

Abstract
Gastric cancer overexpressing the human epidermal growth factor 2 (HER2) protein has a poor outcome, although a combination of chemotherapy and the anti-HER2 antibody trastuzumab has been approved for the treatment of advanced gastric cancer. Vascular endothelial growth factor (VEGF) expression in gastric cancer is correlated with recurrence and poor prognosis; however, the anti-VEGF antibody bevacizumab has shown limited efficacy against gastric cancer in clinical trials. In this study, we evaluated the antitumor effects of trastuzumab; VEGF-Trap binding to VEGF-A, VEGF-B and placental growth factor (PlGF); and a combination of trastuzumab and VEGF-Trap in a gastric cancer xenograft model. Although trastuzumab and VEGF-Trap each moderately inhibited tumor growth, the combination of these agents exerted greater inhibition compared with either agent alone. Immunohistochemical analyses indicated that the reduction in tumor growth was associated with decreased proliferation and increased apoptosis of tumor cells and decreased tumor vascular density. The combined treatment resulted in fewer proliferating tumor cells, more apoptotic cells and reduced tumor vascular density compared with treatment with trastuzumab or VEGF-Trap alone, indicating that trastuzumab and VEGF-Trap had additive inhibitory effects on the tumor growth and angiogenesis of the gastric cancer xenografts. These data suggest that trastuzumab in combination with VEGF-Trap may represent an effective approach to treating HER2-overexpressing gastric cancer.
Keywords: combination treatment, gastric cancer, HER2, trastuzumab, VEGF-Trap
Introduction
Human epidermal growth factor 2 (HER2, also referred to as HER2/neu or ErbB-2) is overexpressed and/or amplified in several human cancers.1 HER2 signaling promotes cell proliferation through the RAS-mitogen-activated protein kinase (MAPK) pathway and inhibits cell death through the phosphatidylinositol 3-kinase (PI3K)-Akt pathway.2 In addition, HER2 signaling upregulates the expression of vascular endothelial growth factor (VEGF), which is critical for tumor angiogenesis.3, 4 HER2 overexpression and amplification have central roles in the initiation, progression and metastasis of breast cancer and gastric cancer and are associated with poor prognosis.5, 6, 7 Trastuzumab, a humanized antibody that binds to the extracellular domain of HER2, has been effectively used as a targeted therapy for HER2-overexpressing metastatic breast cancer.8, 9 The proposed mechanisms of trastuzumab's action involve antiproliferative effects via the inhibition of signaling pathways, downregulation of the HER2 protein, the activation of apoptotic signals in tumor cells, the inhibition of angiogenesis and antibody-dependent cell-mediated cytotoxicity (ADCC).10 However, HER2 inhibition alone cannot completely abolish VEGF expression because this expression is regulated by many other factors.11, 12, 13, 14, 15 Combined targeting of HER2 and VEGF exerts a greater inhibition of breast carcinoma xenograft growth than does either treatment alone through improved inhibition of both tumor cell proliferation and tumor angiogenesis.16, 17 The combined targeting approach has been translated into breast cancer clinical trials, which have provided encouraging results.18, 19, 20
VEGF signaling promotes angiogenesis by stimulating vascular endothelial cell proliferation, migration and tube formation and markedly increases vascular permeability.21 The VEGF family includes five members, VEGF-A, VEGF-B, VEGF-C, VEGF-D and placental growth factor (PlGF), that bind differentially to VEGF receptors (VEGFRs) 1, 2 and 3 and neuropilin, with different specificities.22, 23 VEGF-A and VEGFR2 are primarily responsible for both normal and tumor-related angiogenesis.24 NRP-1 (N (asparagine)-Rich Protein 1) acts as a coreceptor for VEGF-A, increasing the protein's affinity for VEGFR2, and promotes tumor progression and angiogenesis.25 Most human cancers express VEGF-A, and many express abnormally high levels of VEGF-A, which has been associated with poor prognosis.26 VEGF-B and PlGF also have roles in tumor angiogenesis,27, 28, 29, 30, 31, 32 and VEGF-C and VEGF-D have been implicated in lymphangiogenesis.33, 34
Advances in the understanding of tumor angiogenesis have led to the development of antiangiogenic therapies for the treatment of cancers.35 Bevacizumab, a humanized antibody that binds to human VEGF-A and tyrosine kinase inhibitors, such as sunitinib, sorafenib and pazopanib, which primarily target VEGFRs and also inhibit other tyrosine kinases, has been approved for the treatment of cancers.35, 36 Despite their benefits, currently approved antiangiogenic therapies produce initial responses that are followed by a restoration of tumor growth and progression, which is thought to be related to the development of resistance.37 In one approach to combined targeting, a biologic combination of bevacizumab and trastuzumab was clinically feasible and active in HER2-amplified recurrent or metastatic breast cancer in a phase II study.19 However, in recent clinical trials, the addition of bevacizumab to combined therapy with trastuzumab and a chemodrug such as docetaxel or vinorelbine did not significantly improve the therapeutic benefit in patients with HER2-positive metastatic breast cancer.38, 39 VEGF-Trap is a decoy receptor fusion protein that consists of the second immunoglobulin (Ig) domain of VEGFR1 and the third Ig domain of VEGFR2 fused to the Fc of human IgG1.40 VEGF-Trap has a high affinity for human and mouse VEGF-A, VEGF-B and PlGF.41 VEGF-Trap inhibits tumor growth, angiogenesis, metastasis and ascites formation in human xenografts of various cancers (melanoma, glioma, pancreatic cancer, ovarian cancer, glioblastoma and renal cell carcinoma).42 In a phase III trial, a combination of VEGF-Trap and chemotherapy resulted in significant improvements in the overall survival and progression-free survival of patients with metastatic colorectal cancer.40 VEGF-Trap in combination with trastuzumab produced greater inhibition of breast carcinoma xenografts than either individual treatment.16
The clinical success of trastuzumab in breast cancer therapy has led to studies investigating its antitumor activity toward other HER2-overexpressing cancers, including gastric adenocarcinomas.43 Metastatic gastric cancer has a poor outcome.7 HER2 has been implicated as an important regulator of gastric cancer cell growth.6 Recent TOGA trials showed that approximately 22% of gastric cancers overexpressed HER2, and trastuzumab improved the median overall survival from 11.1 to 13.8 months when administered with chemotherapy.44 Thus, trastuzumab was approved for the treatment of advanced gastric cancer. Bevacizumab could also effectively suppress tumor growth and metastasis in a gastric cancer model.45 Recently, in a randomized phase III trial in gastric cancer (AVAGAST), the addition of bevacizumab to chemotherapy as a first-line treatment for advanced gastric cancer was associated with significant increases in progression-free survival; however, this combination did not meet the primary objective.46 Here, we evaluated the antitumor efficacies of trastuzumab, VEGF-Trap and a combination of trastuzumab and VEGF-Trap in an HER2-overexpressing NCI-N87 gastric cancer xenograft model.
Materials and methods
Reagents
Recombinant human VEGF165, VEGF-B and PlGF and recombinant mouse VEGF164 were purchased from R&D Systems (Minneapolis, MN, USA). Recombinant human epidermal growth factor (EGF) was purchased from Roche Diagnostic GmbH (Mannheim, Germany). Trastuzumab (Herceptin; Roche, Basel, Switzerland), bevacizumab (Avastin; Roche) and palivizumab (Synagis; Abbott Laboratories, Berkshire, UK) were purchased from distributors. Palivizumab was used as an IgG1 isotype control for the in vitro and in vivo experiments.
Cell culture
The human gastric cancer cell line NCI-N87, in which HER2 gene amplification has been demonstrated previously,47 was obtained from the Korea Cell Line Bank (Seoul, Korea). The cells were cultured in RPMI-1640 (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (HyClone, Tauranga, New Zealand) and 1 × penicillin–streptomycin (Gibco). NCI-N87Luc+ cells were constructed from NCI-N87 cells at Chuncheon Center, Korea Basic Science Institute, and cultured under the same conditions as the NCI-N87 cells. HEK293T cells were grown in Dulbecco's modified Eagle's medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum. Human umbilical vein endothelial cells (HUVECs) from passage 2 were cultured in endothelial basal medium-2 supplemented with an EGM-2 SingleQuot Kit (Lonza, Walkersville, MD, USA). All cells were cultured in 5% CO2 in a 37 °C humidified incubator.
Construction, expression and purification of VEGF-Trap
VEGF-Trap was constructed as described previously.40 Briefly, a fusion gene encoding the mouse immunoglobulin heavy-chain leader sequence (MEWSWVFLFFLSVTTGVHS; accession number: A0N1R4_MOUSE); domain 2 of human VEGFR1 and domain 3 of human VEGFR2, linked to the lower part of the hinge; and CH2 and CH3 of human IgG1 was synthesized by GeneArt (Regensburg, Germany) and cloned into the pJK-dhfr2 expression vector (Aprogen, Korea). The resulting expression plasmid, pJK-dhfr2-VEGF-Trap, was introduced into HEK293T cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. The transfected cells were cultured in the protein-free medium CD293 (Invitrogen). Protein was purified from the culture supernatant by affinity chromatography on a Protein A column (Millipore, Temecula, CA, USA). The protein concentration was determined with a NanoDrop (Thermo Scientific, Wilmington, DE, USA), based on the molar extinction coefficient. The integrity of the purified protein was measured on an Agilent 2100 Bioanalyser (Agilent Technologies, Waldbronn, Germany).
Flow cytometry
NCI-N87Luc+ cells were incubated with 1 μg of primary antibody in 100 μl of PBA (phosphate-buffered saline with 0.1% bovine serum albumin) for 60 min at 4 °C. After washing three times with phosphate-buffered saline with 0.1% bovine serum albumin, the cells were incubated with a fluorescein isothiocyanate-conjugated anti-Fc antibody (BD Pharmingen, San Diego, CA, USA) for 30 min at 4 °C. Propidium iodide-negative cells were analyzed for antibody binding using a FACSCalibur (Becton Dickinson, Franklin Lakes, NJ, USA). VEGF was detected after cell fixation in methanol, cell permeabilization with 0.1% phosphate-buffered saline-Tween 20, and staining with the anti-VEGF monoclonal antibody bevacizumab.
For cell proliferation and cell cycle analyses, NCI-N87Luc+ cells at 70–80% confluence were serum-starved overnight and mock-incubated or incubated with VEGF165 (100 ng ml−1) or EGF (20 ng ml−1) for 24 h before pulsing with 20 μM bromodeoxyuridine (BrdU) (BD Pharmingen) for 6 h. For antibody treatment, cells at 70–80% confluence were incubated in serum-containing medium overnight and treated with 333 nM IgG, trastuzumab or VEGF-Trap for 48 h before pulsing with 20 μM BrdU. The cells were trypsinized and stained using an APC BrdU Flow Kit (BD Pharmingen) according to the manufacturer's instructions. The number of proliferating cells was analyzed using a FACSAria (Becton Dickinson).
RT-PCR
Total RNA was isolated from HUVECs and NCI-N87Luc+ cells with an Easy-Spin Total RNA Extraction Kit (iNtRON Biotechnology, Seongnam, Korea), followed by cDNA synthesis with a Transcriptor High Fidelity cDNA Synthesis Kit(Roche Diagnostics GmbH, Mannheim, Germany). The polymerase chain reactions (PCRs) were performed in a thermocycler (TaKaRa, Shiga, Japan) with the following cycling parameters: denaturation at 95 °C for 5 min in the first cycle and for 30 s in the second cycle, annealing at 55 °C for 30 s and elongation at 72 °C for 30 s for 30 repetitive cycles. A final extension was performed at 72 °C for 10 min. The primers used for the amplification of the cDNAs were as follows: human VEGF (forward 5′-ATGAACTTTCTGCTCTCTGG-3′ reverse 5′-TCATCTCTCCTATGTGCTGGC-3′), human VEGFR1 (forward 5′-GCACCTTGGTTGTGGGTGAC-3′ reverse 5′-CGTGCTGCTTCCTGGTCC-3′), human VEGFR2 (forward 5′-CATGTGGTCTCTCTGGTTGTG-3′ reverse 5′-TCCCTGGAAGTCCTCCACACT-3′), human NRP-1 (forward 5′-ACGATGAATGTGGCGATACT-3′ reverse 5′-AGTGCATTCAAGGCTGTTGG-3′) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (forward 5′-ATCACCATCTTCCAGGAGCG-3′ reverse 5′-CCTGCTTCACCACCTTCTTG-3′).
In vitro cell proliferation assay
For the WST-1 assay, NCI-N87Luc+ cells were seeded at a density of 5 × 103 cells per well in 100 μl of medium in 96-well plates and incubated at 37 °C in 5% CO2 for 24 h. Various dilutions of isotype-control IgG, trastuzumab or VEGF-Trap were then added to each well, and incubation was continued for 72 h. After incubation, 10 μl of WST-1 (Roche Diagnostics GmbH) was added to each well, and the plate was incubated in 5% CO2 at 37 °C for 4 h. The absorbance was measured at 440 nm with a reference wavelength of 610 nm. The percent viability was calculated by comparison with the IgG-treated control.
Western blot analysis
NCI-N87Luc+ cells were grown to 70–80% confluence and treated with 6.6 nM isotype-control IgG, trastuzumab or VEGF-Trap for 72 h under the cell culture conditions. The cells were lysed in radio immunoprecipitation assay buffer (25 mM Tris-HCl, pH 7.6; 150 mM NaCl; 1% NP-40; 1% sodium deoxycholate; and 0.1% SDS) containing cOmplete Protease Inhibitor Cocktail (Roche Diagnostics GmbH) and PhosSTOP Phosphatase Inhibitor Cocktail (Roche Diagnostics GmbH). The proteins were resolved on denaturing polyacrylamide gels under reducing conditions and transferred to polyvinylidene difluoride membranes. The blots were probed with antibodies (Cell Signaling Technology, Beverly, MA, USA) to phospho-Erk 1/2, phospho-Akt, Erk 1/2, pan-Akt or β-actin, and the immunoreactive bands were detected using appropriate horse radish peroxidase-conjugated secondary antibodies (Cell Signaling Technology) and a WEST-ZOL PLUS chemiluminescent detection system (iNtRON Biotechnology). To detect total Erk 1/2, Akt and β-actin protein, the membranes were stripped with stripping buffer (100 mM 2-mercaptoethanol; 2% sodium dodecyl sulfate; and 62.5 mM Tris-HCl, pH 6.7) for 30 min at 65 °C and reprobed.
ADCC assay
Peripheral blood mononuclear cells were isolated from donor blood by Histopaque (Sigma-Aldrich, St Louis, MD, USA) density centrifugation. NCI-N87Luc+ cells (5 × 103 per well), which were used as target cells, were plated into a 96-well plate in phenol-red-free RPMI-1640 medium containing 1% fetal bovine serum and preincubated for 30 min in a humidified CO2 incubator at 37 °C. After 30 min, freshly prepared peripheral blood mononuclear cells, serving as effector cells, were added to each well at an effector:target (E:T) ratio of 40:1 and incubated for 4 h in a humidified CO2 incubator at 37 °C. The cytotoxicity was determined by measuring the activity of lactate dehydrogenase (LDH) released from the cytosol of the damaged cells using a Cytotoxicity Detection Kit PLUS (LDH) (Roche Diagnostics GmbH). The percentage of cytotoxicity was calculated as: specific lysis (%)=(experimental release−spontaneous release)/(maximum release (by lysis buffer)−spontaneous release) × 100.
In vivo tumor growth inhibition study
NCI-N87Luc+ cells (5 × 106 in 0.1 ml phosphate-buffered saline) were implanted subcutaneously into BALB/c Slc-nu nude mice (Japan SLC, Hamamatsu, Japan). Tumor-bearing mice were randomized into control and treatment groups (n=7 per group). When tumors reached approximately 80–120 mm3 in size, averaging 100 mm3 per group, palivizumab (10 mgkg−1), a humanized antibody to respiratory syncytial virus F protein (as an isotype control), VEGF-Trap (6.6 mg kg−1), trastuzumab (10 mg kg−1) or VEGF-Trap plus trastuzumab (6.6+10 mg kg−1) was intravenously injected into each mouse three times per week for 4 weeks. Body weight and tumor volume were measured two times per week. The tumor volumes were calculated as follows: tumor volume (mm3)=L (mm) × W2 (mm2) × 1/2, where L is the tumor length and W is the perpendicular tumor width.
Immunohistochemistry and immunofluorescence staining
Tumors were fixed in 4% paraformaldehyde and embedded in tissue-freezing medium (Sakura, Tokyo, Japan). Hematoxylin and eosin staining was performed according to standard procedures. To differentiate between viable and necrotic areas, tissue sections were stained with YO-PRO-1 (1:1000; Invitrogen).48 Proliferating cells were identified using a rabbit anti-phospho-histone-H3 (Ser10) antibody (1:200; Millipore). Apoptotic cells were detected using a rabbit anti-activated caspase-3 antibody (1:200; Millipore). Endothelial cells were detected using a rabbit anti-CD31 antibody (1:100; Abcam, Cambridge, UK). The secondary antibody was a goat anti-rabbit IgG (H+L) labeled with Alexa 555 (1:1000; Molecular Probes, Eugene, OR, USA). In control experiments, the primary antibody was omitted. Fluorescent signals were visualized and images were captured using an Olympus FluoView laser scanning microscope (Olympus, Tokyo, Japan).
For hematoxylin and eosin, YO-PRO-1, phospho-histone-H3 and activated caspase-3 staining, 20-μm-thick tissue sections were cut from the middle of a tumor. To differentiate between the viable and necrotic areas of the tumor, multiple low-magnification ( × 4 objective) images, each measuring 3175 μm × 3175 μm, were captured. These images were used to create a composite image in Adobe Photoshop CS5 (Adobe Systems, San Jose, CA, USA). The green fluorescent threshold was adjusted as described previously.48 The images were quantified with the ImageJ software program (http://rsb.info.nih.gov/ij). The total viable (green) and necrotic (black) areas of the tumor were calculated by multiplying the total number of pixels of each color by 38.6 μm2 (the area of each pixel). The area densities of phospho-histone-H3 and activated caspase-3 were measured by capturing five digital images (635 μm × 635 μm each; × 20 objective lens) of the viable (YO-PRO-1-positive) areas of the tumor, including one image from each quadrant of the tumor periphery and one from the center of the tumor. To detect blood vessels, 50-μm-thick tissue sections were used. Density measurements of CD31-positive blood vessels in the tumors were made based on four fields in the intratumoral and peritumoral regions (1270 × 1270 μm2 each; × 10 objective lens). The ImageJ software program was used for all of the density measurements, and the Olympus FluoView v. 1.7a software program was used to capture and process the images.
Statistics
The values are represented as the mean±s.e.m. Significant differences between the means were determined by analysis of variance followed by the Newman–Keuls test. The Student's t-test was used to compare significant differences between the means of two groups. A value of P<0.05 was considered statistically significant.
Results
Production and characterization of VEGF-Trap
VEGF-Trap was transiently expressed in HEK293T cells and purified from the culture supernatant by affinity chromatography on Protein A. Analysis of the purified protein by sodium dodecyl sulfate-polyacrylamide gel electrophoresis indicated that the protein had an apparent molecular mass that was higher than its molecular weight (97 kDa) based on the primary sequence (Supplementary Figure S1a). This result was likely due to the five N-glycosylation sites in each chain of the protein.
The purified VEGF-Trap bound to human and mouse VEGF-A, human VEGF-B and human PlGF (Supplementary Figures S1b and c), as reported previously.41 To test the ability of VEGF-Trap to inhibit VEGF signaling, HUVECs were incubated with human VEGF-A (40 ng ml−1) for 5 min in the presence of IgG, trastuzumab or VEGF-Trap. As shown in Supplementary Figure S1d, VEGF robustly activated VEGFR2, as indicated by the phosphorylation of the residue tyrosine 1175. Moreover, VEGF-Trap inhibited VEGF-induced VEGFR2 phosphorylation, whereas IgG and trastuzumab did not yield this result. To confirm the antiangiogenic activity of VEGF-Trap, we performed WST-1 proliferation assays and transwell migration assays. VEGF-Trap efficiently inhibited VEGF165-induced proliferation and the chemotactic migration of HUVECs (Supplementary Figures S1e and f). Purified VEGF-Trap was used in the subsequent experiments.
Expression of HER2 and VEGF in NCI-N87Luc+ cells
Previous studies have demonstrated that NCI-N87 cells overexpress HER2.47 Indeed, trastuzumab bound to the surface of NCI-N87Luc+ cells at a high level in the flow cytometric analysis (Figure 1a). Because HER2 signaling is known to upregulate VEGF expression, the NCI-N87Luc+ cells were permeabilized and analyzed in the flow cytometric analysis using bevacizumab, which revealed VEGF expression (Figure 1b). Densitometry analysis of the reverse transcriptase-polymerase chain reaction (RT-PCR) results indicated that the level of VEGF expression in NCI-N87Luc+ cells was slightly higher than in HUVECs (Figure 1c).
Figure 1.

Human epidermal growth factor 2 (HER2) and vascular endothelial growth factor (VEGF) expression levels in gastric cancer cells. NCI-N87Luc+ cells express HER2 (a) and VEGF (b), as detected by flow cytometry. (c) Detection of the expression levels of VEGF and VEGF-related receptors (VEGFR) by reverse transcriptase-polymerase chain reaction (RT-PCR). GAPDH, glyceraldehyde 3-phosphate dehydrogenase; HUVEC, human umbilical vein endothelial cell; NRP-1, neuropilin-1.
Because the NCI-N87Luc+ cells expressed VEGF-A, we examined the expression of VEGF-related receptors in the cells by RT-PCR analysis. VEGFR1 and VEGFR2 mRNAs were detected at low levels, whereas the NRP-1 mRNA level was elevated (Figure 1c). Considering that VEGFR2 is the key mediator of VEGF-induced angiogenesis, although NCI-N87Luc+ cells express VEGF, the cells may not depend on VEGF for cell proliferation and survival. The addition of VEGF (100 ng ml−1) to the culture medium did not stimulate the proliferation of NCI-N87Luc+ cells, whereas EGF (20 ng ml−1) stimulated the cells, as indicated by the increased number of S-phase cells (Figures 2a and b).
Figure 2.

Analysis of bromodeoxyuridine (BrdU) incorporation and total DNA content. (a and b) Epidermal growth factor (EGF)-induced cell cycle progression in NCI-N87Luc+ cells. Cells were serum-starved overnight and treated with vascular endothelial growth factor (VEGF) or EGF for 24 h. The cells were then pulsed with BrdU for 6 h, fixed and stained with a allophycocyanin (APC)-coupled anti-BrdU antibody and 7-aminoactinomycin D (7-AAD). (c and d) Cells (70–80% confluence) were treated with immunoglobulin G (IgG), trastuzumab or VEGF-Trap for 48 h and pulsed with BrdU for 6 h. The cells were then fixed and stained with an APC-coupled anti-BrdU antibody and 7-AAD. The stained cells were analyzed by a FACSAria flow cytometer (n=3; *P<0.05 versus control).
Trastuzumab decreased proliferation of NCI-N87Luc+ cells and VEGF expression in vitro
Trastuzumab inhibited the proliferation of NCI-N87Luc+ cells in a dose-dependent manner in the WST-1 assay (Figure 3a). At a concentration of 10 μg ml−1, trastuzumab decreased cell proliferation by approximately 40%. Consistent with this finding, after treatment with trastuzumab, the number of cells in the G0/G1 phase increased by more than twofold (Figures 2c and d), and inhibition of Erk 1/2 and Akt phosphorylation was observed (Figure 3b). In contrast, VEGF-Trap did not inhibit the proliferation of NCI-N87Luc+ cells or cell signaling (Figures 2c, d and 3a, b). To examine whether trastuzumab treatment downregulates VEGF expression in NCI-N87Luc+ cells, the cells were treated with 10 μg ml−1 trastuzumab or IgG, and VEGF mRNA was assessed by RT-PCR. As expected, the level of VEGF mRNA in cells treated with trastuzumab was lower than in the control cells (Figure 3c).
Figure 3.

Antitumor and antiangiogenic activity of trastuzumab (Tr) in vitro. (a) In vitro proliferation (WST-1 assay) of NCI-N87Luc+ cells. (b) Inhibition of Akt and Erk 1/2 phosphorylation by trastuzumab. (c) Measurement of vascular endothelial growth factor (VEGF) mRNA in response to trastuzumab treatment in vitro by reverse transcriptase-polymerase chain reaction (RT-PCR). (d) In vitro trastuzumab-mediated antibody-dependent cell-mediated cytotoxicity (ADCC) against human epidermal growth factor 2 (HER2)-positive gastric cancer cells. GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Trastuzumab-mediated ADCC in vitro
To analyze the ADCC of trastuzumab using NCI-N87Luc+ cells as target cells, a standard LDH assay was performed using human peripheral blood mononuclear cells as effector cells. The NCI-N87Luc+ cells were preincubated with serially diluted trastuzumab, VEGF-Trap or IgG, and then incubated with a 40:1 effector:target (E:T) ratio of human peripheral blood mononuclear cells. Trastuzumab (but not VEGF-Trap or IgG) effectively mediated ADCC in a dose-dependent manner (Figure 3d).
Antitumor activity of trastuzumab, VEGF-Trap or trastuzumab plus VEGF-Trap in vivo
To compare the in vivo antitumor activity of trastuzumab, VEGF-Trap and trastuzumab plus VEGF-Trap in an NCI-N87Luc+ nude mouse xenograft model, each of the treatments or palivizumab (isotype control) was intravenously injected three times per week into nude mice bearing NCI-N87Luc+ xenografts for 28 days. Trastuzumab and VEGF-Trap each moderately inhibited tumor growth without affecting body weight; however, the combination of trastuzumab and VEGF-Trap showed greater inhibition compared with either agent alone (Figures 4a and b). Tumor volume was reduced by 23.5 and 29.9% in the VEGF-Trap and trastuzumab treatment groups, respectively, compared with the isotype control, whereas the combined treatment decreased the tumor volume by 52%. The tumor weight was decreased by 23.6% following the VEGF-Trap treatment and by 16.4% following the trastuzumab treatment, whereas the weight was decreased by 41.2% after the combined treatment (Figure 4c).
Figure 4.
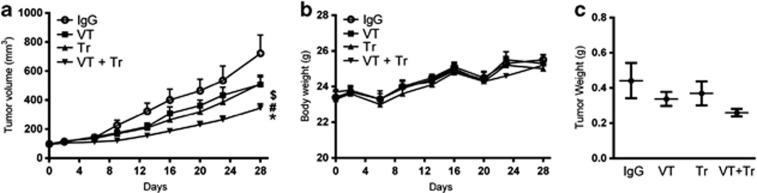
The combined inhibition of human epidermal growth factor 2 (HER2) and vascular endothelial growth factor (VEGF) reduces tumor growth more efficiently than single-agent inhibition. (a) Mice bearing NCI-N87Luc+ tumors were divided into four groups and treated with an isotype-control antibody, VEGF-Trap (VT), trastuzumab (Tr) or a combination of VEGF-Trap and trastuzumab (VT+Tr) for 28 days. (b) Changes in body weight. No significant change in body weight was observed among the treatment groups. (c) Dissected tumor weight after 28 days (n=7; *P<0.05 versus isotype control; #P<0.05 versus VEGF-Trap; $P<0.05 versus trastuzumab). IgG, immunoglobulin G.
To understand the mechanisms by which tumor growth was inhibited, viable areas of the tumors were identified by YO-PRO-1 nuclear staining, which served as a fluorescent equivalent to conventional hematoxylin and eosin staining (Supplementary Figure S2). As shown in Figure 5a, although the control tumors exhibited large viable regions, the tumors treated with VEGF-Trap, trastuzumab or both agents were smaller in overall size and displayed smaller viable regions. The viable area of the tumors was reduced by 50% after treatment with VEGF-Trap or trastuzumab and by 66% after the combined treatment compared with the control tumors (Figure 5b). Necrosis was observed near the center and was reduced at the periphery in all of the tumor groups. However, no significant difference in the total necrotic area was observed among the groups, although the trastuzumab-treated tumors showed a slightly reduced necrotic area (Figure 5c). Thus, the ratio of viable to necrotic area decreased significantly after VEGF-Trap treatment or the combined treatment in comparison with the control tumors, whereas the ratio did not decrease following trastuzumab treatment alone (Figure 5d).
Figure 5.

Viable and necrotic areas of tumors at the end of treatment. (a) Viable (green) and necrotic (black) areas, as differentiated by YO-PRO-1 staining and quantified by the ImageJ software program. The scale bar represents 1 mm. Viable (b) and necrotic tumor areas (c) after 28 days of treatment. (d) Viable and necrotic tumor areas are presented as a ratio (n=7; *P<0.05 versus isotype control; #P<0.05 versus vascular endothelial growth factor (VEGF)-Trap (VT); $P<0.05 versus trastuzumab (Tr)). IgG, immunoglobulin G.
Cell proliferation and apoptosis in the viable tumor regions were assessed by staining for phospho-histone-H3 and activated caspase-3, respectively. The number of phospho-histone-H3-positive cells was reduced by 58.3% after VEGF-Trap treatment, by 57.5% after trastuzumab treatment and by 77% after the combined treatment (Figures 6a and c). The apoptotic cells identified based on activated caspase-3 expression were located throughout the viable regions of the tumors (Figure 6b). The fraction of activated caspase-3 staining, expressed as a percentage of the tumor area, was not significantly increased after VEGF-Trap or trastuzumab treatment but increased by more than fivefold after the combined treatment in comparison with the control tumors (Figure 6d).
Figure 6.

The effect of treatment on tumor cell proliferation and apoptosis. Immunohistochemical staining of phospho (p)-histone-H3 (a and c) and activated caspase-3 (b and d) after 28 days of treatment. The scale bar represents 100 μm. The inset high-magnification ( × 60 objective lens) image shows a YO-PRO-1 (green)-stained nucleus and phospho-histone-H3 or activated caspase-3 (red) (n=7; *P<0.05 versus isotype control; #P<0.05 versus vascular endothelial growth factor (VEGF)-Trap (VT); $P<0.05 versus trastuzumab (Tr)).
Tumor blood vessels were analyzed by staining with an anti-CD31 antibody. The tumor vascular density was significantly decreased in the treatment groups compared with the control group (Figure 7a). In peritumoural regions, an approximately 50% decrease in tumor vascular density was observed after treatment with VEGF-Trap or trastuzumab, and an approximately 80% decrease was noted after the combined treatment (Figure 7b). In intratumoral regions, the decrease in microvessel density was approximately similar in all of the treatment groups, as follows: 85% after VEGF-Trap treatment, 74.5% after trastuzumab treatment and 80.75% after the combined treatment (Figure 7c).
Figure 7.

The effect of treatment on vascular density. Two-dimensional images constructed from Z-stack images showing CD31-positive blood vessels in the peritumoural (a and c) and intratumoral (a and d) regions. The black dotted line indicates the boundary of the tumors. The scale bar represents 100 μm (n=7; *P<0.05 versus isotype control; #P<0.05 versus vascular endothelial growth factor (VEGF)-Trap (VT); $P<0.05 versus trastuzumab (Tr)). IgG, immunoglobulin G.
Discussion
HER2-overexpressing gastric cancer has a poor outcome. In recent clinical trials, trastuzumab plus chemotherapy improved the survival of patients with gastric cancer and was therefore approved for the treatment of advanced gastric cancer. However, bevacizumab has demonstrated limited success in gastric cancer therapy. In the present study, we aimed to evaluate whether a combined blockade of HER2 and VEGF signaling exerts a better inhibition of gastric cancer growth than a blockade of either protein alone by testing the therapeutic efficacies of trastuzumab, VEGF-Trap and a combination of trastuzumab and VEGF-Trap in HER2-expressing NCI-N87Luc+ gastric cancer xenografts. Trastuzumab and VEGF-Trap each moderately inhibited tumor growth; however, the combination of these agents exerted greater inhibition compared with either agent alone. The combined treatment resulted in fewer proliferating tumor cells, more apoptotic cells and reduced tumor vascular density compared with treatment with trastuzumab or VEGF-Trap alone, indicating that trastuzumab and VEGF-Trap had additive inhibitory effects on the tumor growth and angiogenesis of the gastric cancer xenografts. Our study is the first to demonstrate that a combined blockade of HER2 and VEGF signaling exerts greater inhibition of HER2-positive human gastric cancer than individual blockade.
We have speculated on the mechanisms underlying the dual blockade of tumor angiogenesis by trastuzumab and VEGF-Trap. In a study on HER2-overexpressing breast cancer cells, trastuzumab treatment downregulated the expression of VEGF and IL-8 through inhibition of the PI3K-Akt pathway and upregulated the expression of the antiangiogenic factor thrombospondin-1 through activation of the p38 MAPK pathway.4, 13, 49 In the present study, the treatment of gastric cancer cells with trastuzumab resulted in reduced levels of Akt phosphorylation, Erk phosphorylation and VEGF expression. This result suggests that trastuzumab decreased VEGF expression by inhibiting PI3K-Akt signaling in NCI-N87Luc+ cells as in breast cancer cells. However, trastuzumab only affects cancer cells, and the antiangiogenic effect of HER2 blockade is counteracted by the compensatory production of VEGF by tumor stromal cells to rescue angiogenesis. In tumor stroma, various cellular components, such as endothelial cells, tumor-associated fibroblasts and immune cells, can secrete various angiogenic factors that support tumor growth.11, 12 Therefore, by binding to the VEGF and PlGF secreted by stromal cells, VEGF-Trap can exert additional antiangiogenic effects. In the present study, we noted enhanced inhibition of tumor angiogenesis following combined treatment with trastuzumab and VEGF-Trap compared with treatment using either agent individually.
We propose the following possible mechanisms by which the combination of trastuzumab and VEGF-Trap resulted in the enhanced inhibition of tumor growth. The mechanisms of trastuzumab's action include antiproliferative effects on tumor cells by inhibiting signaling pathways and activating apoptotic signals, the inhibition of angiogenesis by downregulation of the endogenous expression of VEGF and other proangiogenic factors, and ADCC. VEGF-Trap has a high affinity for human and mouse VEGF-A, VEGF-B and PlGF, therefore inhibiting tumor angiogenesis and growth by efficiently inhibiting the angiogenic factors secreted by tumor cells and the stroma. Treatment with trastuzumab or VEGF-Trap resulted in a marked decrease in tumor cell proliferation, but not a significant increase in apoptosis, compared with the control, suggesting that the tumor growth inhibition in the mice treated with trastuzumab or VEGF-Trap may be primarily due to reduced tumor cell proliferation. However, the combination of these agents exerted a greater inhibition of tumor cell proliferation compared with either agent alone and also markedly induced apoptosis of the tumor cells, suggesting that the enhanced inhibition of tumor growth in the mice treated with both agents may be due to both reduced proliferation and increased apoptosis of the tumor cells. Taken together, the combination of trastuzumab and VEGF-Trap has additive inhibitory effects on tumor growth and angiogenesis, yielding improved therapeutic efficacy against HER2-overexpressing gastric cancer xenografts compared with either agent alone. Combined treatment with trastuzumab and VEGF-Trap may provide a strategy to delay the emergence of trastuzumab-resistant tumors that primarily occur through the development of alternative mechanisms that produce elevated VEGF levels and/or activate the PI3K-Akt pathway, driving tumor cell proliferation and survival.50, 51
In the case of breast cancer, a biologic combination of bevacizumab and trastuzumab was clinically feasible and active in HER2-amplified recurrent or metastatic breast cancer in a phase II study. However, the addition of bevacizumab to combined therapy with trastuzumab and chemotherapy did not significantly improve the therapeutic benefit in metastatic breast cancer patients in a randomized phase III trial.19, 38 Exploratory analyses of plasma VEGF-A, one of the most promising candidate biomarkers for bevacizumab efficacy, suggested a potentially predictive effect, with more substantial benefit from bevacizumab among patients with high plasma VEGF-A concentrations than among patients with low VEGF-A concentrations.38 The potential predictive role of plasma VEGF-A was also identified in independent biomarker analyses in patients with metastatic gastric cancer and metastatic pancreatic cancer in randomized phase III trials of bevacizumab with chemotherapy.52, 53 Therefore, it may be worthwhile to evaluate whether the addition of bevacizumab to combined therapy with trastuzumab and chemotherapy improves the therapeutic benefit in HER2-positive gastric cancer patients with high plasma VEGF-A concentrations. In addition, the clinical efficacy of VEGF-Trap plus trastuzumab may be different from the efficacy of bevacizumab plus trastuzumab because VEGF-Trap has broader specificity for various ligands, such as VEGF-A, VEGF-B and PlGF. Whether the combination of trastuzumab and VEGF-Trap has enhanced therapeutic efficacy in HER2-positive gastric cancer patients compared with either agent alone remains to be validated in clinical trials.
Acknowledgments
This work was supported by a grant from the National R&D Program for Cancer Control, Ministry of Health and Welfare, Republic of Korea (1020420).
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on Experimental and Molecular Medicine website (http://www.nature.com/emm)
Supplementary Material
References
- Holbro T, Civenni G, Hynes NE. The ErbB receptors and their role in cancer progression. Exp Cell Res. 2003;284:99–110. doi: 10.1016/s0014-4827(02)00099-x. [DOI] [PubMed] [Google Scholar]
- Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol Cell Biol. 2001;21:3995–4004. doi: 10.1128/MCB.21.12.3995-4004.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen XF, Yang G, Mao W, Thornton A, Liu J, Bast RC, Jr, et al. HER2 signaling modulates the equilibrium between pro- and antiangiogenic factors via distinct pathways: implications for HER2-targeted antibody therapy. Oncogene. 2006;25:6986–6996. doi: 10.1038/sj.onc.1209685. [DOI] [PubMed] [Google Scholar]
- Burstein HJ. The distinctive nature of HER2-positive breast cancers. N Engl J Med. 2005;353:1652–1654. doi: 10.1056/NEJMp058197. [DOI] [PubMed] [Google Scholar]
- Gravalos C, Jimeno A. HER2 in gastric cancer: a new prognostic factor and a novel therapeutic target. Ann Oncol. 2008;19:1523–1529. doi: 10.1093/annonc/mdn169. [DOI] [PubMed] [Google Scholar]
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- Cobleigh MA, Vogel CL, Tripathy D, Robert NJ, Scholl S, Fehrenbacher L, et al. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J Clin Oncol. 1999;17:2639–2639. doi: 10.1200/JCO.1999.17.9.2639. [DOI] [PubMed] [Google Scholar]
- Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol. 2002;20:719–726. doi: 10.1200/JCO.2002.20.3.719. [DOI] [PubMed] [Google Scholar]
- Nahta R, Esteva FJ. Herceptin: mechanisms of action and resistance. Cancer Lett. 2006;232:123–138. doi: 10.1016/j.canlet.2005.01.041. [DOI] [PubMed] [Google Scholar]
- Fukumura D, Xavier R, Sugiura T, Chen Y, Park E, Lu N, et al. Tumor induction of VEGF promoter activity in stromal cells. Cell. 1998;94:715–725. doi: 10.1016/s0092-8674(00)81731-6. [DOI] [PubMed] [Google Scholar]
- Harmey J, Dimitriadis E, Kay E, Redmond HP, Bouchier-Hayes D. Regulation of macrophage production of vascular endothelial growth factor (VEGF) by hypoxia and transforming growth factor beta-1. Ann Surg Oncol. 1998;5:271–278. doi: 10.1007/BF02303785. [DOI] [PubMed] [Google Scholar]
- Izumi Y, Xu L, di Tomaso E, Fukumura D, Jain RK. Tumour biology: herceptin acts as an anti-angiogenic cocktail. Nature. 2002;416:279–280. doi: 10.1038/416279b. [DOI] [PubMed] [Google Scholar]
- Jain RK. Molecular regulation of vessel maturation. Nat Med. 2003;9:685–693. doi: 10.1038/nm0603-685. [DOI] [PubMed] [Google Scholar]
- Figg WD, Folkman J. Angiogenesis: An Integrative Approach from Science to Medicine. Springer: New York, NY, USA; 2008. [Google Scholar]
- Le X, Mao W, Lu C, Thornton A, Heymach JV, Sood AK, et al. Specific blockade of VEGF and HER2 pathways results in greater growth inhibition of breast cancer xenografts that overexpress HER2. Cell Cycle. 2008;7:3747–3758. doi: 10.4161/cc.7.23.7212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alami N, Sun Y, De P, Benmassaoud AM, Wang Y, Leyland-Jones B. Combination effects of herceptin, pertuzumab and bevacizumab in a HER2-overexpressing breast cancer xenograft model. Cancer Res. 2009;69:4058(abstract). [Google Scholar]
- Pegram M, Yeon C, Ku N, Gaudreault J, Slamon D. Phase I combined biological therapy of breast cancer using two humanized monoclonal antibodies directed against HER2 proto-oncogene and vascular endothelial growth factor (VEGF) Breast Cancer Res Treat. 2004;88:S124–S125. (abstract 3039). [Google Scholar]
- Pegram M, Chan D, Dichmann R, Tan-Chiu E, Yeon C, Durna L, et al. Phase II combined biological therapy targeting the HER2 proto-oncogene and the vascular endothelial growth factor using trastuzumab (T) and bevacizumab (B) as first line treatment of HER2-amplified breast cancer. Breast Cancer Res Treat. 2006;100:S28–S29. (abstract 301). [Google Scholar]
- Pierga J, Petit T, Delozier T, Ferrero J, Campone M, Gligorov J, et al. Neoadjuvant bevacizumab, trastuzumab, and chemotherapy for primary inflammatory HER2-positive breast cancer (BEVERLY-2): an open-label, single-arm phase 2 study. Lancet Oncol. 2012;13:375–384. doi: 10.1016/S1470-2045(12)70049-9. [DOI] [PubMed] [Google Scholar]
- Dvorak HF. Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J Clin Oncol. 2002;20:4368–4380. doi: 10.1200/JCO.2002.10.088. [DOI] [PubMed] [Google Scholar]
- Yancopoulos GD, Davis S, Gale NW, Rudge JS, Wiegand SJ, Holash J. Vascular-specific growth factors and blood vessel formation. Nature. 2000;407:242–248. doi: 10.1038/35025215. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Gerber H, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer. 2008;8:579–591. doi: 10.1038/nrc2403. [DOI] [PubMed] [Google Scholar]
- Miao H, Lee P, Lin H, Soker S, Klagsbrun M. Neuropilin-1 expression by tumor cells promotes tumor angiogenesis and progression. FASEB J. 2000;14:2532–2539. doi: 10.1096/fj.00-0250com. [DOI] [PubMed] [Google Scholar]
- Lohela M, Bry M, Tammela T, Alitalo K. VEGFs and receptors involved in angiogenesis versus lymphangiogenesis. Curr Opin Cell Biol. 2009;21:154–165. doi: 10.1016/j.ceb.2008.12.012. [DOI] [PubMed] [Google Scholar]
- Donnini S, Machein MR, Plate KH, Weich HA. Expression and localization of placenta growth factor and P1GF receptors in human meningiomas. J Pathol. 1999;189:66–71. doi: 10.1002/(SICI)1096-9896(199909)189:1<66::AID-PATH390>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- André T, Kotelevets L, Vaillant JC, Coudray AM, Weber L, Prévot S, et al. VEGF, VEGF-B, VEGF-C, and their receptors KDR, FLT-1 and FLT-4 during the neoplastic progression of human colonic mucosa. Int J Cancer. 2000;86:174–181. doi: 10.1002/(sici)1097-0215(20000415)86:2<174::aid-ijc5>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Fischer C, Mazzone M, Jonckx B, Carmeliet P. FLT1 and its ligands VEGFB and PlGF: drug targets for anti-angiogenic therapy. Nat Rev Cancer. 2008;8:942–956. doi: 10.1038/nrc2524. [DOI] [PubMed] [Google Scholar]
- Rini BI, Michaelson MD, Rosenberg JE, Bukowski RM, Sosman JA, Stadler WM, et al. Antitumor activity and biomarker analysis of sunitinib in patients with bevacizumab-refractory metastatic renal cell carcinoma. J Clin Oncol. 2008;26:3743–3748. doi: 10.1200/JCO.2007.15.5416. [DOI] [PubMed] [Google Scholar]
- Willett CG, Duda DG, Di Tomaso E, Boucher Y, Ancukiewicz M, Sahani DV, et al. Efficacy, safety, and biomarkers of neoadjuvant bevacizumab, radiation therapy, and fluorouracil in rectal cancer: a multidisciplinary phase II study. J Clin Oncol. 2009;27:3020–3026. doi: 10.1200/JCO.2008.21.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Tang Z, Hou X, Lennartsson J, Li Y, Koch AW, et al. VEGF-B is dispensable for blood vessel growth but critical for their survival, and VEGF-B targeting inhibits pathological angiogenesis. Proc Natl Acad Sci USA. 2009;106:6152–6157. doi: 10.1073/pnas.0813061106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jussila L, Alitalo K. Vascular growth factors and lymphangiogenesis. Physiol Rev. 2002;82:673–700. doi: 10.1152/physrev.00005.2002. [DOI] [PubMed] [Google Scholar]
- Lieu C, Tran H, Jiang Z, Mao M, Overman M, Eng C, et al. The association of alternate VEGF ligands with resistance to anti-VEGF therapy in metastatic colorectal cancer. J Clin Oncol. 2011;29:3533. doi: 10.1371/journal.pone.0077117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebos JM, Kerbel RS. Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat Rev Clin Oncol. 2011;8:210–221. doi: 10.1038/nrclinonc.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, Crouthamel M, Rominger D, Gontarek R, Tummino P, Levin R, et al. Myelosuppression and kinase selectivity of multikinase angiogenesis inhibitors. Br J Cancer. 2009;101:1717–1723. doi: 10.1038/sj.bjc.6605366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8:592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianni L, Romieu GH, Lichinitser M, Serrano SV, Mansutti M, Pivot X, et al. AVEREL: A Randomized Phase III Trial evaluating bevacizumab in combination with docetaxel and trastuzumab as first-line therapy for HER2-positive locally recurrent/metastatic breast cancer. J Clin Oncol. 2013;31:1719–1725. doi: 10.1200/JCO.2012.44.7912. [DOI] [PubMed] [Google Scholar]
- Lin N, Seah D, Gelman R, Desantis S, Mayer E, Isakoff S, et al. A phase II study of bevacizumab in combination with vinorelbine and trastuzumab in HER2-positive metastatic breast cancer. Breast Cancer Res Treat. 2013;139:403–410. doi: 10.1007/s10549-013-2551-9. [DOI] [PubMed] [Google Scholar]
- Holash J, Davis S, Papadopoulos N, Croll SD, Ho L, Russell M, et al. VEGF-Trap: a VEGF blocker with potent antitumor effects. Proc Natl Acad Sci USA. 2002;99:11393–11398. doi: 10.1073/pnas.172398299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos N, Martin J, Ruan Q, Rafique A, Rosconi MP, Shi E, et al. Binding and neutralization of vascular endothelial growth factor (VEGF) and related ligands by VEGF Trap, ranibizumab and bevacizumab. Angiogenesis. 2012;15:171–185. doi: 10.1007/s10456-011-9249-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaya A, Tse V. A preclinical and clinical review of aflibercept for the management of cancer. Cancer Treat Rev. 2012;38:484–493. doi: 10.1016/j.ctrv.2011.12.008. [DOI] [PubMed] [Google Scholar]
- Okines AF, Cunningham D. Trastuzumab in gastric cancer. Eur J Cancer. 2010;46:1949–1959. doi: 10.1016/j.ejca.2010.05.003. [DOI] [PubMed] [Google Scholar]
- Bang Y, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376:687–697. doi: 10.1016/S0140-6736(10)61121-X. [DOI] [PubMed] [Google Scholar]
- Kanai T, Konno H, Tanaka T, Baba M, Matsumoto K, Nakamura S, et al. Anti-tumor and anti-metastatic effects of human-vascular-endothelial-growth-factor-neutralizing antibody on human colon and gastric carcinoma xenotransplanted orthotopically into nude mice. Int J Cancer. 1998;77:933–936. doi: 10.1002/(sici)1097-0215(19980911)77:6<933::aid-ijc23>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Ohtsu A, Shah MA, Van Cutsem E, Rha SY, Sawaki A, Park SR, et al. Bevacizumab in combination with chemotherapy as first-line therapy in advanced gastric cancer: a Randomized, Double-Blind, Placebo-Controlled Phase III Study. J Clin Oncol. 2011;29:3968–3976. doi: 10.1200/JCO.2011.36.2236. [DOI] [PubMed] [Google Scholar]
- Kim SY, Kim HP, Kim YJ, Oh do Y, Im SA, Lee D, et al. Trastuzumab inhibits the growth of human gastric cancer cell lines with HER2 amplification synergistically with cisplatin. Int J Oncol. 2008;32:89–95. [PubMed] [Google Scholar]
- Hashizume H, Falcón BL, Kuroda T, Baluk P, Coxon A, Yu D, et al. Complementary actions of inhibitors of angiopoietin-2 and VEGF on tumor angiogenesis and growth. Cancer Res. 2010;70:2213–2223. doi: 10.1158/0008-5472.CAN-09-1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klos KS, Zhou X, Lee S, Zhang L, Yang W, Nagata Y, et al. Combined trastuzumab and paclitaxel treatment better inhibits ErbB-2-mediated angiogenesis in breast carcinoma through a more effective inhibition of Akt than either treatment alone. Cancer. 2003;98:1377–1385. doi: 10.1002/cncr.11656. [DOI] [PubMed] [Google Scholar]
- Hynes NE, MacDonald G. ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol. 2009;21:177–184. doi: 10.1016/j.ceb.2008.12.010. [DOI] [PubMed] [Google Scholar]
- du Manoir JM, Francia G, Man S, Mossoba M, Medin JA, Viloria-Petit A, et al. Strategies for delaying or treating in vivo acquired resistance to trastuzumab in human breast cancer xenografts. Clin Cancer Res. 2006;12:904–916. doi: 10.1158/1078-0432.CCR-05-1109. [DOI] [PubMed] [Google Scholar]
- Van Cutsem E, de Haas S, Kang YK, Ohtsu A, Tebbutt NC, Ming Xu J, et al. Bevacizumab in combination with chemotherapy as first-line therapy in advanced gastric cancer: a biomarker evaluation from the AVAGAST randomized phase III trial. J Clin Oncol. 2012;30:2119–2127. doi: 10.1200/JCO.2011.39.9824. [DOI] [PubMed] [Google Scholar]
- Van Cutsem E, Jayson G, Dive C, Dilba P, de Haas S, Wild N, et al. Analysis of blood plasma factors in the AVITA Phase III Randomized Study of bevacizumab (bev) with gemcitabine-erlotinib (ge) in patients (pts) with metastatic pancreatic cancer (mPC) Eur J Cancer. 2011;47:(abstract 803). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.