.029. PubMed: 16603397. [DOI] [PubMed] [Google Scholar]
- 32. Ireton K, Payrastre B, Chap H, Ogawa W, Sakaue H et al. (1996) A role for phosphoinositide 3-kinase in bacterial invasion. Science 274: 780-782. doi: 10.1126/science.274.5288.780. PubMed: 8864117. [DOI] [PubMed] [Google Scholar]
- 33. Kwok T, Backert S, Schwarz H, Berger J, Meyer TF (2002) Specific entry of Helicobacter pylori into cultured gastric, epithelial cells via a zipper-like mechanism. Infect Immun 70: 2108-2120. doi: 10.1128/IAI.70.4.2108-2120.2002. PubMed: 11895977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Reddy MA, Prasadarao NV, Wass CA, Kim KS (2000) Phosphatidylinositol 3-kinase activation and interaction with focal adhesion kinase in Escherichia coli K1 invasion of human brain microvascular endothelial cells. J Biol Chem 275: 36769-36774. doi: 10.1074/jbc.M007382200. PubMed: 10973983. [DOI] [PubMed] [Google Scholar]
- 35. Rameh LE, Rhee SG, Spokes K, Kazlauskas A, Cantley LC et al. (1998) Phosphoinositide 3-kinase regulates phospholipase C-γ mediated calcium signaling. J Biol Chem 273: 23750-23757. doi: 10.1074/jbc.273.37.23750. PubMed: 9726983. [DOI] [PubMed] [Google Scholar]
- 36. Eguchi S, Iwasaki H, Ueno H, Frank GD, Motley ED et al. (1999) Intracellular signaling of angiotensin II-induced p70 S6 kinase phosphorylation at Ser411 in vascular smooth muscle cells. J Biol Chem 274: 36843-36851. doi: 10.1074/jbc.274.52.36843. PubMed: 10601235. [DOI] [PubMed] [Google Scholar]
- 37. Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP (2005) Phosphorylation and functional inactivation of TSC2 by Erk: Implications for tuberous sclerosis and cancer pathogenesis. Cell 121: 179-193. doi: 10.1016/j.cell.2005.02.031. PubMed: 15851026. [DOI] [PubMed] [Google Scholar]
- 38. Shi YS, Hsu J-H, Gera J, Lichtenstein A (2002) Signal pathways involved in activation of p70S6K and phosphorylation of 4E-BP1 following exposure of multiple myeloma tumor cells to interleukin-6. J Biol Chem 277: 15712-15720. doi: 10.1074/jbc.M200043200. PubMed: 11872747. [DOI] [PubMed] [Google Scholar]
- 39. Tokuda H, Hatakeyama D, Shibata T, Akamatsu S, Oiso Y et al. (2003) p38 MAP kinase regulates BMP-4-stimulated VEGF synthesis via p70 S6 kinase in osteoblasts. Am J Physiol Endocrinol Metab 284: E1202-E1209. PubMed: 12637256. [DOI] [PubMed] [Google Scholar]
- 40. Wang X, Flynn A, Waskiewicz AJ, Webb BLJ, Vries RG et al. (1998) The phosphorylation of eukaryotic initiation factor eIF4E in response to phorbol esters, cell stresses, and cytokines is mediated by distinct MAP kinase pathways. J Biol Chem 273: 9373-9377. doi: 10.1074/jbc.273.16.9373. PubMed: 9545260. [DOI] [PubMed] [Google Scholar]
- 41. Elkins KL, Winegar RK, Nacy CA, Fortier AH (1992) Introduction of Francisella tularensis at skin sites induces resistance to infection and generation of protective immunity. Microb Pathog 13: 417-421. doi: 10.1016/0882-4010(92)90085-3. PubMed: 1297917. [DOI] [PubMed] [Google Scholar]
- 42. Katz J, Zhang P, Martin M, Vogel SN, Michalek SM (2006) Toll-like receptor 2 is required for inflammatory responses to Francisella tularensis LVS. Infect Immun 74: 2809-2816. doi: 10.1128/IAI.74.5.2809-2816.2006. PubMed: 16622218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang P, Katz J, Michalek SM (2009) Glycogen synthase kinase-3β (GSK3β) inhibition suppresses the inflammatory response to Francisella infection and protects against tularemia in mice. Mol Immunol 46: 677-687. doi: 10.1016/j.molimm.2008.08.281. PubMed: 18929413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang P, Martin M, Michalek SM, Katz J (2005) Role of mitogen-activated protein kinases and NF-κB in the regulation of proinflammatory and anti-inflammatory cytokines by Porphyromonas gingivalis hemagglutinin B. Infect Immun 73: 3990-3998. doi: 10.1128/IAI.73.7.3990-3998.2005. PubMed: 15972486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cowley SC, Meierovics AI, Frelinger JA, Iwakura Y, Elkins KL (2010) Lung CD4-CD8- double negative T cells are prominent producers of IL-17A and IFN-γ during primary respiratory murine infection with Francisella tularensis Live Vaccine Strain. J Immunol 184: 5791-5801. doi: 10.4049/jimmunol.1000362. PubMed: 20393138. [DOI] [PubMed] [Google Scholar]
- 46. Hazlett KRO, Caldon SD, McArthur DG, Cirillo KA, Kirimanjeswara GS et al. (2008) Adaptation of Francisella tularensis to the mammalian environment is governed by cues which can be mimicked in vitro. Infect Immun 76: 4479-4488. doi: 10.1128/IAI.00610-08. PubMed: 18644878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Loegering DJ, Drake JR, Banas JA, McNealy TL, Mc Arthur DG et al. (2006) Francisella tularensis LVS grown in macrophages has reduced ability to stimulate the secretion of inflammatory cytokines by macrophages in vitro. Microb Pathog 41: 218-225. doi: 10.1016/j.micpath.2006.07.007. PubMed: 16996713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hay N, Sonenberg N (2004) Upstream and downstream of mTOR. Genes Dev 18: 1926-1945. doi: 10.1101/gad.1212704. PubMed: 15314020. [DOI] [PubMed] [Google Scholar]
- 49. Inoki K, Guan KL (2006) Complexity of the TOR signaling network. Trends Cell Biol 16: 206-212. doi: 10.1016/j.tcb.2006.02.002. PubMed: 16516475. [DOI] [PubMed] [Google Scholar]
- 50. Gingras A-C, Gygi SP, Raught B, Polakiewics RD, Abraham RT et al. (1999) Regulation of 4E-BP1 phosphorylation : A novel two step mechanism. Genes and Dev 13: 1422-1437. doi: 10.1101/gad.13.11.1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gingras A-C, Raught B, Gygi SP, Niedzwiecka A, Miron M et al. (2001) Hierarchical phosphorylation of the translational inhibitor 4E-BP1. Genes and Dev 15: 2852-2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pyronnet S (2000) Phosphorylation of the cap-binding protein eIF4E by the MAPK-activated protein kinase Mnk1. Biochem Pharmacol 60: 1237-1243. doi: 10.1016/S0006-2952(00)00429-9. PubMed: 11007962. [DOI] [PubMed] [Google Scholar]
- 53. Corradetti MN, Guan K-L (2006) Upstream of the mammalian target of rapamycin: do all roads pass through mTOR? Oncogene 25: 6347-6360. doi: 10.1038/sj.onc.1209885. PubMed: 17041621. [DOI] [PubMed] [Google Scholar]
- 54. Dann SG, Selvaraj A, Thomas G (2007) mTOR Complex1-S6K1 signaling: at the crossroads of obesity, diabetes and cancer. Trends Mol Med 13: 252-259. doi: 10.1016/j.molmed.2007.04.002. PubMed: 17452018. [DOI] [PubMed] [Google Scholar]
- 55. Schmitz F, Heit A, Dreher S, Eisenächer K, Mages J et al. (2008) Mammalian target of rapamycin (mTOR) orchestrates the defense program of innate immune cells. Eur J Immunol 38: 2981-2992. doi: 10.1002/eji.200838761. PubMed: 18924132. [DOI] [PubMed] [Google Scholar]
- 56. Chen X-G, Liu F, Song X-F, Wang Z-H, Dong Z-Q et al. (2010) Rapamycin regulates Akt and ERK phosphorylation through mTORC1 and mTORC2 signaling pathways. Mol Carcinog 49: 603-610. PubMed: 20512842. [DOI] [PubMed] [Google Scholar]
- 57. Li Q, Zhao WD, Zhang K, Fang WG, Hu Y et al. (2010) PI3K-dependent host cell actin rearrangements are required for Cronobacter sakazakii invasion of human brain microvascular endothelial cells. Med Microbiol Immunol 199: 333-340. doi: 10.1007/s00430-010-0168-8. PubMed: 20809254. [DOI] [PubMed] [Google Scholar]
- 58. Gulhati P, Bowen KA, Liu J, Stevens PD, Rychahow PG et al. (2011) mTORC1 and mTORC2 regulate EMT, motility and metastasis of colorectal cancer via RhoA and Rac1 signaling pathways. Cancer Res 7: 3246-3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wong R, Fabian L, Forer A, Brill JA (2007) Phospholipase C and myosin light chain kinase inhibition define a common step in actin regulation during cytokinesis. BMC Cell Biol 8: 15-24. doi: 10.1186/1471-2121-8-15. PubMed: 17509155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kim MJ, Kim E, Ryu SH, Suh P-G (2000) The mechanism of phospholipase Cγ1 regulation. Exp Mol Med 32: 101-109. doi: 10.1038/emm.2000.18. PubMed: 11048639. [DOI] [PubMed] [Google Scholar]
- 61. Rebecchi MJ, Pentyala SN (2000) Structure, function, and control of phosphoinositide-specific phospholipase C. Physiol Rev 80: 1291-1335. PubMed: 11015615. [DOI] [PubMed] [Google Scholar]
- 62. Guay J, Lambert H, Gingras-Breton G, Lavoie JN, Huot J et al. (1997) Regulation of actin filament dynamics by p38 map kinase-mediated phosphorylation of heat shock protein 27. J Cell Sci 110: 357-368. PubMed: 9057088. [DOI] [PubMed] [Google Scholar]
- 63. Rydkina E, Turpin LC, Sahni SK (2008) Activation of p38 mitogen-activated protein kinase module facilitates in vitro host cell invasion by Rickettsia rickettsii . J Med Microbiol 57: 1172-1175. doi: 10.1099/jmm.0.47806-0. PubMed: 18719192. [DOI] [PubMed] [Google Scholar]
- 64. Weng QP, Andrabi K, Kozlowski MT, Grove JR, Avruch J (1995) Multiple independent inputs are required for activation of p70 S6 kinase. Mol Cell Biol 15: 2333-2340. PubMed: 7739516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Li H, Nookala S, Bina RX, Bina JE et al. (2006) Innate immune response to Francisella tularensis is mediated by TLR2 and caspase-1 activation. J Leukoc Biol 80: 766-773. doi: 10.1189/jlb.0406294. PubMed: 16895974. [DOI] [PubMed] [Google Scholar]
- 66. Berven LA, Willard FS, Crouch MF (2004) Role of the p70S6K pathway in regulating the actin cytoskeleton and cell migration. Exp Cell Res 296: 183-195. doi: 10.1016/j.yexcr.2003.12.032. PubMed: 15149849. [DOI] [PubMed] [Google Scholar]
- 67. Ip CKM, Cheung ANY, Ngan HYS, Wong AST (2011) p70 S6 kinase in the control of actin cytoskeleton dynamics and directed migration of ovarian cancer cells. Oncogene 30: 2420-2432. doi: 10.1038/onc.2010.615. PubMed: 21258406. [DOI] [PubMed] [Google Scholar]
- 68. Liu L, Chen L, Chung J, Huang S (2008) Rapamycin inhibits F-actin reorganization and phosphorylation of focal adhesion proteins. Oncogene 27: 4998-5010. doi: 10.1038/onc.2008.137. PubMed: 18504440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ganesan LP, Wei R, Pengal RA, Moldovan L, Moldovan N et al. (2004) The serine/threonine kinase Akt promotes Fcγ receptor-mediated phagocytosis in murine macrophages through the activation of p70S6 kinase. J Biol Chem 279: 54416-54425. doi: 10.1074/jbc.M408188200. PubMed: 15485887. [DOI] [PubMed] [Google Scholar]
- 70. Wlodarski P, Kasprzycka M, Liu X, Marzec M, Robertson ES et al. (2005) Activation of mammalian target of rapamycin in transformed B lymphocytes in nutrient dependent but independent of Akt, mitogen-activated protein kinase/extracellular signal-regulated kinase kinase, insulin growth factor-I, and serum. Cancer Res 65: 7800-7808. PubMed: 16140948. [DOI] [PubMed] [Google Scholar]
- 71. Arvisais EW, Romanelli A, Hou X, Davis JS (2006) AKT-independent phosphorylation of TSC2 and activation of mTOR and ribosomal protein S6 kinase signaling by prostaglandin F2α. J Biol Chem 28: 26904-26913. [DOI] [PubMed] [Google Scholar]
- 72. Genolet R, Araud T, Maillard L, Jaquier-Gubler P, Curran J (2008) An approach to analyse the specific impact of rapamycin on mRNA-ribosome association. BMC Med Genomics 1: 33-44. doi: 10.1186/1755-8794-1-33. PubMed: 18673536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Biondi RM, Kieloch A, Currie RA, Deak M, Alessi DR (2001) The PIF-binding pocket in PDK1 is essential for activation of S6K and SGK, but not PKB. EMBO J 20: 4380-4390. doi: 10.1093/emboj/20.16.4380. PubMed: 11500365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhang Y, Dong Z, Nomura M, Zhong S, Chen N et al. (2001) Signal transduction pathways involved in phosphorylation and activation of p70S6K following exposure to UVA irradiation. J Biol Chem 276: 20913-20923. doi: 10.1074/jbc.M009047200. PubMed: 11279232. [DOI] [PubMed] [Google Scholar]
- 75. Casas-Terradellas E, Tato I, Bartrons R, Ventura F, Rosa JL (2008) ERK and p38 pathways regulate amino acid signaling. Biochim Biophys Acta 1783: 2241-2254. doi: 10.1016/j.bbamcr.2008.08.011. PubMed: 18809440. [DOI] [PubMed] [Google Scholar]
- 76. Roux PP, Shahbazian D, Vu H, Holz MK, Cohen MS et al. (2007) RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates Cap-dependent translation. J Biol Chem 282: 14056-14064. doi: 10.1074/jbc.M700906200. PubMed: 17360704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hu T, Wang Y, Graham WV, Su L, Musch MW et al. (2006) MAPKAPK-2 is a critical signaling intermediate in NHE3 activation following Na+-glucose cotransport. J Biol Chem 281: 24247-24253. doi: 10.1074/jbc.M602898200. PubMed: 16793766. [DOI] [PubMed] [Google Scholar]
- 78. Rane MJ, Coxon PY, Powell DW, Webster R, Klein JB et al. (2001) p38 kinase-dependent MAPKAPK-2 activation functions as 3-phosphoinositide-dependent kinase-2 for Akt in human neutrophils. J Biol Chem 276: 3517-3523. doi: 10.1074/jbc.M005953200. PubMed: 11042204. [DOI] [PubMed] [Google Scholar]
- 79. Menges CW, McCance DJ (2008) Constitutive activation of the Raf-MAPK pathway causes negative feedback inhibition of Ras-PI3K-Akt and cellular arrest through EphA2 receptor. Oncogene 27: 2934-2940. doi: 10.1038/sj.onc.1210957. PubMed: 18059341. [DOI] [PubMed] [Google Scholar]
- 80. Bulavin DV, Amundson SA, Fornace AJ (2002) p38 and Chk1 kinases: different conductors for the G(2)/M checkpoint symphony. Curr Opin Genet Dev 12: 92-97. doi: 10.1016/S0959-437X(01)00270-2. PubMed: 11790561. [DOI] [PubMed] [Google Scholar]
- 81. Casanovas O, Miró F, Estanyol JM, Itarte E, Agell N et al. (2000) Osmotic stress regulates the stability of cyclin D1 in a p38SAPK2-dependent manner. J Biol Chem 275: 35091-35097. doi: 10.1074/jbc.M006324200. PubMed: 10952989. [DOI] [PubMed] [Google Scholar]
- 82. Thornton TM, Rincon M (2009) Non-classical p38 map kinase functions: cell cycle checkpoints and survival. Int J Biol Sci 5: 44-51. PubMed: 19159010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Lennartz MR (1999) Phospholipases and phagocytosis: the role of phospholipid-derived second messengers in phagocytosis. Int J Biochem Cell Biol 31: 415-430. doi: 10.1016/S1357-2725(98)00108-3. PubMed: 10224668. [DOI] [PubMed] [Google Scholar]
- 84. Ko Y, Cho NH, Cho BA, Kim IS, Choi MS (2011) Involvement of Ca2+ signaling in intracellular invasion of non-phagocytic host cells by Orientia tsutsugamushi . Microb Pathog 50: 326-330. doi: 10.1016/j.micpath.2011.02.007. PubMed: 21362468. [DOI] [PubMed] [Google Scholar]
- 85. Janeway CA Jr, Medzhitov R (2002) Innate immune recognition. Annu Rev Immunol 20: 197-216. doi: 10.1146/annurev.immunol.20.083001.084359. PubMed: 11861602. [DOI] [PubMed] [Google Scholar]
- 86. Doyle SE, O'Connell RM, Miranda GA, Vaidya SA, Chow EK et al. (2004) Toll-like receptors induce a phagocytic gene program through p38. J Exp Med 199: 81-90. PubMed: 14699082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Neal MD, Leaphart C, Levy R, Prince J, Billiar TR et al. (2006) Enterocyte TLR4 mediates phagocytosis and translocation of bacteria across the intestinal barrier. J Immunol 176: 3070-3079. PubMed: 16493066. [DOI] [PubMed] [Google Scholar]
- 88. Luther K, Torosantucci A, Brakhage AA, Heesemann J, Ebel F (2007) Phagocytosis of Aspergillus fumigatus conidia by murine macrophages involves recognition by the dectin-1 beta-glucan receptor and Toll-like receptor 2. Cell Microbiol 9: 368-381. doi: 10.1111/j.1462-5822.2006.00796.x. PubMed: 16953804. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
F. tularensis LVS grown in BHI or in MHB show similar bacterial host cell entry. (A) Peritoneal macrophages derived from WT mice were exposed to freshly harvested F. tularensis LVS (MOI=20) grown in Brain Hear Infusion broth (BHI) or in Muller-Hinton broth (MHB) for 90 min to assess bacterial invasion. (B) Peritoneal macrophages were exposed to F. tularensis LVS grown in BHI or MHB for 0-120 min and then lysed. Total p70S6K and phosphorylated p70S6K (Thr389 and Thr421/Ser424), 4E-BP1 (Ser65), S6 (Ser235/236 and Ser240/244) and eI-F4E (Ser209) were assessed by Western analysis. Samples analyzed contained an equal amount of protein. Unstimulated cells (time 0) served as negative controls. The gel is representative of three to five independent experiments.
(TIF)
mTOR siRNA decreased internalization of Francisella and downregulates phosphorylation of mTOR downstream signaling cascade. (A) RAW cells were transfected with control siRNA or with siRNA to mTOR (100 nM), and after 5 days, cells were washed, rested and infected with F. tularensis LVS for 90 min to assess bacterial invasion. Values are the mean ± SEM of 5 independent experiments, each done in triplicate; **p < 0.001; *p < 0.05 compared with infected control transfected with control siRNA. (B) RAW cells were transfected with control siRNA or with siRNA to mTOR (100 nM) or non-transfected, and after 5 days, cells were washed, rested, infected with F. tularensis LVS (MOI=20) for 0-90 min and then lysed. Total p70S6K and Akt, and phosphorylated p70S6K (Thr389 and Thr421/Ser424), 4E-BP1 (Ser65), S6 (Ser235/236 and Ser240/244), eI-F4E (Ser209) and Akt (Ser473) were assessed by Western analysis. Samples contained equal amount of protein. RAW cells transfected with control siRNA were used as negative controls. Gels are representative of three to five independent experiments.
(TIF)
Phosphorylation of p70S6K in RAW cells transfected with siRNA to Akt1/2. RAW cells were transfected or not with siRNA to Akt1/2 (100 nM), and after 5 days, cells were washed, rested, infected with F. tularensis LVS (MOI=20) for 0 and 120 min and lysed. Total and phosphorylated p70S6K (Thr389) and Akt (Ser473) was assessed by Western analysis. Samples analyzed contained equal amount of protein. Unstimulated cells served as negative controls. Gels are representative of three to five independent experiments.
(TIF)
Proposed model of the signaling pathways involved in the phosphorylation of mTOR downstream effector molecules associated with F. tularensis invasion of primary macrophages via TLR2 signaling.
(TIF)