

Abstract
Homo-pentameric Cys-loop receptors contain five identical agonist binding sites, each formed at a subunit interface. To determine the number and locations of binding sites required to generate a stable active state, we constructed a receptor subunit with a mutation that disables the agonist binding site and a reporter mutation that alters unitary conductance and coexpressed mutant and nonmutant subunits. Although receptors with a range of different subunit compositions are produced, patch-clamp recordings reveal that the amplitude of each single-channel opening event reports the number and, for certain subunit combinations, the locations of subunits with intact binding sites. We find that receptors with three binding sites at nonconsecutive subunit interfaces exhibit maximal mean channel open time, receptors with binding sites at three consecutive or two nonconsecutive interfaces exhibit intermediate open time, and receptors with binding sites at two consecutive or one interface exhibit brief open time. Macroscopic recordings after rapid application of agonist reveal that channel activation slows and the extent of desensitization decreases as the number of binding sites per receptor decreases. The overall results provide a framework for defining mechanisms of activation and drug modulation for homo-pentameric Cys-loop receptors.
Introduction
Neurotransmitter receptors of the Cys-loop superfamily mediate rapid synaptic transmission throughout the nervous system and include receptors activated by ACh, GABA, glycine, and serotonin (Lester et al., 2004; Sine and Engel, 2006). They contain five homologous subunits arranged as barrel staves around a central ion-conducting pore (Unwin, 2005). Approximately one-half of each subunit is extracellular, with the remainder comprising four transmembrane domains and a large cytoplasmic domain. The neurotransmitter binding sites are formed at interfaces between extracellular regions of the subunits; one face of each binding site, called the principal face, projects predominantly aromatic residues into the site, whereas the opposing complementary face projects aromatic, hydrophobic, and negatively charged residues (Karlin, 2002; Sine, 2002).
Cys-loop receptors assemble from five copies of one type of subunit, giving rise to homomeric receptors, or, more commonly, from several types of subunits, giving rise to heteromeric receptors. Homomeric receptors are the simplest structural class of Cys-loop receptors and contain five identical agonist binding sites. Most present-day Cys-loop receptors are heteromeric and contain two agonist binding sites, whereas a third site may bind heterotropic ligands (Cromer et al., 2002; Hsiao et al., 2008).
Present-day homomeric Cys-loop receptors likely descended from a homomeric bacterial counterpart (Tasneem et al., 2005) and thus represent a model system to examine structural and mechanistic constraints under which heteromeric receptors evolved to use fewer than five agonist binding sites. Macroscopic dose–response and single-channel kinetic analyses of homomeric Cys-loop receptors suggest that two to five agonist molecules are required for maximal activation (Amin and Weiss, 1996; Palma et al., 1996; Papke et al., 2000; Mott et al., 2001; Gentet and Clements, 2002; Beato et al., 2002, 2004; Solt et al., 2007). However, because dose–response and single-channel measurements cannot directly reveal the number and locations of functional binding sites, this remains a crucial gap in our mechanistic understanding of homomeric Cys-loop receptors.
To fill this gap, we studied a model homomeric Cys-loop receptor composed of the ligand binding domain from the α7 nicotinic receptor and pore and cytoplasmic domains from the 5-HT3A receptor (Palma et al., 1996; Bouzat et al., 2004, 2008; Rayes et al., 2005). To vary the number of agonist binding sites, we installed mutations that prevent agonist binding and, to report the presence of the mutant subunit, installed mutations that alter the single-channel conductance (Kelley et al., 2003). After coexpressing mutant and nonmutant subunits and recording agonist-evoked single-channel currents, the amplitude of each channel opening event reports the number of intact binding sites, whereas the dwell time indicates the stability of the open channel. An analogous approach used coexpression of mutant and nonmutant potassium channel subunits together with macroscopic current recordings to determine the number of subunits underlying inactivation (Mackinnon et al., 1993). Knowing the number of functionally relevant agonist binding sites is essential to understanding the mechanism behind activation of homomeric Cys-loop receptors, whereas the locations of the sites give insight into global conformational changes that open the channel.
Materials and Methods
Site-directed mutagenesis and expression of α7–5HT3A receptors.
Mutant subunits were constructed using the QuikChange site-directed mutagenesis kit (Stratagene) and were confirmed by sequencing the entire cDNA insert. The high conductance form (HC) of the α7–5HT3A chimeric receptor (Eiselé et al., 1993) was constructed as described previously (Rayes et al., 2005). In brief, three arginine residues responsible for the low conductance of the 5HT3A receptor were mutated to glutamine (Q), aspartic acid (D), and alanine (A) (Kelley et al., 2003). BOSC cells were transfected with subunit cDNAs using calcium phosphate precipitation. For a 35 mm dish of cells, the total amount of cDNA was 5 μg, and the ratios of control and mutant subunit cDNAs used in the transfection are described in the text for each specific experiment. Cells were used for single-channel or macroscopic current measurements 1 or 2 d after transfection (Bouzat et al., 1994, 2000, 2002).
Competition of ACh against the initial rate of α-toxin binding.
Transfected cells were harvested by gentle agitation in PBS, centrifuged at 1000 × g for 1 min, and resuspended in potassium Ringer's solution (140 mm KCl, 5.4 mm NaCl, 1.8 mm CaCl2, 1.7 mm MgCl2, 25 mm HEPES, 30 mg/L bovine serum albumin, adjusted to pH 7.4 with 10–11 mm NaOH). Competition of specified concentrations of ACh against the initial rate of 125I-α-bungarotoxin (α-BTX) binding was done as previously described (Sine et al., 1995). The total number of binding sites was determined by incubating cells with 25 nm 125I-α-BTX for 1 h and subtracting a blank determined in the presence of 1 mm d-tubocurarine. After computing fractional occupancy from the initial rates of toxin binding (Sine and Taylor, 1979), the following equation was fitted to the data: 1 − fractional occupancy = 1 − [L]nH/([L]nH + KdnH), where [L] is agonist concentration, Kd is the apparent dissociation constant, and nH is the Hill coefficient.
Single-channel patch-clamp recordings and kinetic analysis.
Recordings were obtained in the cell-attached patch configuration. The bath and pipette solutions contained 142 mm KCl, 5.4 mm NaCl, 0.2 mm CaCl2, and 10 mm HEPES, pH 7.4. Solutions free of magnesium and with low calcium were used to minimize channel block by divalent cations (Rayes et al., 2005). Single-channel currents were recorded and low-pass filtered to 10 kHz using an Axopatch 200 B patch-clamp amplifier (Molecular Devices), digitized at 5 μs intervals, and detected by the half-amplitude threshold criterion using the program TAC (Bruxton Corporation) (Bouzat et al., 2004; Rayes et al., 2005). Open-time histograms were fitted by the sum of exponential functions by maximum likelihood using the program TACFit (Bruxton Corporation).
To define amplitude classes, an analysis of the whole recording was performed by tracking events without any amplitude restriction. Amplitude histograms were then constructed and the different amplitude classes were distinguished. At least 10 different recordings from cells transfected with different ratios of HC and LC subunits were analyzed to define the mean ± SD of each amplitude class.
To determine the mean duration of each amplitude class, the analysis was performed in two different ways. In the first, all opening events were detected without any restriction of amplitude as described above. Open time histograms were then constructed for a given amplitude class by selecting only openings with amplitudes of ±0.6 pA of that of the mean of the amplitude class. This procedure involved the detection of all events in the whole recording in only one step. In the second way, only channel openings whose amplitudes were between ±0.6 pA of that of the mean amplitude class under study were accepted during the detection. This was performed by setting the detection bar to a fixed amplitude corresponding to the mean of the class of interest, then detecting channel events by the half-amplitude threshold criterion, and finally manually discarding the event if the amplitude differed by >0.6 pA of that of the mean amplitude class under study. The corresponding duration histogram was then constructed from the accepted events. Thus, this procedure involves analyzing the recording all the way through for each amplitude class. No significant differences in the mean open and burst durations were observed between the two types of analyses.
To recognize bursts and quantify their durations, a critical closed time (τcrit) was defined as the point of intersection between the second briefest and the succeeding components, and openings separated by closings briefer than this time constitute a burst (Rayes et al., 2005). For most of the recordings, τcrit ranged from 1 to 2 ms. Burst duration histograms are well described by the sum of three exponentials, with the two briefest components corresponding to isolated events and the longest to bursts. Comparison of burst and open duration histograms reveals that, on forming bursts, the mean durations of the brief components are unchanged, but that of the long component increases.
Bursts containing two or more openings were selected on the basis of distributions of mean open channel duration and open probability (Wang et al., 1997; Bouzat et al., 2002; Rayes et al., 2005). Bursts with mean open durations and open probabilities within 2 SDs of the mean of the major component were selected for additional analyses (Rayes et al., 2005). Typically, >70% of bursts were selected. No significant changes were observed in the burst duration calculated before or after the selection procedure.
The kinetic analysis was restricted to events within bursts of channel openings. The resulting open and closed intervals from selected bursts were analyzed according to a kinetic scheme (scheme 1) using an interval-based maximum likelihood algorithm that corrects for missed events and computes error limits of the fitted rate constants (Qin et al., 1996) (QuB Suite; www.qub.buffalo.edu; State University of New York, Buffalo, NY). A dead time of 30 μs was imposed in all recordings. Probability density functions of open and closed durations were calculated from the fitted rate constants and instrumentation dead time and superimposed on the experimental dwell time histogram as described by Qin et al. (1996).
Simulation of single-channel and macroscopic currents were performed using QuB software based on kinetic schemes using the experimentally determined rate constants.
Macroscopic current recordings.
For whole-cell recordings, the perfusion system consisted of solution reservoirs, manual switching valves, a solenoid-driven pinch valve, and two tubes (inner diameter, 0.3 mm) oriented at 90° inserted into the culture dish [modified from Liu and Dilger (1991)]. One tube contained extracellular solution (ECS) without agonist (normal solution) and the other contained ECS with different concentrations of ACh (test solution). A series of 2 s pulses of ECS containing ACh were applied at 15 s intervals. The pipette solution contained 134 mm KCl, 5 mm EGTA, 1 mm MgCl2, and 10 mm HEPES, pH 7.3. The extracellular solution contained 150 mm NaCl, 5.6 mm KCl, 0.5 mm CaCl2, and 10 mm HEPES, pH 7.3. Macroscopic currents were recorded at an applied potential: −50 mV, filtered at 5 kHz, and digitized at 20 kHz. Data analysis was performed using the IgorPro software (Wavemetrics). Individual records were aligned at the point at which the current reached 50% of maximum, and the resulting records were aligned and summed. The solution exchange time was estimated by placing an open pipette at the cell position, and switching from normal bath solution to a diluted (1:1 with water) bath solution. Typical times vary between 1 and 2 ms.
k-means analysis.
Mean rise times of macroscopic currents were analyzed using the k-means clustering analysis (MacQueen, 1967). Briefly, the algorithm starts from a random initial clustering and then iterates by calculating the cluster centroids and reassigning elements to the cluster with the closest centroid until no more reassignments are possible. The aim is to find the clustering solution that minimizes the intracluster sum of distances. We performed this analysis considering a potential number of clusters ranging from 1 to 7. The optimal number of cluster (k) corresponds to the lowest number that allows a minimal intracluster SSQ (sum of squared deviations).
Results
Discrete single-channel amplitude classes report subunit stoichiometry
In principle, one can alter the number of intact agonist binding sites in a homo-pentameric receptor by installing a mutation that disables the binding site and coexpressing the mutant with a nonmutant subunit. The resulting population will consist of receptors with zero to five intact binding sites, but the individual receptors will be indistinguishable. What is needed is a means to directly register the stoichiometry of mutant and nonmutant subunits in individual receptors.
To determine subunit stoichiometry of individual receptors, we used an electrical fingerprinting strategy in which a mutation that disables the agonist binding site is generated in a subunit that also contains a mutation that alters the single-channel current amplitude. By combining mutant and nonmutant subunits, and monitoring single-channel current amplitude, the number of functional binding sites in the receptor that elicited each channel opening event can be determined.
We took advantage of our previously described high-conductance (HC) form of the chimeric α7–5HT3A homomeric receptor (Bouzat et al., 2004; Rayes et al., 2005) that opens to a large and uniform single-channel current amplitude with only rare transitions to subconductance states (Fig. 1a). This chimera contains mutations of three Arg residues within the cytoplasmic domain (Kelley et al., 2003) that increase the unitary current amplitude from undetectable to ∼10 pA at a membrane potential of −120 mV.
Figure 1.
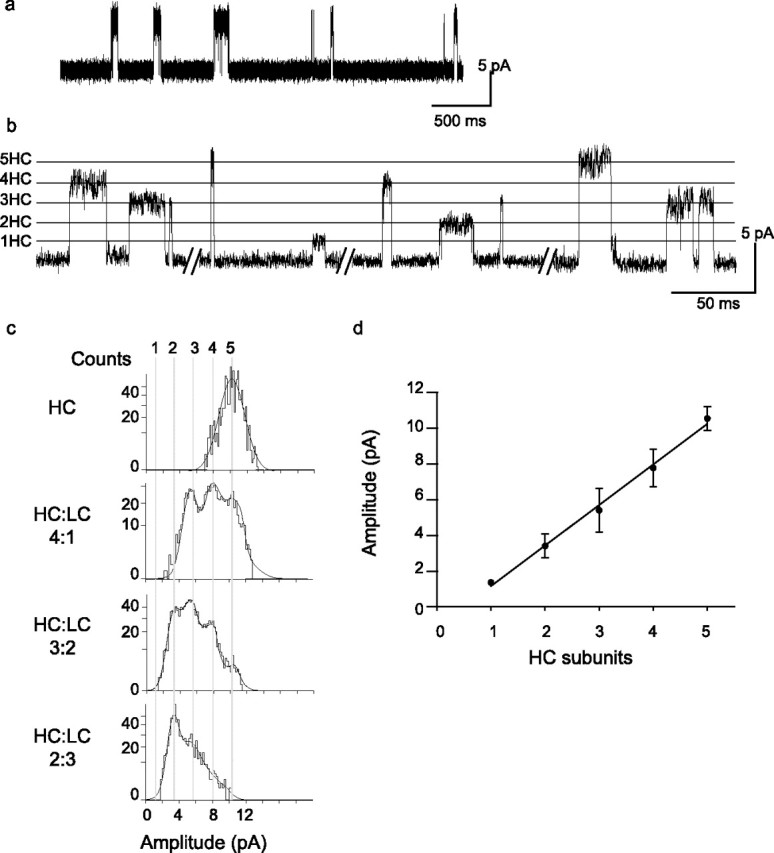
Discrete single-channel amplitude classes arise from receptors with different numbers of HC and LC subunits. a, Currents through the HC form of the α7–5HT3A chimeric receptor show a single amplitude class. b, Recordings from cells transfected with both HC and LC subunits. The traces are excerpts from the same recording. Each horizontal line indicates the number of HC subunits per receptor. c, Representative amplitude histograms obtained after detecting all opening events in a given recording. Changing the transfection ratios of HC and LC subunits changes the fraction of events in a given amplitude class. Vertical lines indicate the number of HC subunits in each amplitude class. Mean current amplitudes ± SD for the different amplitude classes are as follows: 10.4 ± 0.4, 8.2 ± 0.2, 5.6 ± 0.4, 3.6 ± 0.5, and 1.3 pA (n = 10 patches). For the smallest amplitude class, the mean amplitude was obtained from the sum of tables corresponding to 10 different recordings, and therefore it lacks SD. d, Plot of mean current amplitude (±SD) against number of HC subunits. A line with a slope of 2.27 pA/HC subunit is fitted to the data (r2 = 0.99). [ACh], 0.5–2 mm; membrane potential, −120 mV. The results are shown as mean ± SD of at least 10 patches for each condition.
After coexpressing high and low conductance forms of the α7–5HT3A chimera (designated HC and LC, respectively) in BOSC 23 cells, a saturating concentration of ACh elicits channel opening events with five discrete and equally spaced current amplitudes, one for each possible number of HC subunits per pentameric receptor (Fig. 1b–d). The observation of equally spaced current amplitudes indicates that the number of mutant subunits, but not the location of the subunits within the pentamer, governs the unitary current amplitude.
A second requirement is to ensure that the reporter mutations that decrease unitary current amplitude do not affect the stability of the open channel. We therefore classified each channel opening event based on its current amplitude and generated open duration histograms for each of the four largest amplitude classes (Fig. 2). Each amplitude class exhibits three exponential components of open times, but neither the time constants nor the relative areas of the components differ among amplitude classes (Fig. 2b). To ensure agonist occupied all intact binding sites, throughout this work we applied from 0.5 to 20 mm ACh to each test receptor and confirmed that the kinetic classes of channel openings remained unchanged.
Figure 2.
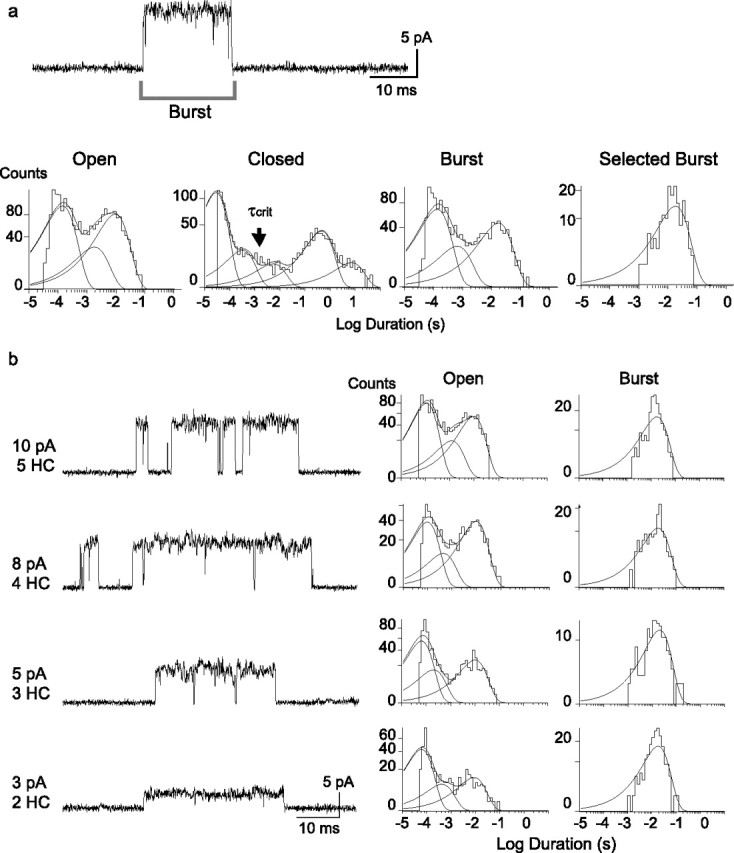
Single-channel lifetime is constant for different amplitude classes. a, Single-channel currents from cells transfected with the HC form of α7–5HT3A subunit were recorded from cell-attached patches at −120 mV. Channel openings occur as either isolated events or bursts of several events in quick succession. A representative burst activated by 2 mm ACh is shown. The open time histogram is fitted by three exponential components: O1 = 140 ± 30 μs, O2 = 1.5 ± 0.9 ms, and O3 = 8.9 ± 1.9 ms. The critical closed time (τcrit) for determining bursts is indicated by the arrow. The distribution of bursts consisting of one or more openings is well described by the sum of three exponentials, with the two briefest components corresponding to isolated events and the longest to bursts. The distribution of bursts selected to contain two or more openings is well described by a single exponential; mean durations of selected and nonselected bursts were 19.2 ± 4 and 16.5 ± 2.6 ms, respectively. b, Single-channel recordings from cells transfected with the HC form of α7–5HT3A (5 HC) or with different LC-to-HC subunit ratios. The traces correspond to bursts of channel openings for the indicated number of HC subunits and amplitude class. Open time histograms were constructed after selecting events corresponding to a given amplitude class. Burst duration histograms were constructed after selecting bursts as described in a. [ACh], 0.5–2 mm. Membrane potential, −120 mV.
The traces in Figures 1a and 2a show that single-channel openings appear as either isolated events or as bursts of several events in quick succession. Because the vast majority of current flows during bursts, we considered these to be the most functionally relevant, and examined mean burst durations for each amplitude class. To determine the mean burst duration, openings flanked by closings shorter than a defined critical time (τcrit) were identified, the resulting composite event was deemed a burst (Fig. 2a) (see Materials and Methods), and the duration of each burst was entered in a dwell time histogram. For the four largest amplitude classes, burst duration histograms are described by a single exponential function. Moreover, the mean burst durations are similar for each amplitude class (Table 1, lines b–d; Fig. 2b). Thus, although the reporter mutations alter the unitary current amplitude, the mean open and burst durations are unaffected.
Table 1.
Open channel lifetime and burst duration for receptors containing different numbers of binding sites
| No. sites | HC | LC | pA | O1 (ms) (area) | O2 (ms) (area) | O3 (ms) (area) | τburst (ms) | |
|---|---|---|---|---|---|---|---|---|
| a | 5 | C | 10 | 0.14 ± 0.03 (0.43 ± 0.07) | 1.5 ± 0.9 (0.18 ± 0.10) | 8.9 ± 1.9 (0.39 ± 0.10) | 16.5 ± 2.6 | |
| b | C | C | 10 | 0.15 ± 0.05 (0.36 ± 0.14) | 1.6 ± 0.6 (0.12 ± 0.02) | 7.4 ± 1.7 (0.55 ± 0.15) | 17.3 ± 1.9 | |
| c | C | C | 8 | 0.15 ± 0.01 (0.40 ± 0.07) | 1.4 ± 0.3 (0.17 ± 0.07) | 9.1 ± 2.0 (0.44 ± 0.03) | 18.7 ± 2.9 | |
| d | C | C | 5 | 0.13 ± 0.01 (0.62 ± 0.02) | 2.0 ± 0.8 (0.16 ± 0.06) | 9.8 ± 2.2 (0.27 ± 0.09) | 23.9 ± 2.6 | |
| e | 4 | C | Y190T | 8 | 0.14 ± 0.03 (0.31 ± 0.05) | 1.6 ± 0.4 (0.18 ± 0.08) | 7.2 ± 0.8 (0.50 ± 0.10) | 18.5 ± 2.1 |
| f | 3 | C | Y190T | 5 | 0.19 ± 0.04 (0.40 ± 0.08) | 1.4 ± 0.8 (0.25 ± 0.06) | 7.7 ± 0.6 (0.37 ± 0.03) | 20.8 ± 4.6 |
| g | 3 consecutive | C | Y190T/W55T | 8 | 0.15 ± 0.06 (0.30 ± 0.11) | 1.5 ± 0.8 (0.32 ± 0.23) | 5.3 ± 1.7 (0.39 ± 0.31) | 9.7 ± 2.7 |
| h | 2 consecutive or 1 | C | Y190T/W55T | 5 | 0.08 ± 0.02 (0.97 ± 0.02) | 0.8 ± 0.4 (0.04 ± 0.01) | nd | nd |
| i | 2 | Y190T | C | 5 | 0.15 ± 0.04 (0.45 ± 0.22) | 1.1 ± 0.4 (0.40 ± 0.17) | 4.5 ± 2.2 (0.14 ± 0.05) | 7.4 ± 2.2 |
| j | 1 | Y190T | C | 8 | 0.22 ± 0.02 (0.87 ± 0.09) | 2.2 ± 0.8 (0.11 ± 0.07) | nd | nd |
| k | 2 nonconsecutive | Y190T/W55T | 5.6a | 0.20 ± 0.01 (0.70 ± 0.16) | 1.10 ± 0.20 (0.26 ± 0.15) | 3.8 ± 2.2 (0.10 ± 0.05) | 5.1 ± 1.2 |
O1, O2, and O3 correspond to the open time components determined from fitting the sum of exponentials to open time histograms constructed from events corresponding to a given amplitude class. Only channels corresponding to 10, 8, or 5 pA class were analyzed. τburst is the burst duration calculated from the identified bursts (see Materials and Methods) (Fig. 2). C corresponds to the control subunit lacking binding site mutations. nd, Not detected. Line k corresponds to recordings performed at −70 mV with the indicated mutant HC subunits. Results are shown as mean ± SD of at least five different patches for each amplitude class.
aThe current amplitude was determined at −70 mV.
Mutations that disable agonist binding sites
A third requirement is to disable the agonist binding site to which the reporter subunit contributes, while allowing ACh to occupy the remaining unaltered binding sites. Structure–function studies of muscle and neuronal nicotinic receptors established that Tyr 190 (O'Leary and White, 1992; Sine et al., 1994) at the principal face and Trp 55 (Chiara et al., 1998) at the complementary face are essential for agonist binding. We therefore generated mutations of these residues and measured the ability of ACh to elicit single-channel and macroscopic currents, as well as to inhibit the initial rate of α-bungarotoxin binding. For receptors bearing the mutation W55T, concentrations of ACh up to 20 mm do not elicit single-channel or macroscopic currents (Fig. 3), and concentrations up to 10 mm do not inhibit binding of α-bungarotoxin (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). For receptors bearing the mutation Y190T, concentrations of ACh up to 20 mm again do not elicit single-channel or macroscopic currents (Fig. 3), and α-bungarotoxin does not show specific binding. The subunit bearing Y190T is nevertheless assembly competent because when it is coexpressed with the control LC subunit, ACh elicits single-channel openings with amplitudes of 8, 5, and 3 pA (see below); the lack of α-bungarotoxin binding therefore results from a change in the binding site but not from an inability to assemble with other subunits and express on the cell surface. In the following experiments, we use Y190T to disable the binding site from the principal face, and W55T to disable the binding site from the complementary face.
Figure 3.

The mutations Y190T and W55T inactivate the binding site. BOSC23 cells were transfected with the high conductance forms of control (HC), HC-Y190T, HC-W55T, or HC-Y190T/W55T subunits. a, Single-channel recordings from cells expressing receptors containing the indicates subunits. Transfected cells were identified by the presence of green fluorescent protein. Membrane potential, −120 mV. For the mutant receptors, no channel activity is detected in the presence of either 1 or 20 mm ACh. b, Whole-cell currents elicited by ACh applied to cells expressing the specified receptor, as identified by presence of green fluorescent protein. Applied potential, −50 mV. For the control (HC), 1 mm ACh elicits robust macroscopic currents, whereas for the mutant receptors no currents are elicited by 1 or 20 mm ACh.
Receptors with differnt number and locations of functionally binding sites
Given the means to disable agonist binding sites and noninvasively register subunit stoichiometry, we generated receptors with different numbers of intact binding sites, and from the conductance signature of each channel opening episode, identified the number of intact binding sites in the corresponding receptor. After coexpressing the control HC subunit with the LC-Y190T subunit, and applying 2–10 mm ACh, channel opening episodes corresponding to all five amplitude classes were observed. To most accurately determine durations of the channel opening events, we analyzed only the 10, 8, and 5 pA amplitude classes, corresponding to receptors with five, four, and three HC subunits, respectively. To increase the frequency of events of a specific amplitude class, we transfected cells with different ratios of HC to LC subunits (Fig. 4). For example, transfecting with a 3:1 ratio promotes the 8 pA class, whereas transfecting with a 1:1 ratio promotes the 5 pA class.
Figure 4.

Activation of receptors with four or three intact agonist binding sites. Cells were cotransfected with HC and LC-Y190T subunit cDNAs at the indicated ratios. Arrangements of subunits for receptors giving rise to the amplitude classes 10 pA (5 binding sites), 8 pA (4 binding sites), and 5 pA (3 binding sites) are shown (top panel). For each transfection ratio, recordings in the presence of 2 mm ACh at a potential of −120 mV are shown at two different timescales with the amplitude class of interest marked by the red line. Open and burst duration histograms were constructed after selecting events corresponding to 8 pA (3:1 ratio) and 5 pA amplitude classes (1:1 ratio). Burst duration histograms were constructed as described in Materials and Methods.
The 8 pA class of channel openings corresponds to receptors with four intact binding sites, and exhibits normal open and burst durations (Fig. 4; Table 1, line e), indicating at least one of the five binding sites is dispensable. The 5 pA class of channel openings corresponds to receptors with three intact binding sites, located at either consecutive or nonconsecutive subunit interfaces (Fig. 4), but this class also exhibits normal open and burst durations (Table 1, line f). Thus, at least two binding sites per receptor can be disabled and still achieve maximal open channel lifetime, although this experiment could not distinguish between the two possible arrangements of three intact binding sites.
To distinguish between arrangements of the three intact binding sites, we constructed a subunit with mutations that disable both the principal and the complementary face of the binding site, Y190T/W55T. When incorporated into a pentamer, the double-mutant subunit disables the two binding sites to which it contributes (Fig. 5). As observed for receptors with a mutation at either face of the binding sites, receptors with mutations at both faces exhibit no single-channel or macroscopic currents in response to 20 mm ACh (Fig. 3). After coexpressing the HC subunit with the LC-Y190T/W55T subunit and applying 2–10 mm ACh, channel opening episodes with amplitudes of 10, 8, 5, and 3 pA are observed. To most accurately determine durations of the channel opening events, we again analyzed events belonging to the three largest amplitude classes (Fig. 5). The 8 pA class of openings corresponds to receptors with three binding sites at consecutive subunit interfaces (Fig. 5) and exhibits a briefer mean burst duration (τburst = 9.7 ms) (Table 1, line g) compared with control (τburst = 16.5 ms) (Table 1, line a). Thus, confining the three binding sites to consecutive subunit interfaces reduces the stability of the open channel, and the remaining possibility, three binding sites at nonconsecutive subunit interfaces, provides maximal stability.
Figure 5.

Activation of receptors with three consecutive, two consecutive, or one intact agonist binding site. Cells were cotransfected with HC and LC-Y190T/W55T subunit cDNAs at the indicated ratios. Arrangements of subunits for receptors giving rise to the amplitude classes 10 pA (5 functional binding sites), 8 pA (3 consecutive binding sites), and 5 pA (2 consecutive or 1 binding site) are shown (top panel). For each transfection ratio, recordings in the presence of 2 mm ACh at −120 mV are shown at two different timescales with the main amplitude class of interest marked by the red line. Open and burst duration histograms were constructed after selecting events corresponding to the 8 pA (3:1 ratio) and 5 pA amplitude class (3:2 ratio). Burst duration histograms were constructed as described in Materials and Methods. Events in the 5 pA amplitude class did not occur in bursts.
In the recordings just described, channel openings of the 5 pA class correspond to receptors with binding sites at two consecutive or one subunit interface (Fig. 5), and appear as isolated events with a brief mean duration (τopen = 0.08 ms) (Table 1, line h). A second population of isolated brief openings with small relative area is also observed (τopen = 0.85 ms) (Table 1, line h), but we cannot determine whether these arise from receptors with two consecutive or one binding site. Nevertheless, this experiment shows that confining two binding sites to consecutive subunit interfaces enables only isolated brief channel openings.
To further examine receptors with one or two intact binding sites, we coexpressed the LC subunit with the HC-Y190T subunit, and applied 2–10 mm ACh. The 10 pA amplitude class was not observed, as this would correspond to receptors with five HC subunits and five disabled binding sites. The remaining four amplitude classes were observed, but to examine receptors with one or two intact binding sites, we focus here on the 8 and 5 pA classes. Channel opening episodes with 8 pA amplitude correspond to receptors with one intact binding site and appear as isolated events with a brief mean duration (Fig. 6) (τopen = 0.2 ms) (Table 1, line j). A second kinetic class of isolated openings with small relative area is also observed (τopen = 2.2 ms) (Table 1, line j), showing that receptors with only one intact binding site give rise to two kinetic classes of brief openings. In the same recording, openings of the 5 pA amplitude class correspond to receptors with two binding sites at either consecutive or nonconsecutive subunit interfaces and appear as bursts with a mean duration that is briefer (τopen = 4.5 ms; τburst = 7.4 ms) (Table 1, line i) than observed for receptors with three binding sites at nonconsecutive subunit interfaces. Because receptors with two binding sites at consecutive subunit interfaces elicit isolated brief openings (Fig. 5), these bursts of openings with intermediate durations arise from receptors with two binding sites at nonconsecutive subunit interfaces. Thus, receptors with one intact binding site or two consecutive binding sites produce isolated brief channel openings, whereas receptors with two intact binding sites at nonconsecutive subunit interfaces produce bursts of openings with a mean duration about one-half that of the control homo-pentamer.
Figure 6.

Activation of receptors with two nonconsecutive, two consecutive, or one intact agonist binding site. Cells were cotransfected with HC-Y190T and control LC subunits cDNAs at the indicated ratios. Arrangements of subunits for receptors giving rise to the amplitude classes 8 pA (1 functional binding site) and 5 pA (2 functional binding sites) at −120 mV are shown (top panel). Recordings performed in the presence of 2 mm ACh at −120 mV are shown at two different timescales with the main amplitude class for each ratio marked by the red line. Open and burst duration histograms were constructed after selecting events corresponding to the 8 pA (2:1 ratio) and 5 pA (1:1 ratio) amplitude classes. Burst duration histograms were constructed as described in Materials and Methods. Events in the 8 pA amplitude class did not occur in bursts.
To confirm that receptors with two nonconsecutive binding sites produce bursts of openings with intermediate duration of the control HC receptor, we coexpressed HC-Y190T and HC-W55T subunits and recorded single-channel currents at a membrane potential of −70 mV in the presence of 2–10 mm ACh. The change in membrane potential does not significantly affect mean open and burst durations (at −70 mV, τopen = 7.4 ± 1.9 ms; τburst = 17.6 ± 1.5 ms; at −120 mV, τopen = 8.9 ± 1.9 ms; τburst = 16.5 ± 2.6 ms) (Table 1). In this experiment, eight arrangements of subunits are possible, two of which contain no intact binding sites (Fig. 7). Of the other six arrangements, two correspond to receptors with two intact binding sites at nonconsecutive subunit interfaces, and four correspond to receptors with one intact binding site. Although isolated brief openings predominated, presumably because of a greater probability of forming receptors with one intact binding site, we also observed bursts of openings with intermediate mean duration (τopen = 3.8 ms; τburst = 5.1 ms) (Table 1, line k). Thus, receptors with two binding sites at nonconsecutive subunit interfaces produce bursts of openings with a mean duration one-half to one-third of that produced by the control homo-pentamer with five binding sites. Summing up, receptors with three intact binding sites at nonconsecutive subunit interfaces elicit bursts of channel openings with maximal mean open time.
Figure 7.
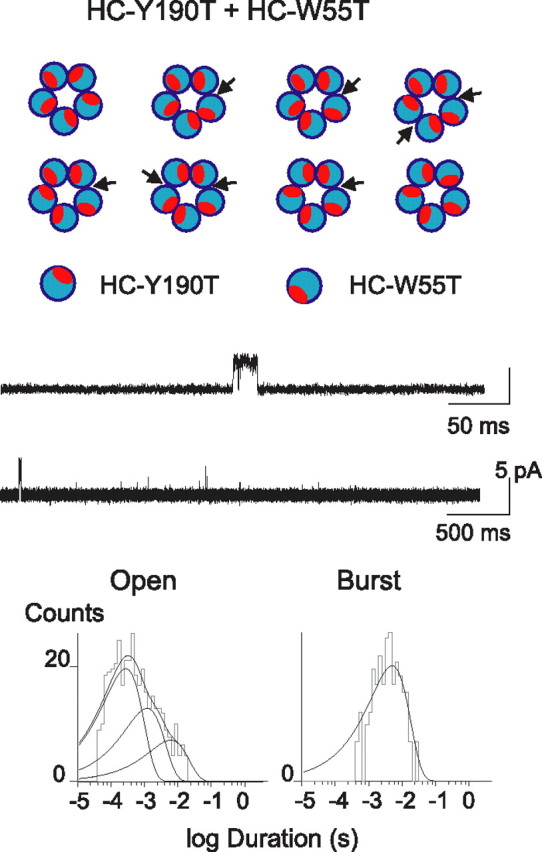
Activation of receptors with two nonconsecutive or one intact agonist binding site. The HC-Y190T and HC-W55T subunits were coexpressed at a 1:1 ratio resulting in eight different arrangements of subunits (top panel). All eight arrangements give rise to a single amplitude class with a mean current of 5.6 pA at −70 mV. Recordings were obtained in the presence of 2 or 10 mm ACh. Traces are shown at two different timescales. Open and burst duration histograms are shown.
To further compare receptors with three intact binding sites with those with five intact binding sites, we fitted a kinetic scheme to sequences of openings and closings within bursts. Because bursts comprise two exponential components of openings and two of closings (Rayes et al., 2005), we fitted the following scheme to the data:

Here, the Cn are closed states, On are open states, and the Greek letters are rate constants for transition between the states. We previously showed that scheme 1 describes sequences of openings and closings within bursts from the HC form of the α7–5HT3A chimera (Rayes et al., 2005); because a high concentration of ACh was used, each state is considered fully occupied by agonist. However, because closed and open times within bursts were independent of ACh concentration, data obtained across a range of ACh concentrations could not yield rate constants for the agonist binding steps leading to the states in scheme 1. Maximum likelihood fitting of scheme 1 to closed and open dwell times within bursts describes the experimental data for the control HC α7–5HT3A chimera (Table 2, HC; supplemental Fig. 2, available at www.jneurosci.org as supplemental material), as observed by Rayes et al. (2005).
Table 2.
Kinetic parameters of receptors containing different number of binding sites

Values are results of simultaneous fits of scheme 1 to data from at least three different patches at 2–10 mm ACh at −120 mV (see Materials and Methods). The log likelihood was 12,984, 13,178, and 13,237 for columns 1, 2, and 3, respectively. For the references of the subunit arrangements, see Figures 4–6.
Macroscopic currents simulated on the basis of scheme 1, but with a desensitized state connected to O2, mimic macroscopic currents recorded after rapid application of ACh (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Thus, for the control HC form of the α7–5HT3A chimera, scheme 1 describes both single-channel and macroscopic currents.
Scheme 1 also describes dwell time sequences for the 5 pA class of receptors with five intact binding sites (Table 2, HC + LC), and those for the 5 pA amplitude class with three intact binding sites at consecutive or nonconsecutive subunit interfaces [HC + LC-Y190T in Fig. 4, Table 2, supplemental Fig. 2 (available at www.jneurosci.org as supplemental material)]. Moreover, the fitted rate constants are similar for all three of these variant receptors (Table 2). However, scheme 1 did not describe dwell time sequences for the 8 pA amplitude class corresponding to receptors with three intact binding sites at consecutive subunit interfaces (obtained by coexpression of HC and LC-Y190T/W55T subunits in Fig. 5). Thus, receptors with intact binding sites at five or three nonconsecutive subunit interfaces are kinetically indistinguishable.
Rate of activation and extent of desensitization depend on the number of intact binding sites per receptor
To determine whether the number of agonist binding sites per receptor affects the rate of activation and the extent of desensitization, we coexpressed different mole ratios of the HC and HC-Y190T/W55T subunits, rapidly applied ACh, and recorded whole-cell macroscopic currents. Because macroscopic currents result from the overall population of receptors in the cell, this experiment cannot distinguish the arrangements of subunits within individual receptors. However, by varying the mole ratio of the subunits, the average number of intact binding sites per receptor can be systematically changed. For cells expressing the control HC α7–5HT3A chimeric receptor, a step pulse of 20 mm ACh elicits a rapid increase of current that, in the continued presence of ACh, decays because of desensitization, reaching a steady-state current of 5–20% of the peak current (Fig. 8a, top). The rate of the current increase does not change between 20 and 40 mm ACh, suggesting that agonist association and dissociation steps do not contribute to the observed rise time (Hu et al., 2003). Simulations of macroscopic currents show that the channel opening rate, and not the closing rate or the rate of desensitization, contributes appreciably to the rise time (supplemental Fig. 4, available at www.jneurosci.org as supplemental material).
Figure 8.
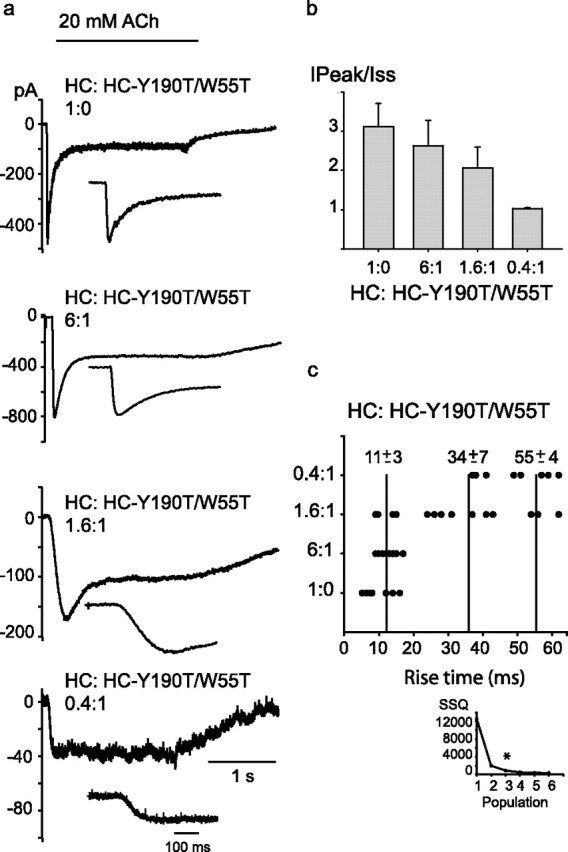
Dependence of activation rate and extent of desensitization on the number of intact binding sites per receptor. a, Cells were transfected with the control HC subunit alone (top trace) or with the HC-Y190T/W55T mutant subunit at the indicated subunit ratios. Macroscopic currents were recorded in the whole-cell configuration at an applied potential: −50 mV. b, The ratios of the peak to the steady-state currents (IPeak/Iss) are shown for each transfection ratio. Data are shown as mean ± SD (n = 5). c, Rise times of the currents were plotted for individual cells for the indicated transfection ratios. The vertical lines indicate the mean ± SD of the rise time for each cluster. A k-means analysis (MacQueen, 1967) reveals three clusters of rise times (inset). *Indicates the lowest number of populations that allows a minimal intracluster SSQ.
Macroscopic current recordings reveal that, as the average number of intact binding sites per receptor decreases, the rise time increases, suggesting a decreased rate of channel opening. Also, the peak current relative to the steady-state current decreases, indicating a reduced extent of desensitization (Fig. 8). The reduced extent of desensitization may reflect slower onset and/or faster recovery from desensitization. Also, a reduction in the opening rate or an increase in the closing rate may reduce the extent of desensitization (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). Although we cannot distinguish among these possibilities, our findings show that the extent of desensitization is less pronounced in receptors containing fewer agonist binding sites.
Comparison of currents elicited by 20 and 40 mm ACh again showed no differences. Although the rise times for a given mole ratio of subunits varied among different cells, the overall data grouped into three distinct populations. The population with longest rise time increases as the fraction of receptors with only one or two intact binding sites increases.
After transfection with a subunit ratio of 0.4:1 (HC to HC-Y190T/W55T), ACh-elicited currents rise slowly to a stable plateau, but do not show a transient peak (Fig. 8a). This subunit ratio predicts 70% of receptors have no intact binding sites, 21% have one binding site, 6% have two consecutive sites, and 2% have three consecutive binding sites (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Because the vast majority of functional receptors contain two consecutive or one binding site, we conclude that two or more binding sites at nonconsecutive subunit interfaces are required to achieve rapid activation and extensive steady-state desensitization.
Finally, we find that sensitivity to agonist decreases as the number of intact binding sites per receptor decreases. For control receptors composed of five HC subunits, the peak macroscopic current evoked by 1 mm ACh is equivalent to that evoked by 20 mm ACh (Fig. 9). However, as the average number of intact binding sites per receptor decreases, the peak current evoked by 1 mm ACh progressively decreases compared with that evoked by 20 mm ACh.
Figure 9.
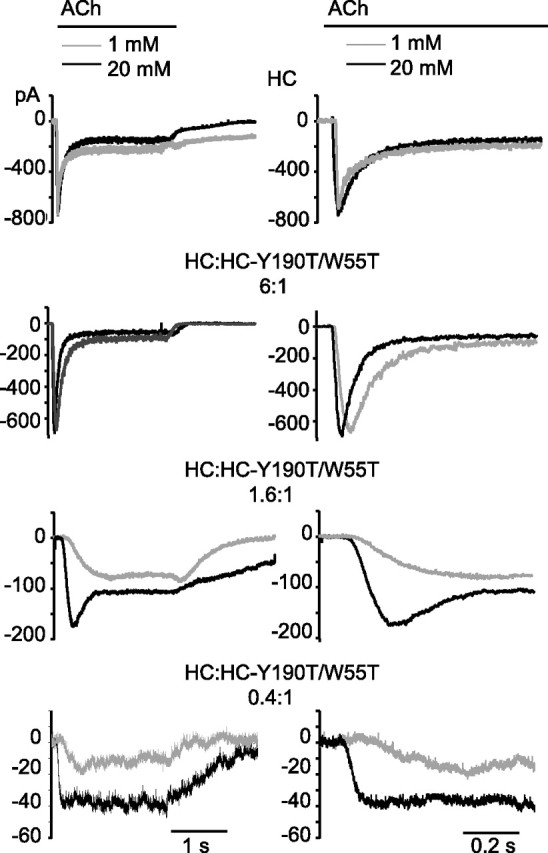
Dependence of peak currents elicited by 1 and 20 mm ACh on the number of intact binding sites per receptor. Cells were transfected with the HC subunit alone (top traces) or with the HC-Y190T/W55T mutant subunit at the indicated subunit ratios (bottom traces). Macroscopic currents were recorded in the whole-cell configuration at an applied potential of −50 mV. Currents were elicited by 1 mm (gray) or 20 mm (black) ACh and shown at two different timescales. For receptors composed of HC subunits, the rise time does not change significantly between 1 and 20 mm ACh; for this receptor, the ratio of the 20–80% rise times at 1 and 20 mm ACh (Rt1 mm/Rt20 mm) is 1.3 ± 0.3. Differences in rise times between 1 and 20 mm ACh become apparent as the relative amount of the HC-Y190T/W55T subunit increases. The Rt1 mm/Rt20 mm ratios are 2.1, 3.1, and 7.1 for subunit ratios of 6:1; 1.6:1, and 0.4:1, respectively. For each transfection condition, currents in response to 40 mm ACh were also measured; in all cases, peak currents elicited by 20 and 40 mm ACh were not significantly different, indicating that saturation was achieved.
Discussion
The ancestral Cys-loop receptor was likely homomeric and contained five identical binding sites (Ortells and Lunt, 1995; Le Novère et al., 2002; Tasneem et al., 2005). Present-day homomeric receptors, such as neuronal α7 and 5-HT3A receptors, diverged least from the ancestral receptor and likely share functional features among members of the superfamily. Evolution led to the appearance of new subunits that lost the ability to form agonist binding sites, giving rise to heteromeric receptors with fewer than five binding sites (Tsunoyama and Gojobori, 1998; Le Novère et al., 2002). The prototypic heteromeric receptors, muscle ACh and GABAA receptors, contain only two agonist binding sites.
Because heteromeric receptors contain only two agonist binding sites, whereas homomeric receptors contain five, a long-standing question has been how many of the five neurotransmitter binding sites in homomeric receptors are required to efficiently open the channel. The answer to this question will provide a mechanistic framework for describing the activation process and give insight into interactions between subunits that enable global conformational changes required for channel opening. At present, only indirect approaches based on kinetic modeling of macroscopic and single-channel currents have addressed this issue, and the results have not been definitive. Indeed, from two to five binding sites have been proposed to activate different homomeric Cys-loop receptors (Legendre, 1998; Papke et al., 2000; Beato et al., 2002). Nevertheless, recent kinetic modeling studies for 5-HT3A receptors (Solt et al., 2007) (J. Corradi, F. Gumilar, and C. Bouzat, unpublished observations) and glycine receptors (Beato et al., 2004; Lape et al., 2008) suggest three binding sites are required for optimal activation, in agreement with our direct experimental evidence.
The chimeric α7–5HT3A receptor has served as a prototype for investigating the pharmacology of α7 receptors (Eiselé et al., 1993; Nicke et al., 2003). The high conductance and uniform open channel current of the HC form of the chimeric receptor are valuable attributes in the electrical fingerprinting strategy used here. Furthermore, the mutations that affect conductance have the advantage that they do not alter the intrinsic kinetics of channel gating (Rayes et al., 2005; Bouzat et al., 2008); a recent study of 5-HT3A receptors shows that deletion of the entire M3-M4 loop affects mainly channel conductance of 5-HT3A (Jansen et al., 2008).
The α7–5HT3A receptor has additional advantages in defining the number and locations of binding sites required for channel opening. First, residues contributing to the binding site are well known (Brejc et al., 2001; Sine, 2002). Among these residues, we chose to mutate Y190 from the principal face and W55 from the complementary face (O'Leary and White, 1992; Sine et al., 1994; Chen et al., 1995; Chiara et al., 1998; Costa et al., 2003); mutations of either residue alone or both residues together completely inhibit the response to agonist. Second, the single-channel current amplitudes span from undetectable with five LC subunits to 10 pA at −120 mV with five HC subunits, allowing distinction of channel opening episodes from receptors with different numbers of high- and low-conductance subunits. The observation of discrete and equally spaced amplitude classes indicates that the number of subunits carrying conductance mutations but not their disposition in the pentamer determines current amplitude.
The vast majority of current through the HC form of the α7–5HT3A chimera flows during bursts openings in quick succession. The kinetics of bursts does not depend on agonist concentration (Rayes et al., 2005), as observed for native 5-HT3A, α7, and GABAA receptors (Akk et al., 2004; Lema and Auerbach, 2006; Bouzat et al., 2008) (J. Corradi, F. Gumilar, and C. Bouzat, unpublished observations).
The lack of concentration-dependent kinetics suggests that brief and long channel openings arise from fully liganded receptors and that channel openings are terminated mainly by desensitization (Rayes et al., 2005; Bouzat et al., 2008). This concentration-independent burst structure seems to be common to excitatory homomeric Cys-loop receptors, but differs from that of muscle AChRs and homomeric and heteromeric glycine receptors, for which closed intervals within bursts depend on agonist concentration (Sine and Steinbach, 1987; Sine et al., 1990; Bouzat et al., 2000, 2002; Beato et al., 2004; Lape et al., 2008). GABAA receptors also show concentration-independent bursts but these coalesce into clusters that include concentration-dependent interburst closings (Steinbach and Akk, 2001; Akk et al., 2004).
We find that only one functional binding site per receptor allows activation by agonist, but the openings are brief and solitary. Kinetic analyses of muscle AChRs, GABAA, and glycine receptors also showed that openings activated by occupancy by a single agonist are brief compared with openings activated by occupancy by two agonists (Colquhoun and Sakmann, 1985; Ohno et al., 1996; Fisher and Macdonald, 1997; Haas and Macdonald, 1999; Bouzat et al., 2002; Greenfield et al., 2002; Beato et al., 2004).
A novel finding of this work is that two intact binding sites at consecutive subunit interfaces enables only brief solitary openings, which mimic those of receptors with only one intact binding site. However, two intact binding sites at nonconsecutive interfaces enable bursts of openings with a much longer-lived open state appropriate for efficient activation. Thus, as observed for the muscle AChR and GABAA receptors, two binding sites separated by an intervening subunit enable global structural changes that produce a stable open state.
Receptors with intact binding sites at three nonconsecutive interfaces maximize the stability of openings within bursts and exhibit a kinetic signature indistinguishable from that of receptors with five intact binding sites. Thus, a third agonist binding site allows positive modulation by the same neurotransmitter and resembles allosteric modulation observed in heteromeric receptors. In heteromeric receptors, the third site allows potentiation by heterotropic regulatory ligands, such as benzodiazepines for GABAA receptors (Cromer et al., 2002) or zinc for neuronal nicotinic receptors (Hsiao et al., 2008). Moreover, the arrangement of GABA and benzodiazepine binding sites in GABAA receptors (Cromer et al., 2002) is analogous to that shown here for functionally relevant binding sites in α7–5HT3A receptors.
Our findings also raise new questions regarding the mechanism of activation of homomeric receptors. For example, there may be a specific temporal sequence in which occupancy of the three functionally relevant binding sites produces a stable open state. We can speculate that two nonconsecutive binding sites are first occupied, and successive occupancy of the third site potentiates the response. Also, at a saturating concentration of agonist, the two supernumerary binding sites should be occupied and may contribute to some other facet of receptor function even though they do not contribute to open channel stability.
In whole-cell recordings, macroscopic currents result from the overall population of receptors within the same cell. Although arrangements of subunits in individual receptors cannot be determined, the probability of each arrangement can be calculated. We show that, whereas two nonconsecutive binding sites allow rapid activation in response to agonist, as well as desensitization in its continued presence, two consecutive binding sites allow only slow channel activation and no desensitization. By comparing macroscopic responses at different agonist concentrations, we found that the response saturates at higher concentrations with receptors containing reduced numbers of intact binding sites, indicating that the availability of more than two binding sites enhances agonist sensitivity. The enhanced sensitivity, together with the benefit of a single self-assembling gene product, could have been important as far back as prokaryotes (Tasneem et al., 2005) and before the appearance of structurally efficient synapses. In present-day homomeric receptors, high agonist sensitivity could be important in the function of extrasynaptic and presynaptic receptors (Jones and Wonnacott, 2004).
The overall results show that, as the structures of the principal and complementary faces of the binding site evolved, heteromeric assembly of the subunits resulted in receptors with fewer than five agonist binding sites, but without the penalty of a reduced maximal response to agonist. However, three mechanistic constraints came into play. First, to elicit a global conformational change that produced a stable active state, at least two binding sites at a distance from each other were required (Jackson, 1989), as in the prototypic ACh receptor at the motor endplate. Second, two binding sites at nonconsecutive subunit interfaces enabled the channel to open rapidly after binding of agonist, and protected against overstimulation by agonist by preserving desensitization. Finally, in many heteromeric receptors, a third site enables allosteric modulation by ligands other than the natural neurotransmitter. For example, in heteromeric GABAA receptors, agonist binds to sites at the two interfaces formed by α and β subunits, whereas benzodiazepines bind to a site at the single interface formed by α and γ subunits (Amin et al., 1997; Campo-Soria et al., 2006). Moreover, it was suggested that homology between the two types of binding sites can be traced to a gene duplication event resulting in a modified subunit that contributes to a modulatory site (Smith and Olsen, 1995). Positive allosteric modulators have emerged as a therapeutic strategy for disorders associated with receptor deficit (Gotti et al., 2006). Our study suggests such modulatory sites evolved from a site for the natural neurotransmitter.
Footnotes
This work was supported by National Institutes of Health Grant NS053521 (S.M.S., C.B.) and grants from Universidad Nacional del Sur, Agencia Nacional de Promoción Científica y Tecnológica, Consejo Nacional de Investigaciones Científicas y Técnicas, Loreal–United Nations Educational, Scientific and Cultural Organization, and Fundación Fiorini (C.B.). We thank Dr. G. Spitzmaul for contributions in the initial development of this work and C. Free, H. De Genaro, and R. Gonzalez for technical contributions.
References
- Akk G, Bracamontes J, Steinbach JH. Activation of GABA(A) receptors containing the alpha4 subunit by GABA and pentobarbital. J Physiol. 2004;15:387–399. doi: 10.1113/jphysiol.2003.058230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin J, Weiss DS. Insights into the activation mechanism of rho1 GABA receptors obtained by coexpression of wild type and activation-impaired subunits. Proc Biol Sci. 1996;263:273–282. doi: 10.1098/rspb.1996.0042. [DOI] [PubMed] [Google Scholar]
- Amin J, Brooks-Kayal A, Weiss DS. Two tyrosine residues on the alpha subunit are crucial for benzodiazepine binding and allosteric modulation of gamma-aminobutyric acidA receptors. Mol Pharmacol. 1997;51:833–841. doi: 10.1124/mol.51.5.833. [DOI] [PubMed] [Google Scholar]
- Beato M, Groot-Kormelink PJ, Colquhoun D, Sivilotti LG. Openings of the rat recombinant alpha 1 homomeric glycine receptor as a function of the number of agonist molecules bound. J Gen Physiol. 2002;119:443–466. doi: 10.1085/jgp.20028530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beato M, Groot-Kormelink PJ, Colquhoun D, Sivilotti LG. The activation mechanism of α1 homomeric glycine receptors. J Neurosci. 2004;24:895–906. doi: 10.1523/JNEUROSCI.4420-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouzat C, Bren N, Sine SM. Structural basis of the different gating kinetics of fetal and adult acetylcholine receptors. Neuron. 1994;13:1395–1402. doi: 10.1016/0896-6273(94)90424-3. [DOI] [PubMed] [Google Scholar]
- Bouzat C, Barrantes F, Sine S. Nicotinic receptor fourth transmembrane domain: hydrogen bonding by conserved threonine contributes to channel gating kinetics. J Gen Physiol. 2000;115:663–672. doi: 10.1085/jgp.115.5.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouzat C, Gumilar F, del Carmen Esandi M, Sine SM. Subunit-selective contribution to channel gating of the M4 domain of the nicotinic receptor. Biophys J. 2002;82:1920–1929. doi: 10.1016/S0006-3495(02)75541-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouzat C, Gumilar F, Spitzmaul G, Wang HL, Rayes D, Hansen SB, Taylor P, Sine SM. Coupling of agonist binding to channel gating in an ACh binding protein linked to an ion channel. Nature. 2004;430:896–900. doi: 10.1038/nature02753. [DOI] [PubMed] [Google Scholar]
- Bouzat C, Bartos M, Corradi J, Sine SM. The interface between extracellular and transmembrane domains of homomeric Cys-loop receptors governs open-channel lifetime and rate of desensitization. J Neurosci. 2008;28:7808–7819. doi: 10.1523/JNEUROSCI.0448-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brejc K, van Dijk WJ, Klaassen RV, Schuurmans M, van Der Oost J, Smit AB, Sixma TK. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature. 2001;411:269–276. doi: 10.1038/35077011. [DOI] [PubMed] [Google Scholar]
- Campo-Soria C, Chang Y, Weiss DS. Mechanism of action of benzodiazepines on GABAA receptors. Br J Pharmacol. 2006;148:984–990. doi: 10.1038/sj.bjp.0706796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Zhang Y, Akk G, Sine S, Auerbach A. Activation kinetics of recombinant mouse nicotinic acetylcholine receptors: mutations of α-subunit tyrosine 190 affect both binding and gating. Biophys J. 1995;69:849–859. doi: 10.1016/S0006-3495(95)79959-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiara DC, Middleton RE, Cohen JB. Identification of tryptophan 55 as the primary site of [3H]nicotine photo-incorporation in the gamma-subunit of the Torpedo nicotinic acetylcholine receptor. FEBS Lett. 1998;423:223–226. doi: 10.1016/s0014-5793(98)00093-3. [DOI] [PubMed] [Google Scholar]
- Colquhoun D, Sakmann B. Fast events in single-channel currents activated by acetylcholine and its analogues at the frog muscle end-plate. J Physiol. 1985;369:501–557. doi: 10.1113/jphysiol.1985.sp015912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa V, Nistri A, Cavalli A, Carloni P. A structural model of agonist binding to the α3β4 neuronal nicotinic receptor. Br J Pharmacol. 2003;140:921–931. doi: 10.1038/sj.bjp.0705498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cromer BA, Morton CJ, Parker MW. Anxiety over GABA(A) receptor structure relieved by AChBP. Trends Biochem Sci. 2002;27:280–287. doi: 10.1016/s0968-0004(02)02092-3. [DOI] [PubMed] [Google Scholar]
- Eiselé JL, Bertrand S, Galzi JL, Devillers-Thiéry A, Changeux JP, Bertrand D. Chimaeric nicotinic-serotonergic receptor combines distinct ligand binding and channel specificities. Nature. 1993;366:479–483. doi: 10.1038/366479a0. [DOI] [PubMed] [Google Scholar]
- Fisher JL, Macdonald RL. Single channel properties of recombinant GABAA receptors containing gamma 2 or delta subtypes expressed with alpha 1 and beta 3 subtypes in mouse L929 cells. J Physiol. 1997;505:283–297. doi: 10.1111/j.1469-7793.1997.283bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentet LJ, Clements JD. Binding site stoichiometry and the effects of phosphorylation on human alpha1 homomeric glycine receptors. J Physiol. 2002;544:97–106. doi: 10.1113/jphysiol.2001.015321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotti C, Zoli M, Clementi F. Brain nicotinic acetylcholine receptors: native subtypes and their relevance. Trends Pharmacol Sci. 2006;27:482–491. doi: 10.1016/j.tips.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Greenfield LJ, Jr, Zaman SH, Sutherland ML, Lummis SC, Niemeyer MI, Barnard EA, Macdonald RL. Mutation of the GABAA receptor M1 transmembrane proline increases GABA affinity and reduces barbiturate enhancement. Neuropharmacology. 2002;42:502–521. doi: 10.1016/s0028-3908(01)00196-4. [DOI] [PubMed] [Google Scholar]
- Haas KF, Macdonald RL. GABAA receptor subunit gamma2 and delta subtypes confer unique kinetic properties on recombinant GABAA receptor currents in mouse fibroblasts. J Physiol. 1999;514:27–45. doi: 10.1111/j.1469-7793.1999.027af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao B, Mihalak KB, Magleby KL, Luetje CW. Zinc potentiates neuronal nicotinic receptors by increasing burst duration. J Neurophysiol. 2008;99:999–1007. doi: 10.1152/jn.01040.2007. [DOI] [PubMed] [Google Scholar]
- Hu XQ, Zhang L, Stewart RR, Weight FF. Arginine 222 in the pre-transmembrane domain 1 of 5-HT3A receptors links agonist binding to channel gating. J Biol Chem. 2003;278:46583–46589. doi: 10.1074/jbc.M308974200. [DOI] [PubMed] [Google Scholar]
- Jackson MB. Perfection of a synaptic receptor: kinetics and energetics of the acetylcholine receptor. Proc Natl Acad Sci U S A. 1989;86:2199–2203. doi: 10.1073/pnas.86.7.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen M, Bali M, Akabas MH. Modular design of Cys-loop ligand-gated ion channels: functional 5-HT3 and GABA ρ1 receptors lacking the large cytoplasmic M3M4 loop. J Gen Physiol. 2008;131:137–146. doi: 10.1085/jgp.200709896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones IW, Wonnacott S. Precise localization of α7 nicotinic acetylcholine receptors on glutamatergic axon terminals in the rat ventral tegmental area. J Neurosci. 2004;24:11244–11252. doi: 10.1523/JNEUROSCI.3009-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin A. Emerging structure of the nicotinic acetylcholine receptors. Nat Rev Neurosci. 2002;3:102–114. doi: 10.1038/nrn731. [DOI] [PubMed] [Google Scholar]
- Kelley SP, Dunlop JI, Kirkness EF, Lambert JJ, Peters JA. A cytoplasmic region determines single-channel conductance in 5-HT3 receptors. Nature. 2003;424:321–324. doi: 10.1038/nature01788. [DOI] [PubMed] [Google Scholar]
- Lape R, Colquhoun D, Sivilotti LG. On the nature of partial agonism in the nicotinic receptor superfamily. Nature. 2008;454:722–727. doi: 10.1038/nature07139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre P. A reluctant gating mode of glycine receptor channels determines the time course of inhibitory miniature synaptic events in zebrafish hindbrain neurons. J Neurosci. 1998;18:2856–2870. doi: 10.1523/JNEUROSCI.18-08-02856.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lema GM, Auerbach A. Modes and models of GABA(A) receptor gating. J Physiol. 2006;572:183–200. doi: 10.1113/jphysiol.2005.099093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Novère N, Corringer PJ, Changeux JP. The diversity of subunit composition in nAChRs: evolutionary origins, physiologic and pharmacologic consequences. J Neurobiol. 2002;53:447–456. doi: 10.1002/neu.10153. [DOI] [PubMed] [Google Scholar]
- Lester HA, Dibas MI, Dahan DS, Leite JF, Dougherty DA. Cys-loop receptors: new twists and turns. Trends Neurosci. 2004;27:329–336. doi: 10.1016/j.tins.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Liu Y, Dilger JP. Opening rate of acetylcholine receptor channels. Biophys J. 1991;60:424–432. doi: 10.1016/S0006-3495(91)82068-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackinnon R, Aldrich R, Lee A. Functional stoichiometry of Shaker potassium channel inactivation. Science. 1993;62:757–759. doi: 10.1126/science.7694359. [DOI] [PubMed] [Google Scholar]
- MacQueen J. Some methods for classification and analysis of multivariate observations. In: Le Cam LM, Neyman J, editors. Fifth Berkeley Symposium on Mathematical Statistics and Probability. Vol. 1. Berkekey, CA: University of California; 1967. pp. 281–297. Statistics. [Google Scholar]
- Mott DD, Erreger K, Banke TG, Traynelis SF. Open probability of homomeric murine 5-HT3A serotonin receptors depends on subunit occupancy. J Physiol. 2001;535:427–443. doi: 10.1111/j.1469-7793.2001.00427.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicke A, Samochocki M, Loughnan ML, Bansal PS, Maelicke A, Lewis RJ. Alpha-conotoxins EpI and AuIB switch subtype selectivity and activity in native versus recombinant nicotinic acetylcholine receptors. FEBS Lett. 2003;554:219–223. doi: 10.1016/s0014-5793(03)01161-x. [DOI] [PubMed] [Google Scholar]
- Ohno K, Wang HL, Milone M, Bren N, Brengman JM, Nakano S, Quiram P, Pruitt JN, Sine SM, Engel AG. Congenital myasthenic syndrome caused by decreased agonist binding affinity due to a mutation in the acetylcholine receptor epsilon subunit. Neuron. 1996;17:157–170. doi: 10.1016/s0896-6273(00)80289-5. [DOI] [PubMed] [Google Scholar]
- O'Leary ME, White MM. Mutational analysis of ligand-induced activation of the Torpedo acetylcholine receptor. J Biol Chem. 1992;267:8360–8365. [PubMed] [Google Scholar]
- Ortells MO, Lunt GG. Evolutionary history of the ligand-gated ion-channel superfamily of receptors. Trends Neurosci. 1995;18:121–127. doi: 10.1016/0166-2236(95)93887-4. [DOI] [PubMed] [Google Scholar]
- Palma E, Bertrand S, Binzoni T, Bertrand D. Neuronal nicotinic α7 receptor expressed in Xenopus oocytes presents five putative binding sites for methyllycaconitine. J Physiol. 1996;491:151–161. doi: 10.1113/jphysiol.1996.sp021203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papke RL, Meyer E, Nutter T, Uteshev VV. Alpha7 receptor-selective agonists and modes of alpha7 receptor activation. Eur J Pharmacol. 2000;393:179–195. doi: 10.1016/s0014-2999(00)00009-1. [DOI] [PubMed] [Google Scholar]
- Qin F, Auerbach A, Sachs F. Estimating single-channel kinetic parameters from idealized patch-clamp data containing missed events. Biophys J. 1996;70:264–280. doi: 10.1016/S0006-3495(96)79568-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayes D, Spitzmaul G, Sine SM, Bouzat C. Single channel kinetic analysis of chimeric α7–5HT3A receptors. Mol Pharmacol. 2005;68:1475–1483. doi: 10.1124/mol.105.015438. [DOI] [PubMed] [Google Scholar]
- Sine SM. The nicotinic receptor ligand binding domain. J Neurobiol. 2002;53:431–446. doi: 10.1002/neu.10139. [DOI] [PubMed] [Google Scholar]
- Sine SM, Engel AG. Recent advances in Cys-loop receptor structure and function. Nature. 2006;440:448–455. doi: 10.1038/nature04708. [DOI] [PubMed] [Google Scholar]
- Sine SM, Steinbach JH. Activation of acetylcholine receptors on clonal mammalian BC3H-1 cells by high concentrations of agonist. J Physiol. 1987;385:325–359. doi: 10.1113/jphysiol.1987.sp016496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sine SM, Taylor P. Functional consequences of agonist-mediated state transitions in the cholinergic receptor. J Biol Chem. 1979;254:3315–3325. [PubMed] [Google Scholar]
- Sine SM, Claudio T, Sigworth FJ. Activation of Torpedo acetylcholine receptors expressed in mouse fibroblasts. Single channel current kinetics reveal distinct agonist binding affinities. J Gen Physiol. 1990;96:395–437. doi: 10.1085/jgp.96.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sine SM, Quiram P, Papanikolaou F, Kreienkamp HJ, Taylor P. Conserved tyrosines in the alpha subunit of the nicotinic acetylcholine receptor stabilize quaternary ammonium groups of agonists and curariform antagonists. J Biol Chem. 1994;269:8808–8816. [PubMed] [Google Scholar]
- Sine SM, Kreienkamp HJ, Bren N, Maeda R, Taylor P. Molecular dissection of subunit interfaces in the acetylcholine receptor: identification of determinants of alpha-conotoxin M1 selectivity. Neuron. 1995;15:205–211. doi: 10.1016/0896-6273(95)90077-2. [DOI] [PubMed] [Google Scholar]
- Smith GB, Olsen RW. Functional domains of GABAA receptors. Trends Pharmacol Sci. 1995;16:162–168. doi: 10.1016/s0165-6147(00)89009-4. [DOI] [PubMed] [Google Scholar]
- Solt K, Ruesch D, Forman SA, Davies PA, Raines DE. Differential effects of serotonin and dopamine on human 5-HT3A receptor kinetics: interpretation within an allosteric kinetic model. J Neurosci. 2007;27:13151–13160. doi: 10.1523/JNEUROSCI.3772-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbach JH, Akk G. Modulation of GABA(A) receptor channel gating by pentobarbital. J Physiol. 2001;15:715–733. doi: 10.1111/j.1469-7793.2001.00715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasneem A, Iyer LM, Jakobsson E, Aravind L. Identification of the prokaryotic ligand-gated ion channels and their implications for the mechanisms and origins of animal Cys-loop ion channels. Genome Biol. 2005;6:R4. doi: 10.1186/gb-2004-6-1-r4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunoyama K, Gojobori T. Evolution of nicotinic acetylcholine receptor subunits. Mol Biol Evol. 1998;15:518–527. doi: 10.1093/oxfordjournals.molbev.a025951. [DOI] [PubMed] [Google Scholar]
- Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4 Å resolution. J Mol Biol. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- Wang HL, Auerbach A, Bren N, Ohno K, Engel AG, Sine SM. Mutation in the M1 domain of the acetylcholine receptor alpha subunit decreases the rate of agonist dissociation. J Gen Physiol. 1997;109:757–766. doi: 10.1085/jgp.109.6.757. [DOI] [PMC free article] [PubMed] [Google Scholar]