

Abstract
We show that cerebral endothelial cells secrete trophic factors that support the survival and proliferation of rat oligodendrocyte precursor cells (OPCs). This OPC-supportive phenomenon was mediated by Akt and Src signaling pathways. Noncytotoxic levels of oxidative stress downregulate trophic factor production and disrupt the ability of cerebral endothelial cells to support OPCs. These data suggest that a novel oligovascular niche may be important for sustaining oligodendrocyte renewal and homeostasis in mammalian brain.
Introduction
Emerging data now suggest that the cerebrovascular system does not merely provide inert plumbing to deliver blood for the brain. Instead, cerebral endothelial cells may comprise a rich source of molecular signaling that contributes to brain homeostasis and function (Iadecola and Nedergaard, 2007; Lok et al., 2007; Zlokovic, 2008; Guo and Lo, 2009). The most well documented example of these endothelial–brain interactions comprise the neurovascular niche. It is now accepted that cell–cell signaling between cerebral endothelium and neuronal precursor cells helps mediate and sustain pockets of ongoing neurogenesis and angiogenesis in adult brain (Iadecola, 2004; Greenberg and Jin, 2005; Chopp et al., 2007; Zacchigna et al., 2008; Zlokovic, 2008).
Cross talk between the vascular and neuronal compartments in the neurovascular niche is mediated by an exchange of soluble signals. This phenomenon is partly mediated by the ability of cerebral endothelium to secrete a rich repertoire of trophic factors (Leventhal et al., 1999; Shen et al., 2004; Guo et al., 2008). Because many trophic factors may also affect non-neuronal cells, we asked whether a corresponding “oligovascular niche” might also exist, wherein cerebral endothelial cells support the survival and proliferation of oligodendrocyte precursor cells (OPCs).
Materials and Methods
Cell culture.
OPCs were prepared as previously described with some modifications (Ness et al., 2005; van Leyen et al., 2008). Briefly, cerebral cortices from 1- to 2-d-old Sprague Dawley rats were dissected, minced, and digested. Dissociated cells were plated in poly-d-lysine-coated 75 cm2 flasks, and maintained in DMEM containing 20% heat-inactivated fetal bovine serum and 1% penicillin/streptomycin. After the cells were confluent (∼10 d), the flasks were shaken for 1 h on an orbital shaker (220 rpm) at 37°C to remove microglia. They are then changed to new medium and shaken overnight (∼20 h). The medium was collected and plated on noncoated tissue culture dishes for 1 h at 37°C to eliminate contaminating astrocytes and microglia. The nonadherent cells were collected and replated in Neurobasal medium containing glutamine, 1% penicillin/streptomycin, 10 ng/ml PDGF, 10 ng/ml FGF, and 2% B27 supplement onto poly-dl-ornithine-coated plates. Four to five days after plating, the OPCs were used for the experiments. To differentiate OPCs to myelin basic protein-positive oligodendrocytes, the culture medium was switched to DMEM containing 1% penicillin/streptomycin, 10 ng/ml CNTF, 15 nm T3, and 2% B27 supplement. Human brain microendothelial cell (hBMEC, purchased from CSC Systems), a human brain endothelial cell line (hECL, from M. Stins, Johns Hopkins University, Baltimore, MD), and a rat brain microendothelial cell line (RBE.4, from J. Madri, Yale University, New Haven, CT) were maintained in EBM-2 containing EGM-2MV SingleQuots kit onto collagen-coated 25 cm2 flasks. To test the hypothesis that cerebral endothelial cells provide trophic support for OPCs, we performed two different types of experiments. First, we cocultured OPCs and cerebral endothelial cells together in Neurobasal medium containing glutamine, 1% penicillin/streptomycin, and 2% AO-free B27 supplement. And in our second set of experiments, we transferred conditioned media from endothelial cells onto separately cultured OPCs.
Endothelial-conditioned media.
To prepare endothelial-conditioned media, we used cells at 90–95% confluence, grown in Neurobasal medium containing glutamine, 1% penicillin/streptomycin, and 2% AO-free B27 supplement for 24 h. Conditioned medium was collected and filtered using 0.20 μm filter before use in OPC studies. The endothelial-conditioned medium was used without dilution in all experiments except for the experiments in Figure 1 d.
Figure 1.
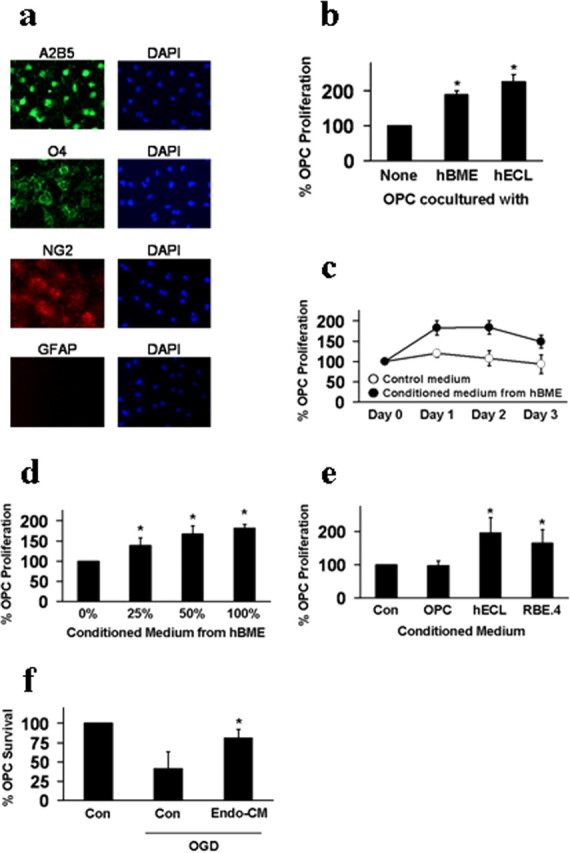
a, Immunostaining demonstrated that OPCs expressed A2B5 (green), O4 (green), NG2 (red), and lacked GFAP (red). Nuclei were stained with DAPI (blue). b, Coculturing with cerebral endothelial cells promoted OPC proliferation. Two different types of endothelial cells were tested, primary human brain microvascular endothelial cells (hBME) and a human brain endothelial cell line (hECL). c, Conditioned media from hBME amplified OPC proliferation over 1–3 d in culture. d, Different dilutions of hBME-conditioned media showed a “dose–response” effect on OPC proliferation. e, Conditioned media from a rat brain endothelial cell line (RBE.4) equally promoted OPC proliferation compared with media from a human brain endothelial cell line (hECL). No effects were seen with conditioned media from matching OPC cultures. f, Conditioned media from hBME also showed OPC-protective effects against oxygen–glucose deprivation (OGD)-induced stress. *p < 0.05.
Oxygen–glucose deprivation.
OPCs were replaced with glucose-free medium followed by hypoxia induced with a modular chamber (Billups-Rothenberg) perfused with 90% N2/5% CO2/5% H2. The chamber was sealed and placed at 37°C for 4 h hypoxia. After hypoxia, cells were removed from the chamber and the glucose-free medium was changed to either control media or endothelial-conditioned media, and then maintained in the regular incubator for 20 h. Control cultures were incubated under normoxic conditions for equivalent durations.
Immunocytochemistry.
After cells reached 70–80% confluence, they were washed with ice-cold PBS, pH 7.4, followed by 4% paraformaldehyde for 30 min. After being further washed three times in PBS containing 0.1% Triton X-100, they were incubated with 1% bovine serum albumin in PBS for 1 h. Then cells were incubated with primary antibodies against the OPC markers (Ness et al., 2005) A2B5 (1:50), O4 (1:50), NG2 (1:50), or the astrocytic marker glial fibrillary acidic protein (GFAP; 1:400) at 4°C overnight. After washing with PBS, they were incubated with secondary antibodies conjugated with fluorescein isothiocyanate for 1 h at room temperature. Finally, nuclei were counterstained with 4, 6-diamidino-2-phenylindole (DAPI), and cells were rinsed and coverslips were placed. Immunostaining was analyzed with a fluorescence microscope (Olympus BX51) interfaced with a digital charge-coupled device camera and an image analysis system.
Western blotting.
Cultures were rinsed twice with ice-cold PBS, and the cells were collected into cell lysis buffer (20 mm Tris, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, 2.5 mm sodium pyrophosphate, 1 mm glycerol phosphate, 1 mm Na3VO4, 1 g/ml leupeptin, and 1 mm PMSF). Cell lysates were then homogenized and centrifuged at 10,000 × g for 10 min at 4°C, and protein concentration in the supernatant was determined with the Bradford assay (Bio-Rad). Samples were heated with equal volumes of SDS sample buffer (Novex) and 10 mm DTT at 95°C for 5 min, then each sample (20 ng per lane) was loaded onto 4–20% Tris-glycine gels. After electrophoresis and transferring to polyvinylidene difluoride membranes (Novex), the membranes were blocked in Tris-buffered saline containing 0.1% Tween 20 and 0.2% I-block (Tropix) for 90 min at room temperature. Membranes were then incubated overnight at 4°C with monoclonal anti-MBP antibody (1:1000, Abcam), monoclonal anti-p-Akt antibody (1:1000, Cell Signaling Technology, New England Biolabs), or anti-Akt antibody (1:1000, Cell Signaling Technology) after incubation with peroxidase-conjugated secondary antibodies and visualization by enhanced chemiluminescence (GE Healthcare).
Determination of cell proliferation/survival.
Cell proliferation/survival was assessed by WST reduction assay (Dojindo). This assay is based on the detection of dehydrogenase activity of viable cells. The cells were incubated with 10% WST solution for 1 h at 37°C. Then the absorbance of the culture medium was measured with a microplate reader at a test wavelength of 450 nm and a reference wavelength of 630 nm.
Statistical analysis.
All of experiments were performed in duplicate, repeated 3–6 times independently. Quantitative data were analyzed by using ANOVA followed by Tukey's honestly significant difference tests. Data are expressed as mean ± SD. A value of p < 0.05 was considered significant.
Results
OPCs were grown from neonatal rat brains. Immunostaining confirmed that these cells were positive for the OPC markers A2B5, O4, and NG2 (Fig. 1 a). The lack of GFAP-positive cells suggested that our preparations were not contaminated by astrocytes. And our OPCs were functional since they can be matured into myelin basic protein-positive oligodendrocytes over time (supplemental Fig. S1a, available at www.jneurosci.org as supplemental material). A Transwell system (supplemental Fig. S2a, available at www.jneurosci.org as supplemental material) was used to coculture rat OPCs together with human brain endothelial cells. The presence of brain endothelial cells almost doubled the rate of OPC proliferation in these coculture systems (Fig. 1 b). Since there was no direct cell–cell contact in our Transwell coculture system, these data suggested that cerebral endothelial cells may secrete potent oligo-supportive factors.
To extend these coculture observations, we used a media transfer approach that was previously used to dissect soluble factor coupling in the neurovascular niche (Shen et al., 2004; Guo et al., 2008). Cerebral endothelial cells were grown separately and endothelial-conditioned media was then obtained and added to OPCs (supplemental Fig. S2b, available at www.jneurosci.org as supplemental material). Control OPCs were treated with the same medium derived from empty wells without endothelial cells. Treatment with endothelial-conditioned media significantly amplified OPC proliferation rates over 1–3 d in culture without any obvious changes in cell size or morphology (Fig. 1 c; supplemental Fig. S1b, available at www.jneurosci.org as supplemental material). Furthermore, 48 h treatment of endothelial-conditioned media did not induce myelin basic protein expression in OPCs, suggesting that no significant augmentation of differentiation occurred (supplemental Fig. S1c, available at www.jneurosci.org as supplemental material). Using different dilutions of endothelial-conditioned media allowed us to demonstrate a “dose-dependent” effect, further suggesting a role for soluble oligo-supportive factors that were being produced by the cerebral endothelial cells (Fig. 1 d). There was no significant species effect since conditioned media from the rat brain endothelial cell line RBE.4 was equally oligo-supportive (Fig. 1 e). In addition to promotion of OPC proliferation under normal conditions, we also asked whether cerebral endothelial cells could defend OPCs against stress under injurious conditions. OPCs were subjected to oxygen–glucose deprivation, and the protective effects of endothelial-conditioned media were examined. Endothelial-conditioned media significantly improved OPC survival under these metabolic stress conditions (Fig. 1 f).
If cerebral endothelial cells produced soluble trophic factors that supported OPCs, it should be possible to detect the activation of specific OPC signaling pathways as a result of this type of intercellular coupling. Because the Akt pathway is known to contribute to OPC survival and proliferation (Flores et al., 2000), we asked whether this type of signaling could be detected in our system. OPCs were exposed to endothelial-conditioned media, and Akt activation was biochemically and pharmacologically assessed. Western blots showed that endothelial-conditioned media rapidly induced Akt phosphorylation in OPCs (Fig. 2 a). Blockade of Akt signaling with the PI3-kinase inhibitor LY294002 suppressed phospho-Akt levels (Fig. 2 b) and prevented conditioned media from amplifying OPC proliferation (Fig. 2 c). Next, we assessed the role of Src signaling, which is known to link upstream trophic factors to downstream Akt pathways. If endothelial trophic factors were responsible for activating Akt in the recipient OPCs, then we should be able to document Src signaling in our OPC system as well. PP2, an inhibitor for Src tyrosine kinase, blocked Akt phosphorylation in a dose-dependent manner (Fig. 2 d). In contrast, no effects were seen with the inactive drug PP3 (Fig. 2 e). Correspondingly, PP2 also prevented endothelial-conditioned media from amplifying OPC proliferation (Fig. 2 f).
Figure 2.

a, Conditioned media from human brain endothelial cells (Endo-CM) increased phospho-Akt levels in recipient OPCs. b, Blockade of Akt signaling with the PI3-kinase inhibitor LY294002 prevented Akt from being phosphorylated in OPCs exposed to endothelial-conditioned media (Endo-CM). c, LY294002 blocked the ability of Endo-CM to amplify OPC proliferation. d, e, The Src tyrosine kinase inhibitor PP2 reduces Akt phosphorylation in OPCs exposed to cerebral endothelial-conditioned media. No effects were seen with the inactive drug PP3. f, Correspondingly, PP2 decreases the OPC-proliferative properties of endothelial-conditioned media, and the inactive drug PP3 had no effect. *p < 0.05.
Thus far, the data suggest that cerebral endothelial cells secrete potent soluble mediators that trigger Src and Akt signals within OPCs that support their survival and proliferation. But what mediators might be involved? Cerebral endothelial cells produce a wide range of trophic factors, including FGF and BDNF (Dugas et al., 2008; Guo et al., 2008). To show that these factors were important, cerebral endothelial cells were exposed to various concentrations of sodium nitroprusside (SNP) for 24 h, which triggers nitrosative and oxidative stress. Between 10 and 100 μm levels of SNP, no direct endothelial cell death was induced (Fig. 3 a). However, even in the absence of cytotoxicity, endothelial secretion of both FGF and BDNF were significantly suppressed (Fig. 3 b,c). The final link to oligovascular signaling was then demonstrated using our media transfer protocols again (supplemental Fig. S2c, available at www.jneurosci.org as supplemental material). Various concentrations of SNP (0–100 μm) were added to wells containing either cerebral endothelial cells or empty media, and 24 h later, the conditioned media was removed and added to OPCs. Conditioned media from oxidatively stressed endothelial cells clearly lost the ability to promote OPC proliferation (Fig. 3 d), consistent with the loss of trophic factors such as FGF and BDNF.
Figure 3.
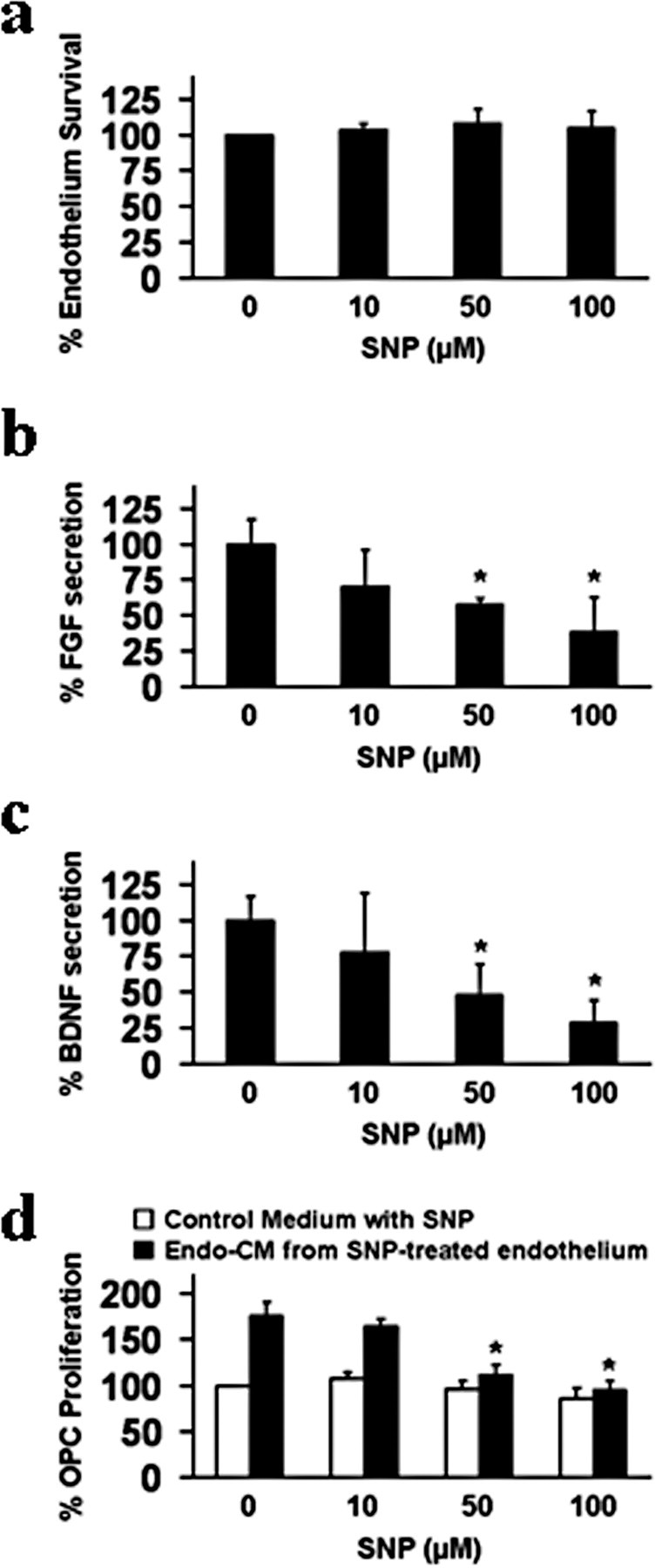
a, Sodium nitroprusside (SNP, 10–100 μm, exposure for 24 h) was not directly cytotoxic to primary human brain endothelial cells. b, c, But at these nonlethal concentrations, SNP significantly downregulated FGF and BDNF production (ELISA) by human brain endothelial cells. d, Conditioned media from primary human brain endothelial cells injured by nonlethal SNP no longer promoted OPC proliferation. Control OPC cultures were treated with empty media with matching levels of SNP. *p < 0.05.
Discussion
The concept of a neurovascular niche is now well accepted. Cerebral endothelial cells produce molecular signals that help sustain the renewal and differentiation of neuronal precursors within pockets of ongoing neurogenesis in adult brain (Dugas et al., 2008; Guo et al., 2008; Guo and Lo, 2009). In the present study, we showed that (1) coculturing OPCs together with cerebral endothelial cells promoted OPC survival and proliferation, (2) conditioned media from cerebral endothelial cells similarly augmented OPCs, (3) endothelial-conditioned media promoted OPC survival via Akt and Src signaling pathways, and (4) subjecting endothelial cells to noncytotoxic levels of oxidative stress (i) downregulated the production of trophic factors like BDNF and FGF and (ii) correspondingly blocked the ability of endothelial cells to support OPC function. Together, these findings support a novel concept of an oligovascular niche, wherein trophic factors released from cerebral endothelium activate Src and Akt signaling to sustain OPC proliferation (Fig. 4).
Figure 4.

A proposed oligovascular niche; cerebral endothelial cells release trophic factors that activate Src, PI3 kinase, and Akt signaling to promote OPC proliferation. Oxidative stress or blockade of Src and Akt signaling prevents endothelial trophic factors from supporting OPCs.
OPCs are widely distributed in adult brain, so the oligovascular niche should provide a mechanism for promoting white matter maintenance. Furthermore, a recent study suggested that OPCs may also serve as a precursor pool for cortical projection neurons (Rivers et al., 2008). Thus, endothelial–OPC interactions may be important for both white and gray matter homeostasis. The ability of cerebral endothelium to produce a large spectrum of trophic factors should be broadly important for sustaining the entire brain parenchyma. Nevertheless, there are a few issues that warrant further studies in the future. First, our cell culture experiments only focus on endothelial–OPC interactions. In vivo, other glial types may also contribute, including pericytes and astrocytes. How trophic signals mediate multicellular interactions should be carefully dissected and validated in vivo. Second, we only show endothelial effects on OPC proliferation by Akt signaling. But Akt may also mediate myelin synthesis in mature oligodendrocytes (Flores et al., 2008). In this study, 48 h treatment of endothelial-conditioned media did not induce myelin basic protein expression in OPC cultures. So at least in our experimental in vitro system, we were unable to detect any obvious ability of endothelial cells to augment myelin synthesis. However, these cell cultures are highly dependent on choice of media conditions. To execute our coculture and media transfer protocols, we had to select “compromise” conditions that allowed both endothelial cells and OPCs to survive. We cannot be certain that different effects may occur under different cell culture conditions. Furthermore, will the endothelial-conditioned media help maintain myelination in mature white matter? These issues can only be clarified in a true in vivo context. Third, our data are limited to measurements of viability and proliferation in OPCs. But Akt signaling is a potent widely applicable prosurvival pathway. It is likely that these Akt pathways might also block complex cell death pathways after oligodendrocytes are damaged by various insults. Although we showed that endothelial-conditioned media protected OPCs against oxygen–glucose deprivation, we cannot rule out the possibility that endothelial-conditioned media merely promoted the proliferation of surviving OPCs under metabolic stress conditions. Without individual cell labeling and tracking, these two interacting scenarios cannot be truly distinguished. More detailed signaling studies to assess oligo-protection by endothelium should be very interesting. Finally, our oligovascular niche hypothesis would naturally suggest that deficiencies in vascular trophic coupling could underlie white matter disease. In our model system, nonlethal levels of oxidative stress downregulates FGF and BDNF, without inducing outright endothelial cell death. Hence, it is possible that sick endothelial cells may lead to sick white matter, even in the absence of ischemia. A clinically relevant correlate in this regard might represent the development of white matter disease and vascular dementia. Is it possible that diseased microvessels and the corresponding oligovascular disruptions are broadly responsible for white matter dysfunction in neurodegenerative disorders? What range of trophic factors are affected? And are there ways to treat white matter disease by rescuing oligovascular signaling?
In recent years, it has become clear that the cerebral endothelium comprises much more than just inert plumbing for the brain. Matrix and trophic interactions between endothelial cells and neurons sustain neurogenesis (Leventhal et al., 1999; Shen et al., 2004) and may protect neurons against oxidative and metabolic insults (Dugas et al., 2008; Guo et al., 2008). Analogous interactions within a widely distributed oligovascular niche may provide a similar mechanism for sustaining white matter renewal and integrity. Further studies are warranted to dissect how these mechanisms function in normal brain, and how disruptions in oligovascular signaling may underlie white matter disease and neurodegeneration.
Footnotes
This work was supported in part by the Deane Foundation, American Heart Association, and National Institutes of Health Grants R37-NS37074, R01-NS48422, R01-NS53560, P50-NS10828, and P01-NS55104. We thank Drs. Paul Rosenberg, Xiaoying Wang, Klaus van Leyen, Shuzhen Guo, Guang Jin, Jianfeng Xu, and Josephine Lok for many helpful discussions.
References
- Chopp M, Zhang ZG, Jiang Q. Neurogenesis, angiogenesis, and MRI indices of functional recovery from stroke. Stroke. 2007;38:827–831. doi: 10.1161/01.STR.0000250235.80253.e9. [DOI] [PubMed] [Google Scholar]
- Dugas JC, Mandemakers W, Rogers M, Ibrahim A, Daneman R, Barres BA. A novel purification method for CNS projection neurons leads to the identification of brain vascular cells as a source of trophic support for corticospinal motor neurons. J Neurosci. 2008;28:8294–8305. doi: 10.1523/JNEUROSCI.2010-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores AI, Mallon BS, Matsui T, Ogawa W, Rosenzweig A, Okamoto T, Macklin WB. Akt-mediated survival of oligodendrocytes induced by neuregulins. J Neurosci. 2000;20:7622–7630. doi: 10.1523/JNEUROSCI.20-20-07622.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores AI, Narayanan SP, Morse EN, Shick HE, Yin X, Kidd G, Avila RL, Kirschner DA, Macklin WB. Constitutively active Akt induces enhanced myelination in the CNS. J Neurosci. 2008;28:7174–7183. doi: 10.1523/JNEUROSCI.0150-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg DA, Jin K. From angiogenesis to neuropathology. Nature. 2005;438:954–959. doi: 10.1038/nature04481. [DOI] [PubMed] [Google Scholar]
- Guo S, Lo EH. Dysfunctional cell-cell signaling in the neurovascular unit as a paradigm for central nervous system disease. Stroke. 2009;40(Suppl 3):S4–S7. doi: 10.1161/STROKEAHA.108.534388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S, Kim WJ, Lok J, Lee SR, Besancon E, Luo BH, Stins MF, Wang X, Dedhar S, Lo EH. Neuroprotection via matrix-trophic coupling between cerebral endothelial cells and neurons. Proc Natl Acad Sci U S A. 2008;105:7582–7587. doi: 10.1073/pnas.0801105105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat Rev Neurosci. 2004;5:347–360. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci. 2007;10:1369–1376. doi: 10.1038/nn2003. [DOI] [PubMed] [Google Scholar]
- Leventhal C, Rafii S, Rafii D, Shahar A, Goldman SA. Endothelial trophic support of neuronal production and recruitment from the adult mammalian subependyma. Mol Cell Neurosci. 1999;13:450–464. doi: 10.1006/mcne.1999.0762. [DOI] [PubMed] [Google Scholar]
- Lok J, Gupta P, Guo S, Kim WJ, Whalen MJ, van Leyen K, Lo EH. Cell-cell signaling in the neurovascular unit. Neurochem Res. 2007;32:2032–2045. doi: 10.1007/s11064-007-9342-9. [DOI] [PubMed] [Google Scholar]
- Ness JK, Valentino M, McIver SR, Goldberg MP. Identification of oligodendrocytes in experimental disease models. Glia. 2005;50:321–328. doi: 10.1002/glia.20206. [DOI] [PubMed] [Google Scholar]
- Rivers LE, Young KM, Rizzi M, Jamen F, Psachoulia K, Wade A, Kessaris N, Richardson WD. PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat Neurosci. 2008;11:1392–1401. doi: 10.1038/nn.2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Q, Goderie SK, Jin L, Karanth N, Sun Y, Abramova N, Vincent P, Pumiglia K, Temple S. Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science. 2004;304:1338–1340. doi: 10.1126/science.1095505. [DOI] [PubMed] [Google Scholar]
- van Leyen K, Arai K, Jin G, Kenyon V, Gerstner B, Rosenberg PA, Holman TR, Lo EH. Novel lipoxygenase inhibitors as neuroprotective reagents. J Neurosci Res. 2008;86:904–909. doi: 10.1002/jnr.21543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacchigna S, Lambrechts D, Carmeliet P. Neurovascular signalling defects in neurodegeneration. Nat Rev Neurosci. 2008;9:169–181. doi: 10.1038/nrn2336. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]