

Abstract
Both insulin resistance (type II diabetes) and β-amyloid (Aβ) oligomers are implicated in Alzheimer's disease (AD). Here, we investigate the role of Aβ oligomer-induced c-Jun N-terminal kinase (JNK) activation leading to phosphorylation and degradation of the adaptor protein insulin receptor substrate-1 (IRS-1). IRS-1 couples insulin and other trophic factor receptors to downstream kinases and neuroprotective signaling. Increased phospho-IRS-1 is found in AD brain and insulin-resistant tissues from diabetics. Here, we report Aβ oligomers significantly increased active JNK and phosphorylation of IRS-1 (Ser616) and tau (Ser422) in cultured hippocampal neurons, whereas JNK inhibition blocked these responses. The omega-3 fatty acid docosahexaenoic acid (DHA) similarly inhibited JNK and the phosphorylation of IRS-1 and tau in cultured hippocampal neurons. Feeding 3xTg-AD transgenic mice a diet high in saturated and omega-6 fat increased active JNK and phosphorylated IRS-1 and tau. Treatment of the 3xTg-AD mice on high-fat diet with fish oil or curcumin or a combination of both for 4 months reduced phosphorylated JNK, IRS-1, and tau and prevented the degradation of total IRS-1. This was accompanied by improvement in Y-maze performance. Mice fed with fish oil and curcumin for 1 month had more significant effects on Y-maze, and the combination showed more significant inhibition of JNK, IRS-1, and tau phosphorylation. These data indicate JNK mediates Aβ oligomer inactivation of IRS-1 and phospho-tau pathology and that dietary treatment with fish oil/DHA, curcumin, or a combination of both has the potential to improve insulin/trophic signaling and cognitive deficits in AD.
Introduction
Alzheimer's disease (AD) is the most common form of neurodegenerative dementia in the aged. AD is neuropathologically characterized by abnormal accumulations of extracellular amyloid plaques and intracellular neurofibrillary tangles throughout cortical and limbic brain regions. Cognitive deficits in AD are widely believed to result from progressive synaptic dysfunction and neurodegeneration initiated by soluble aggregated β-amyloid peptide 1–42 (Aβ42) and further involving aggregates of hyperphosphorylated tau, a principal component of intracellular neurofibrillary tangles. Epidemiologists find high-fat diet/obesity or diabetes that cause insulin resistance are risk factors for AD (Ott et al., 1996, 1999; Leibson et al., 1997). In AD brain, insulin and insulin signaling are decreased when compared with healthy control subjects (Craft et al., 1998, 2003; Rivera et al., 2005; Steen et al., 2005). Insulin/insulin receptor (IR) signaling normally plays a pivotal role in regulation of peripheral glucose metabolism and energy homeostasis. In the brain, the insulin/IR complex is abundantly distributed in synaptic membranes of the cerebral cortex and hippocampus (Heidenreich et al., 1983, 1988; Matsumoto and Rhoads, 1990; Zhao et al., 1999) and functions to regulate synaptic activities that are required for learning and memory (Hendricks et al., 1984; Lowe et al., 1986; Biessels et al., 1996; Wan et al., 1997; Zhao and Alkon, 2001). In β-amyloid precursor (APP) transgenic (Tg2576) AD model mice, insulin resistance develops, suggesting some link between Aβ and insulin signaling (Pedersen and Flynn, 2004).
Trophic factor or insulin receptors are tyrosine kinases, which autophosphorylate after activation by ligands, resulting in recognition by insulin receptor substrate (IRS-1 and IRS-2) adaptors (White, 2002). IRS is then phosphorylated at tyrosines, permitting recruitment of SH2 (Src homology 2)-domain-containing proteins, including phosphatidylinositol (PI) 3-kinase (Sun et al., 1995). This activates signaling pathways responsible for the pleiotropic actions of insulin (Virkamäki et al., 1999). With insulin resistance in diabetics and models, IRS-1 is phosphorylated at Ser312 by insulin-stimulated or stress-activated kinases, including c-Jun N-terminal kinase (JNK), which uncouples IRS-1 (Aguirre et al., 2002) and triggers rapid IRS-1 degradation (Sun et al., 1999), yielding a deficient signal transduction response (Pederson et al., 2001; Rui et al., 2001). IRS deficits contribute to insulin resistance in animal models and diabetics (Saad et al., 1992; Rondinone et al., 1997). Significantly reduced IRS-1 and IRS-2 occur in AD brain, accompanied by elevated cytosolic phospho-IRS-1 (Ser312 and Ser616). These phosphoserine epitopes colocalize with neurofibrillary tangles, suggesting a possible link between “insulin resistance” and tau pathology (Moloney et al., 2008).
IRS-1 is phosphorylated at different serine/threonine residues by multiple kinases, including JNK1 (Aguirre et al., 2000; Lee et al., 2003), Rho kinase (Begum et al., 2002), protein kinase Cζ (PKCζ) (Liu et al., 2001), PI3-K/Akt/mTOR (mammalian target of rapamycin) (Ozes et al., 2001), and inhibitor κB kinase (Gao et al., 2002), but JNK is most implicated in AD. Activated JNK is induced by reactive oxygen species (Shen and Liu, 2006) and observed in neurons and dystrophic neurites of AD model mice and AD brain in which it progressively overlaps tau-positive neurofibrillary pathology, but remains scarce in age-matched normal controls (Shoji et al., 2000; Zhu et al., 2001). Active JNK localizes to plaque neurites containing phosphorylated tau in AD mouse models Tg2576 (Puig et al., 2004) and APPsw × PS1P264L (Savage et al., 2002) in which JNK activation coincided with age-dependent amyloid deposition, tau phosphorylation, and synaptophysin loss. Aβ peptides induce JNK activation and neurotoxicity in cultured primary cortical neurons (Shoji et al., 2000); conversely, JNK inhibition in PC12 reduced Aβ toxicity (Troy et al., 2001). Thus, we hypothesized Aβ oligomers mediate aberrant IRS-1 and tau phosphorylation via JNK signaling. To test this, we used treatments that limited JNK activation including docosahexaenoic acid (DHA) (22:6n-3) in cultured neurons and in 3xTg-AD mice, an AD transgenic mouse model that progressively develops Aβ and tau pathology (Oddo et al., 2003). Treatments included DHA-rich fish oil and curcumin, a turmeric component that attenuated cognitive deficits, neuroinflammation, and amyloid and JNK activation in animal studies (Frautschy et al., 2001; Yang et al., 2005; Garcia-Alloza et al., 2007; Begum et al., 2008).
Materials and Methods
Chemicals and reagents.
All chemicals were purchased from Sigma-Aldrich, unless otherwise stated. Anti-phospho-JNK (Thr183/Tyr185) mouse monoclonal antibody (mAb) was purchased from Cell Signaling. Anti-phospho-IRS-1 (Ser616, Ser312), anti-IRS-1, and anti-phospho-tau (Ser422) rabbit polyclonal antibodies (pAb) were purchased from BioSource International. Anti-β-actin mAb was from Millipore Bioscience Research Reagents. Anti-Aβ-pAb (DAE) was developed in rabbits to Aβ amino acids 1–13 and characterized in our laboratory (Yang et al., 1994). Anti-Aβ mAb 6E10 was obtained from Signet Laboratories. Aβ1–42 peptide was from American Peptide. JNK inhibitor II [anthra[1,9-cd]pyrazol-6(2H)-one (SP600125)] was obtained from Calbiochem.
Preparation of oligomers.
Aβ1–42 peptide (Aβ42) was dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) in the vial and incubated at room temperature until dissolved completely. The HFIP was removed by gentle streaming of N2. Aβ42 was then dissolved with 10 mm HEPES, pH 7.4, to a final concentration of 110 μm. This solution was incubated at 37°C with a micro stir bar at 500 rpm for 12 h and centrifuged at 14,000 × g for 5 min to remove fibrillar and other large Aβ aggregates. The presence of Aβ oligomers was confirmed by 6E10 anti-Aβ antibody after its separation on 10–20% Tris-Tricine gradient gels (Ma et al., 2006). Protein concentration was determined using the Bio-Rad DC protein assay. For cell culture experiments, the oligomer-rich supernatant preparation was diluted to 100 nm in Neurobasal media without glutamate and B27.
Primary neuronal culture and treatment.
Neuronal cultures were prepared from embryonic day 18 Sprague Dawley rat fetuses as described previously (Ma et al., 2007). Before treatment with Aβ oligomer preparations, cells were rinsed once with Neurobasal media without glutamate and B27 for primary neurons. Cells were exposed to 100 nm Aβ42 oligomers for 5 h at 37°C.
Cell lysate preparation.
Primary neurons were placed on ice, washed with cold PBS once, scraped with 1 ml of PBS, and transferred to Microfuge tubes. After centrifugation at 1000 rpm for 5 min, cells were washed with cold PBS twice and dissolved in Tris-buffered saline (TBS) buffer with a mixture of protease and phosphatase inhibitors. After brief sonication, the lysates were kept at 4°C for 30 min and centrifuged at 55,000 rpm for 20 min, and the supernatants were collected for TBS fraction (cytosol fraction). The pellet was dissolved in lysis buffer with a mixture of protease and phosphatase inhibitors, sonicated, and centrifuged at 55,000 rpm for 20 min to extract lysis fractions (membrane pellet fractions).
Human AD and controls.
Eight autopsy-confirmed AD and six age-matched healthy individuals were used in this study based on autopsy reports from Dr. Harry Vinters (University of California Los Angeles Alzheimer's Disease Research Center, Los Angeles, CA). For the pathologic diagnosis of AD, plaque density was assessed using the Consortium to Establish a Registry for Alzheimer's Disease (CERAD) criteria (Mirra et al., 1991) and neurofibrillary tangles were evaluated using the criteria by Braak and Braak (Braak and Braak, 1991). Autopsy-confirmed AD stage was typically intermediate [CERAD (Mirra et al., 1991), moderate plaques and Braak stage III∼IV]. The ages of the AD patients were at 64∼93 years. The postmortem interval before autopsy ranged from 3.5–19 h (Table 1).
Table 1.
Cases of autopsy-confirmed AD and healthy control subjects used in the immunofluorescent staining
| Postmortem cases | No. of cases | Age (range) (years) | Postmortem interval (h) | Senile plaques | Braack stage | Other pathologic changes |
|---|---|---|---|---|---|---|
| Healthy controls | 6 | 50∼90 | 11∼19 | None | I | One case has a small aneurysm and shows only mild atherosclerotic change |
| AD | 8 | 64∼93 | 3.5∼15 | Moderate to abundant plaques are mainly in dense core or diffuse | III∼IV | None |
Animals.
Five-month-old 3xTg-AD mice (n = 8–11 for each group) were used in this study. The mice were fed with or without a high-fat bad diet (HFBD) consisting of an omega-3-depleting, high n-6 safflower oil base diet previously described (Calon et al., 2004) that was supplemented with high 1500 ppm cholesterol and high saturated fat as coconut oil (21% fat, 16% coconut oil, 5% safflower oil; TD 07701; Harlan Teklad). Table 2 shows the high-fat bad-diet composition in detail. The coconut and safflower oil-based HFBD mice were subgrouped to be fed with or without fish oil (TD.07705; modification of TD.07701 to achieve ∼0.6% DHA via fish oil; Nordic Naturals), curcumin (TD.07702; modification of TD.07701 to add 500 ppm curcumin), and fish oil plus curcumin (TD.07706; modification of TD.07701 to add ∼0.6% DHA via fish oil, and 500 ppm curcumin) for 4 months. The fish oil is added at the expense of coconut oil. Surgical and animal procedures were approved by the local Institutional Animal Care Use Committee and performed with strict adherence to the current guidelines set out in the National Institutes of Health Guide for the Care and Use of Laboratory Animals at the Association for Assessment and Accreditation of Laboratory Animal Care-accredited Greater Los Angeles Veterans Affairs Healthcare System.
Table 2.
Component of mouse diet
| Formula (g/kg) | High-fat bad diet | 0.6% DHA diet | Curcumin diet | 0.6% DHA plus curcumin diet |
|---|---|---|---|---|
| Casein, ″vitamin-free″ test | 200 | 200 | 200 | 200 |
| dl-Methionine | 3 | 3 | 3 | 3 |
| Sucrose | 329.1887 | 329.1887 | 329.1887 | 329.1887 |
| Maltodextrin | 50 | 50 | 50 | 50 |
| Corn starch | 100 | 100 | 100 | 100 |
| Coconut oil | 160 | 136 | 160 | 136 |
| Safflower oil, linoleic | 50 | 50 | 50 | 50 |
| Fish oil (25% DHA) | 0 | 24 | 0 | 24 |
| Curcumin | 0 | 0 | 0.5 | 0.5 |
| Cholesterol | 1.5 | 1.5 | 1.5 | 1.5 |
| Cellulose | 50 | 50 | 50 | 50 |
| Mineral mix, AIN-93G-MX (94046) | 42 | 42 | 42 | 42 |
| Calcium phosphate, dibasic | 4 | 4 | 4 | 4 |
| Vitamin mix, without choline, A, D, E (83171) | 6 | 6 | 6 | 6 |
| Vitamin A palmitate (500,000 IU/g) | 0.048 | 0.048 | 0.048 | 0.048 |
| Vitamin D3, cholecalciferol (500,000 IU/g) | 0.0053 | 0.0053 | 0.0053 | 0.0053 |
| Vitamin E, dl-α -tocopheryl acetate (500 IU/g) | 0.054 | 0.054 | 0.054 | 0.054 |
| Choline dihydrogen citrate | 4.2 | 4.2 | 4.2 | 4.2 |
| TBHQ, antioxidant | 0.004 | 0.004 | 0.004 | 0.004 |
Note: Prolab RMH 2500 5P14 was used for normal control standard diet, which supplied for 4.5% fat and 101 ppm of cholesterol. TBHQ, tert-Butylhydroquinone.
Y-maze.
As previously described (Rosario et al., 2006), mice were tested for 7 min for spontaneous alternation behavior (SAB) in a Y-maze constructed from black-colored Plexiglas (short arms A and B, 6 × 5 × 3 in; long arm C, 8 × 5 × 3 in) based on San Diego Instruments Y-maze model 2005. The total number of arm choices and alternations were recorded and SAB score was calculated by the proportion of alternations (an arm choice differing from the previous two choices) to the total number of alternation opportunities (total arm entries − 2) (King and Arendash, 2002).
Tissue preparation.
Tissue samples were sequentially processed by sonication in TBS and ultracentrifugation (55,000 × 20 min) to obtain supernatants (TBS, soluble-cytosol fraction). Pellets were then sonicated in immunoprecipitation lysis buffer containing 1% Triton X-100, 0.5% sodium deoxycholate, and 0.5% SDS, and recentrifuged to obtain lysis extract supernatants (membrano-cytoskeletal extract). All buffers contained previously described protease inhibitor and phosphatase inhibitor mixtures (Calon et al., 2004). For immunocytochemical staining (ICC), cryostat sections from paraformaldehyde-fixed mouse brain hemispheres or AD and control hippocampus (University of California Los Angeles and University of Southern California Alzheimer Center Pathology cores) were cut and stained for confocal microscopy as previously described (Calon et al., 2004).
Immunocytochemical staining and image analysis.
Hippocampal neuron [9 d in vitro (DIV)] poly-d-lysine-coated coverslip cultures were Aβ or vehicle treated as described in the figure legends. After treatment, cultured cells were PBS washed, 4% paraformaldehyde fixed (10 min), blocked 30 min with 3% goat serum, 10% horse serum, 0.3% Triton X-100 in PBS, and double labeled with anti-phospho-IRS-1 Ser616 (1:100) or anti-phospho-tau (Ser422)/anti-rabbit mouse rhodamine (1:200) and anti-Aβ mAb 6E10 (1:200)/anti-mouse rabbit FITC (1:200) and mounted with DAPI (4–6-diamidino-2-phenylindole)-containing medium (Vector Laboratories). Slides were viewed with a fluorescent microscope. Immunofluorescent phospho-IRS-1 Ser616 or phospho-tau Ser422 staining density was quantified by digitally photographing with equal magnification and exposure time, a minimum of 100 neurons per treatment, and quantifying with automated analysis using 5 Image-Pro Plus software to quantify the phospho-IRS-1 Ser616 or phospho-tau Ser422 fluorescence levels. For in vivo tissue ICC, cryostat sections from paraformaldehyde-fixed mouse brain hemispheres or AD and control hippocampus were stained by anti-phospho-IRS-1 Ser616 or Ser312 or anti-phospho-tau PHF-1 and imaged by confocal microscopy as previously described (Calon et al., 2004).
Western immunoblotting.
For Western immunoblotting, protein concentration was determined using the Bio-Rad DC protein assay. Equal amounts of protein per sample were added to Laemmli loading buffer and boiled for 3 min. Except for Aβ oligomer detection, 30 μg of protein per well was electrophoresed on 10% Tris-glycine gels and transferred to Immobilon-P polyvinylidene difluoride membranes (Millipore). For Aβ species separation, the protein samples were electrophoresed on 10–20% Tris-Tricine gradient gels (Ma et al., 2006).
Statistical analysis.
Statistical analyses were performed with StatView 5.0 software. Differences among means were assessed by ANOVA followed by Tukey–Kramer post hoc tests. Square root transformation to establish homogeneity of variance was used as needed for data analysis.
Results
Phosphorylated IRS-1 was redistributed in AD and in a 3xTg-AD mouse model
Confocal imaging indicated that neurons in the hippocampus of aged cognitively normal patients show that IRS-1 pS312 is localized predominantly to nuclei, whereas in AD patients the staining pattern is altered. It is sometimes found in granules in the cytosol (Fig. 1A, B). Overall, the immunostaining patterns of IRS-1 pS312 and pS616 in AD brain are variable. Some neurons show increased IRS-1 pS312 staining in neuritic plaques, colocalizing with PHF-1 (Fig. 1C); IRS-1 pS616 staining labels tangles in many neurons and some dystrophic neurites (Fig. 1D, E). IRS-1 pS312 also typically shows colocalization with PHF-1 or AT8 ptau in neurofibrillary tangles (Figs. 1F, 2A). In addition, some other neurons show that IRS-1 pS312 staining is partially colocalized with apparent granulovacuolar degeneration (GVD) (Fig. 2B, C), in which pIRS-1 occasionally colocalized with phosphorylated tau stained by AT8 (Fig. 2C). Elevated IRS-1 pS312 and pS616 and its partial colocalization with PHF tau-immunoreactive neurofibrillary lesions in AD brain have been reported in two AD populations: matched pairs of 29 AD and 29 control cases from the University of Pennsylvania; and a separate sample of 30 elders with no cognitive impairment, 30 with mild cognitive impairment, and 30 with mostly early AD from the Religious Orders Study (Rush University, Chicago, IL) (Talbot et al., 2006). Similarly, IRS-1 pS312 staining of hippocampal neurons from 3-month-old 3xTg-AD-negative mice shows also predominantly nuclear localization (Fig. 1G), whereas in the 3-month-old 3xTg-AD transgenic-positive mice, more cytosolic IRS-1 pS312 is visible (Fig. 1H). Colocalization of IRS-1 pS312 with PHF-1 becomes more prominent at 7–8 months of age, and then by 18 months the colocalization with ptau in dendrites becomes more extensive (Fig. 1I, J). The transgene effect is also seen in animals on a high omega-6/saturated fat diet used to exacerbate pathology. Increased cytosolic phosphpho-IRS-1 was observed by 9 months of age using IRS-1 pS616 antibody (Fig. 1K, L). Figure 3 demonstrates quantification of total IRS-1, showing that IRS-1 in detergent-extracted hippocampal membrane pellet is significantly reduced by transgene in 9 month 3xTg mice (*p < 0.05), confirming that defects in this model resemble AD (Moloney et al., 2008).
Figure 1.
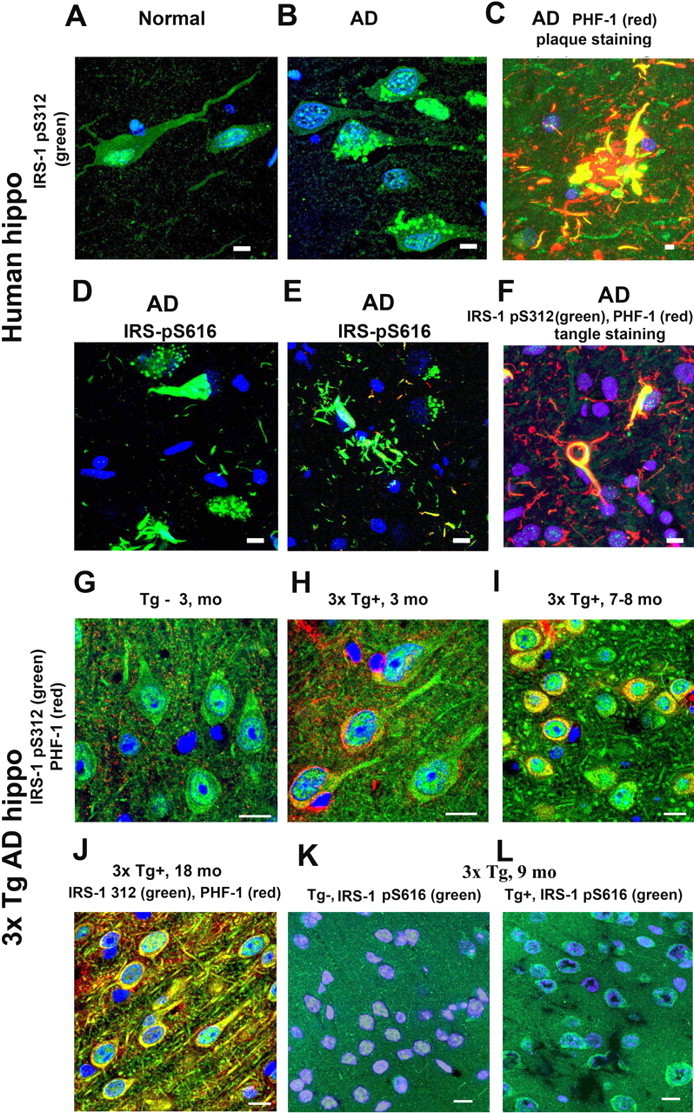
Redistribution of phosphorylated IRS-1 in AD and in 3xTg-AD mice and its enhancement by high-fat bad diet in 3xTg-AD mice. Using an antibody to the pS312 phosphoepitope of IRS-1, confocal immunocytochemistry illustrated that neurons from control human hippocampus (hippo) expressed high levels in the nucleus (A), whereas in AD patients, it was redistributed in the cytosol, sometimes in a granulovacuolar pattern (B). Around plaques, increased IRS-1 staining colocalized with dystrophic neurites (C). Antibodies to phosphorylated IRS-1 Ser616 typically stained tangles (D) and dystrophic neurites (E). IRS-1 pS312 colocalized with PHF-1 (F). Compared with 3-month-old control mice, which showed hippocampal CA1 nuclear staining of IRS-1 pS312 (G), 3xTg-AD mice had elevated IRS-1 pS312 in cytosol (H), which increased at 7–8 months of age (I). By 18 months of age, there was extensive colocalization of IRS-1 ps312 with PHF-1 (J). This transgene effect was also seen in animals using another IRS-1 antibody (pS616) in an experiment in which 5-month-old 3xTg-AD mice were fed with a HFBD (see text) for 4 months, and the phosphorylation of IRS-1 Ser616 was examined. Compared with Tg− control mice (K), 3xAD-Tg+ mice showed more cytosolic pS616 IRS-1 labeling and minimal nuclear labeling at 9 months of age (L). Scale bars, 20 μm.
Figure 2.
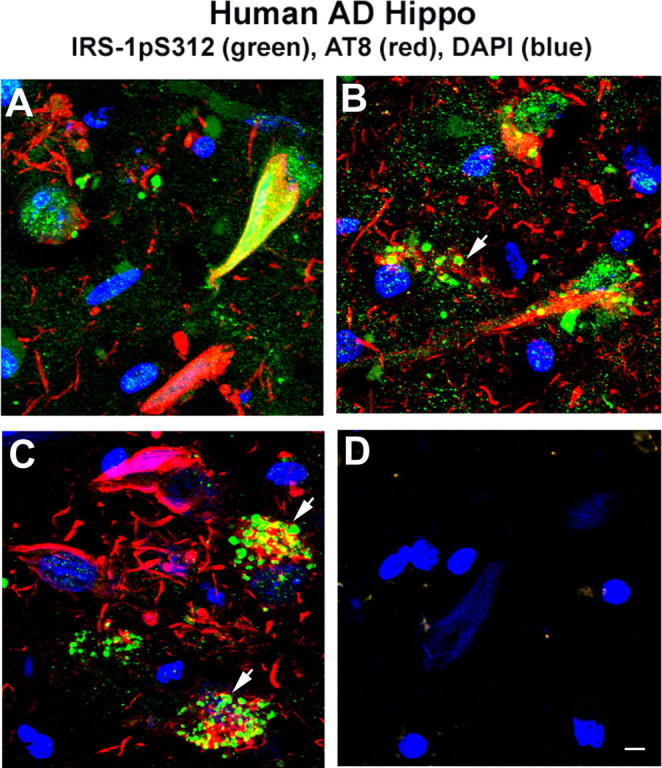
Immunostaining of phosphorylated IRS-1 in AD brain was variable and partially colocalized with the GVD. A, Immunostaining of phospho-IRS-1 Ser312 showed increased IRS-1 staining (green) typically in tangle-bearing neurons (AT8; red), and this colocalized with AT8. B, C, pIRS-1 Ser312 labeled apparent GVD. pIRS-1 was occasionally colocalized with phosphorylated tau in GVD (arrowheads indicate neurons containing granules). D, No phospho-IRS-1 and tau staining were observed in the absence of primary antibodies. Scale bar, 20 μm.
Figure 3.

Total IRS-1 was reduced in membrane extract of hippocampus in 3xTg-AD compared with Tg− control mice. Detergent lysis buffer extracts of hippocampal membrane pellets from 3xTg-AD (n = 8 for standard diet; n = 9 for high-fat bad diet) and Tg− controls (n = 8 for each group) were evaluated by Western for total IRS-1 (tIRS-1). Total IRS-1 was significantly reduced in transgene-positive mice (4.324 ± 0.248 for standard diet group; 5.015 ± 0.483 for high-fat bad diet group) compared with transgene-negative controls (6.232 ± 0.537 for standard group; 6.816 ± 0.576 for high-fat bad diet group) (p < 0.01, p < 0.05, respectively). Shown are mean ± SE. The Western immunoblotting was repeated three times. No change was observed in IRS-1 in TBS fractions (data not shown).
Aβ42 oligomers induced aberrant inactivation of IRS-1 in cultured hippocampal neurons
To determine whether the changes of IRS-1 observed in vivo in AD and AD models could be modeled in vitro, we examined the impact of Aβ oligomers on IRS-1 phosphorylation in cultured hippocampal neurons using an antibody to the phosphoepitope Ser616, which is implicated in insulin signaling defects and which worked better for Western blot than IRS-1 Ser312. Aβ oligomers used in this experiment were characterized as previously reported (Ma et al., 2006). Briefly, analysis of our soluble Aβ“oligomer” preparations showed ∼58% of Aβ42 converted to 12- and 24-mer, 34% of Aβ42 converted to 3- or 4-mer, whereas only 8% of Aβ42 remained in monomer status, which was characterized by silver stain and 4G8 anti-Aβ (Ma et al., 2006) antibody. Hippocampal neurons cultured 9 DIV were treated with 100 nm soluble Aβ42 oligomer and IRS-1Ser616 was examined. After 5 h treatment, the intensity of phospho-IRS-1, assessed by immunocytochemistry, was significantly elevated and redistributed from diffuse cytosol to nucleus and neurites (data not shown). Punctate anti-Aβ antibody staining decorated neuronal cell bodies and processes, similar to the synaptic pattern obtained with soluble Aβ oligomers extracted from human AD brain (Lacor et al., 2004), and quantification of immunofluorescence demonstrated that Aβ oligomers significantly increased intensity of staining (data not shown). Western blotting analysis produced similar results. IRS-1 Ser616 levels were significantly increased in membrane when compared with controls (**p = 0.01) (Fig. 4).
Figure 4.

Aβ42 oligomers induced aberrant inactivation of IRS-1 Ser616 in cultured hippocampal neurons. Western immunoblot analysis of pIRS-1 induced by Aβ42 oligomers in primary hippocampal neurons. Hippocampal neurons cultured for 9 DIV were treated with 100 nm Aβ oligomers. Harvested cells were then sonicated in lysis buffer and Western blotted with anti-pIRS-1Ser616 antibody. pIRS-1 levels were significantly increased in the membrane fraction after 5 h of Aβ42 oligomer-treatment (17.218 ± 0.922) when compared with controls (7.170 ± 0.674) (p = 0.013). β-Actin was used to normalize protein loading. Shown are mean ± SE. The experiment was repeated three times.
Aβ42 oligomers induced aberrant activation of JNK-sensitive tau Ser422 in cultured hippocampal neurons
In insulin-resistant diabetes, JNK has been identified as a kinase capable of phosphorylating and inactivating (uncoupling) IRS-1, notably at the serine 312 site. In AD, the MAP (mitogen-activated protein) kinase JNK can phosphorylate tau at Ser422, which is postulated to be involved in end-stage neurodegeneration (Hardy and Selkoe, 2002; Ma et al., 2006). Therefore, we tested whether Aβ oligomers could directly trigger tau Ser422 phosphorylation in cultured neurons and the impact of JNK inhibitors on this effect. Hippocampal neurons cultured 9 DIV were treated with 100 nm soluble Aβ42 oligomer preparations and tau Ser422 were examined by an anti-tau Ser422 antibody. After 5 h treatment, the phospho tau Ser422 epitope was assessed by immunostaining and observed to be significantly elevated and this phosphorylated tau was mostly concentrated in cytosol. However, some neurons showed that phospho-tau was redistributed from diffuse cytosol to membrane and neurites (data not shown). Punctate anti-Aβ antibody staining decorated neuronal cell bodies and processes similar to our previous report (Ma et al., 2008), and quantification of immunofluorescence showed increased intensity (data not shown). Western blotting analysis supported the ICC results, with phosphorylated tau significantly increased in cytosol (TBS) fractions compared with controls (**p < 0.01) (Fig. 5). To evaluate whether JNK is an upstream signaling event for phospho-IRS-1 Ser616 and tau Ser422 induced by Aβ oligomers, we used a specific JNK inhibitor, SP600125, which significantly blocked Aβ oligomer-induced aberrant inactivation of IRS-1 Ser616 (**p < 0.01) (Fig. 6A, B) and phospho-tau Ser422 assessed by Western analysis and densitometry (**p < 0.001) (Fig. 6A, C).
Figure 5.

Aβ42 oligomers induced aberrant activation of JNK-sensitive tau Ser422 in cultured hippocampal neurons. Western immunoblot analysis of ptau induced by Aβ42 oligomers in primary hippocampal neurons. Lysis fractions from 9 DIV hippocampal neurons treated with or without 100 nm Aβ oligomers were Western blotted with an anti-ptau-1Ser422 antibody. ptau levels were significantly increased in the cytosol fraction after 5 h in Aβ42 oligomer-treated neurons (21.161 ± 2.407) when compared with controls (5.488 ± 1.000) (p = 0.004). β-Actin was used for normalization. Shown are mean ± SE. The experiment was repeated three times.
Figure 6.

JNK inhibitor SP600125 blocked Aβ oligomer-induced aberrant inactivation of IRS-1 Ser616 and JNK-sensitive tau Ser422 phosphorylation in cultured hippocampal neurons. Hippocampal neurons were grown for 9 DIV and then pretreated with 10 μm JNK inhibitor, SP600125, for 30 min, followed by 100 nm Aβ42 oligomers for 5 h. After harvesting cells, pIRS-1 and ptau were evaluated by Western blot. A, B, Western immunoblot analysis of IRS-1 phosphorylation revealed induction by Aβ42 oligomers, which was antagonized by JNK inhibitor SP600125. pIRS-1 protein levels in membrane fractions were significantly increased by Aβ (B) (**p < 0.01), whereas JNK inhibitor SP600125 blocked the phosphorylated IRS-1 when compared with control (CTRL) (B) (**p < 0.01). A, C, Western immunoblot showed ptau was elevated by Aβ42 oligomers and JNK inhibitor SP600125 blocked this. ptau protein levels were significantly increased in cytosol (C) (***p = 0.001), whereas JNK inhibitor SP600125 significantly blocked the elevated ptau when compared with CTRL (C) (***p = 0.001). β-Actin was used for normalization. Error bars indicate SEM.
Like JNK inhibitor, DHA suppressed Aβ oligomer-induced JNK phosphorylation/inactivation of IRS-1 and JNK-sensitive tau Ser422 phosphorylation in cultured hippocampal neurons
IRS-1 has been proposed as a target of free fatty acids in insulin resistance. Omega-6 (n-6) linoleic acid reduces IRS-1 and insulin-induced glucose uptake in 3T3-L1 adipocytes, a cell model that mimics insulin resistance in mice (Gao et al., 2004). Our previous studies reported that the depletion of dietary omega-3 (n-3) polyunsaturated fatty acids (n-3 PFAs) by a safflower oil-based high n-6 linoleic acid diet in Tg2576 mice, an AD transgenic mouse model, resulted in large losses of the p85 subunit of phosphatidylinositol 3-kinase, whereas treatment of these mice with DHA (22:6n-3), a long-chain n-3 PFA, significantly protected PI3-K loss and ameliorated behavior (Calon et al., 2004). Other groups have also reported that DHA was able to protect insulin resistance and alterations of insulin signaling in rodents in vivo (Delarue et al., 2004). Therefore, to investigate the role of n-3 PFAs for IRS-1>p85-PI3-K mediated CNS insulin signaling, we tested whether DHA has the ability to suppress Aβ oligomer-induced phosphorylation and inactivation of IRS-1 as well as tau Ser422 phosphorylation in cultured neurons andcompared with effect of a JNK inhibitor. Intriguingly, after pretreatment of cultured neurons with DHA for 48 h, the Aβ oligomer-induced inactivating phosphorylation of IRS-1 (indexed with phospho-Ser616 antibody), as well as phospho-tau Ser422, were both significantly suppressed (***p < 0.001) (Fig. 7A–C). These results demonstrated DHA has effects similar to JNK inhibitor on the inhibition of Aβ oligomer-induced uncoupling phosphorylation of IRS-1 and tau phosphorylation.
Figure 7.
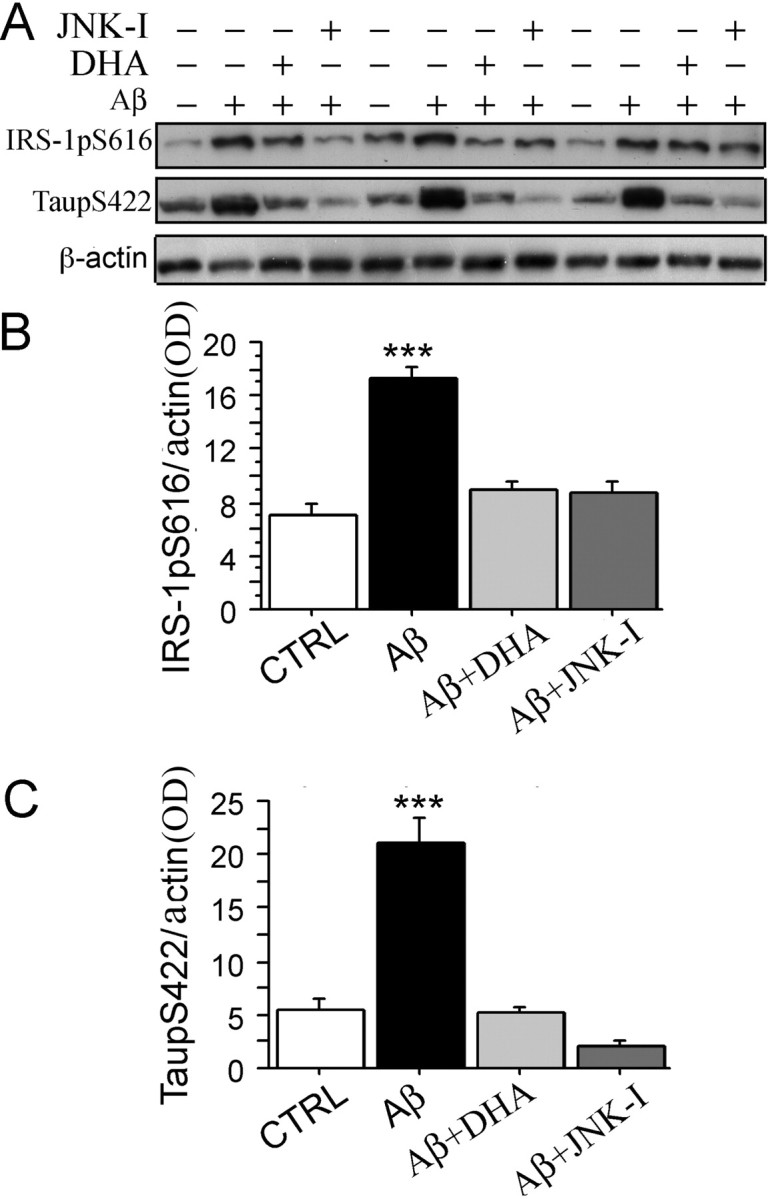
DHA suppressed Aβ oligomer-induced aberrant inactivation of IRS-1 Ser616 and JNK-sensitive tau Ser422 phosphorylation in cultured hippocampal neurons. Hippocampal neurons were cultured for 9 DIV and then pretreated either with 2.5 μm DHA for 48 h or with the JNK inhibitor SP600125 (10 μm) for 30 min, followed by 100 nm Aβ42 oligomers for 5 h, after which pIRS-1 and ptau were evaluated by Western blot. A, B, Western immunoblot analysis of pIRS-1 showed induction by Aβ42 oligomers, a response antagonized by either DHA or JNK inhibitor (SP600125). pIRS-1 protein levels were significantly increased in membrane fractions (B) (***p < 0.001) by Aβ42 oligomers, but not if DHA or JNK inhibitor SP600125 was also present (B). A, C, Western immunoblot analysis shows that ptau was induced by Aβ42 oligomers, a response that was blocked by JNK inhibitor SP600125. ptau protein levels were significantly increased in cytosol (C) (***p < 0.001), but not if DHA or JNK inhibitor SP600125 was also present. β-Actin was used for normalization for protein loading. Error bars indicate SEM.
High-fat bad diet increased phosphorylation of JNK, IRS-1, and tau in 3xTg-AD transgenic-positive mice
We used a line of 3xTg-AD mice that appeared to have less aggressive Aβ and tau pathology and no water maze behavior defects at early ages (5- to 9-month-old mice) (Rosario et al., 2006; Hirata-Fukae et al., 2008), in contrast to the original report. To accelerate pathogenesis, we used a HFBD based on an omega-3-depleting high n-6 linoleic acid diet (Calon et al., 2004) supplemented with 16% saturated fat (coconut oil) and 1500 ppm cholesterol (Table 1, Harlan Teklad test diet). Western blot analysis showed the levels of pJNK, pIRS-1 Ser616, and ptau Ser422 were significantly increased in membrane fractions from HFBD 3xTg-AD mice when compared with control standard diet mice (Fig. 8A) (pJNK, *p < 0.05) (Fig. 8B) (pIRS-1, *p = 0.05) (Fig. 8C) (ptau, **p < 0.01).
Figure 8.

Effect of HFBD, fish oil, curcumin, and the combination on phosphorylation of JNK, IRS-1, and tau in 3xTg-AD mice. A–C, Five-month-old 3xTg-AD transgenic-positive mice were fed standard mouse chow or with a HFBD (see text) for 4 months, and the phosphorylation of JNK, IRS-1 Ser616, and tau Ser422 were evaluated from detergent lysis buffer-extracted hippocampal membrane pellet fractions by Western blot. The levels of pJNK, pIRS-1 Ser616, and ptau Ser422 were significantly increased from HBFD 3xTg-AD mice when compared with control standard diet mice (A; pJNK, *p = 0.022) (B; pIRS-1, *p = 0.053) (C; ptau, **p = 0.004). For treatments, 5-month-old 3xTg-AD mice were fed with HFBD or HBFD plus 2.4% fish oil (FO), or HBFD plus 500 ppm curcumin or HBFD plus both FO and curcumin for 4 months, and the phosphorylation of JNK, IRS-1 Ser616, and tau Ser422 were evaluated from detergent lysis buffer-extracted hippocampal membrane pellet fractions by Western blot. The levels of pJNK were significantly reduced from fish oil-treated HFBD 3xTg-AD mice (A; *p = 0.025), the serine phosphorylated (inactivated) IRS-1 (B; p = 0.073), and phospho-tau (C; p = 0.078) showed a trend toward reduction when compared with HFBD alone control mice. The activated JNK (A; p = 0.059) showed a trend to be reduced by curcumin treatment in 3xTg-AD, whereas IRS-1 Ser616 (B; *p = 0.05) and ptau Ser422 (C; *p = 0.027) were significantly reduced when compared with 3xAD-Tg mice on HFBD alone. The combination of fish oil and curcumin significantly suppressed all three endpoints, pJNK (A; **p = 0.007), pIRS-1 (B; *p = 0.04), and ptau (C; *p = 0.016) in 3xTg-AD mice when compared with 3xAD-Tg mice on HFBD alone. β-Actin was used for normalization. Error bars indicate SEM.
Fish oil suppressed JNK and showed a trend to suppress IRS-1 and tau phosphorylation in 3xTg-AD mice
Our previous studies found that Tg2576 mice fed with safflower oil-based, n-3 fatty acid-depleted diet showed a significant decrease in brain DHA concentration, excitatory synaptic markers including drebrin, PSD-95, and CaMKIIa, and cognitive function, whereas DHA supplementation significantly protected from these losses and ameliorated water maze deficits (Calon et al., 2004, 2005). In the current study, HFBD-fed 3xTg-AD transgenic mice were used. Five-month-old HFBD 3xTg-AD mice were administrated chow with or without 2.4% fish oil (calculated to deliver 0.6% DHA) for 4 months, behaviorally tested, and killed at 9 months. The phospho-JNK, IRS-1 Ser616, and tau Ser422 were examined by Western blot. Fish oil significantly suppressed the activation of JNK (*p = 0.025) (Fig. 8A), similar to DHA in vitro, and showed a trend to suppress phosphorylation of IRS-1 (Fig. 8B) (p = 0.073) and tau (Fig. 8C) (p = 0.078), which were both elevated in HFBD 3xTg-AD mice compared with the vehicle mice. These results parallel the observation that DHA blocks Aβ oligomer-induced inactivation of IRS-1 and tau phosphorylation in cultured primary neurons in vitro.
Curcumin suppressed JNK-sensitive tau Ser422 and showed a trend for suppression of JNK activation and IRS-1 inactivation in 3xTg-AD mice
Curcumin, the yellow pigment in the curry spice turmeric, suppresses tumor initiation and tumor promotion. Curcumin is also a potent inhibitor for AP-1, nuclear factor κB (NF-κB), and JNK activation. Curcumin inhibits JNK activation by various agonists, including PMA plus ionomycin, anisomycin, UV-C, gamma irradiation, tumor necrosis factor (TNF), and sodium orthovanadate in cultured cell lines (Chen and Tan, 1998). Both JNK and NF-κB inhibitory activities may contribute to the antiinflammatory and anticancer properties of curcumin (Ruby et al., 1995; Singh and Aggarwal, 1995). We previously reported that curcumin suppressed JNK activation in aged Tg2576 mice (Begum et al., 2008), in which the safflower oil-based diet 17-month-old Tg2576 mice were fed with or without 500 ppm curcumin (Yang et al., 2005). Our group has also found that curcumin is an effective antiamyloid and anti-Aβ oligomer agent in vivo and in vitro (Yang et al., 2005). In the current study, we tested whether, like DHA, curcumin also inhibits JNK signaling to IRS-1 and tau in 3xTg-AD mice. These mice, which contain a mutant human tau transgene and develop tau pathology, were fed with or without 500 ppm curcumin for 4 months, behaviorally tested, and then killed. The phosphorylation of JNK, IRS-1 at Ser616, and tau at Ser422 were evaluated by Western blot. Curcumin showed a trend to suppress JNK (p = 0.059) (Fig. 8A) in the 3xTg-AD mice and significantly suppressed IRS-1 phosphorylation (*p = 0.05) (Fig. 8B) and the JNK-sensitive phospho-tau Ser422 epitope (*p < 0.05) (Fig. 8C).
Combination of fish oil and curcumin enhanced the effects on the suppression of JNK, IRS-1, and tau phosphorylation in 3xTg-AD mice
DHA (22:6n-3) is one of the most peroxidizable lipids in the brain because of its six double bonds (Montine et al., 2004). DHA could be depleted by the elevated lipid peroxidation in AD brain and Tg2576 mice (Aksenov et al., 2001; Praticò et al., 2001; Montine et al., 2004). In contrast, curcumin, a potent antioxidant and antiinflammatory compound should protect DHA from lipid peroxidation. Since both agents also reduce Aβ accumulation but by different mechanisms (Lim et al., 2005; Yang et al., 2005) and reduce JNK activation, we speculated that the combination of fish oil/DHA and curcumin diet could have the synergistic effects on the inhibition of activated JNK, IRS-1, and tau in 3xTg-AD mice. To test this hypothesis, high-fat bad-diet fed 5-month-old 3xTg-AD mice were treated either with fish oil, curcumin alone, or fish oil plus curcumin for 4 months, behaviorally tested, and killed, and the phosphorylation of JNK, IRS-1, and tau were examined by Western blot. As expected, the mice fed with fish oil plus curcumin diet had significantly lower levels of phosphorylated JNK (**p < 0.01), IRS-1 (*p < 0.05), and JNK-sensitive tau phospho-Ser422 (*p < 0.05) than the mice fed only with curcumin or fish oil compared with the vehicle diet mice (Fig. 8A–C)). Overall, the combination of curcumin and fish oil was more effective than either agent alone.
Fish oil and curcumin prevented the degradation of total IRS-1 in 3xTg-AD mice
With insulin resistance in diabetics and models, IRS-1 is phosphorylated by insulin-stimulated or stress-activated kinases, which uncouples IRS-1 (Aguirre et al., 2002) and triggers rapid IRS-1 degradation (Sun et al., 1999), yielding a deficient signal transduction response (Pederson et al., 2001; Rui et al., 2001). Decreased levels of total IRS-1 have been observed in these 3xTg-AD transgenic-positive mice (Moloney et al., 2008) and confirmed in our work here (Fig. 2). To evaluate whether fish oil or curcumin treatment protects IRS-1 from degradation, we examined total IRS-1 protein levels by Western blot (Fig. 9) and found that all three treatment groups showed significant effects on total IRS-1 levels. Fish oil increased total IRS-1 by 17.3% (**p < 0.01); curcumin or the combination increased total IRS-1 by 11.2% (*p < 0.05).
Figure 9.

Effect of HFBD, fish oil, curcumin, and the combination on total IRS-1 in 3xTg-AD mice. Five-month-old 3xTg-AD mice were fed with HFBD or HBFD plus fish oil or curcumin alone or in combination for 4 months and total IRS-1 (tIRS-1) was evaluated by Western blot. The levels of IRS-1 were significantly increased in membrane fractions from all treatment groups; fish oil increased levels by 17.3% (**p < 0.01), curcumin by 11.2% (*p < 0.05), and the combination of them by 11.2% (*p < 0.05) when compared with HFBD alone in 3xTg-AD mice. Error bars indicate SEM.
Combination of fish oil and curcumin ameliorated behavioral decline in 3xTg-AD mice
To compare the effect of fish oil, curcumin, and the combination of fish oil and curcumin on behavior, we tested SAB in the Y-maze, a short-term memory test, which showed deficits in our line of 5- to 9-month-old 3xTg-AD mice despite the absence of Morris water maze (MWM) deficits. The four different treatment groups of 3xTg-AD mice were evaluated after 1 or 2 months on diets (Fig. 9). Although both fish oil and curcumin alone showed either significant effects or strong trends for effects on the inhibition of JNK, IRS-1, and tau phosphorylation in 3xTg-AD mice, they did not produce a significant improvement in Y-maze performance (SAB) after 1 month on diets. However, mice fed with both fish oil and curcumin together for 1 month showed significant improvement of SAB scores indicating reduced cognitive decline in 3xTg-AD mice (**p < 0.01). After 2 months on diets (7 months of age), testing was repeated and deficits compared with wild-type controls and transgenic mice performance at 1 month on diet were enhanced (**p < 0.01). Furthermore, after 2 months, fish oil alone (**p < 0.01), curcumin alone (**p < 0.01), and the combination (*p < 0.05) all prevented decline in the 3xTg-AD mouse model (Fig. 10). Testing was also repeated at 3 months, yielding similar results with all three treatments maintaining protection (data not shown).
Figure 10.

Curcumin and/or fish oil prevent cognitive deficits. Spontaneous alternation tests of 3xTg-AD mice compared with wild-type (WT) controls of the same age and background strain using Y-maze were performed after 1 or 2 months of treatment. WT mice had no decline with age on the HFBD. In contrast, the 3xTg-AD mice exhibited a significant behavior deficit developing from 5 to 7 months of age, which was more evident in the HFBD (significant from WT after 1 month on diet, **p < 0.01) (6 months of age). Only fish oil plus curcumin (Cur) added to HFBD base diet corrected the deficit at 1 month (**p < 0.01), but all three treatments (fish oil, curcumin, fish oil plus curcumin) prevented additional cognitive decline after 2 months of treatment (**p < 0.01 for fish oil and curcumin, *p < 0.05 for fish oil plus curcumin). Error bars indicate SEM.
Discussion
In this study, we used in vitro and in vivo models to identify an Aβ-dependent mechanism contributing to insulin/trophic factor pathway defects (increased phospho-IRS-1, decreased total IRS-1) that are observed in AD brain and similar to reports on insulin-resistant peripheral tissue. In primary neurons, Aβ oligomers induced JNK activation and IRS-1 phosphorylation, which is known to uncouple insulin signaling. The n-3 fatty acid DHA prevented this. Diet with high levels of “bad” fats (saturated fat, cholesterol, n-6 linoleic acid) and low n-3 increased phosphorylation of JNK, IRS-1, and tau in 3xTg-AD transgenic mice. Dietary fish oil (a DHA source) or the polyphenolic antiinflammatory agent, curcumin, prevented these changes alone, worked better in combination, and ameliorated behavioral decline.
Insulin/IR signaling regulates glucose metabolism and energy homeostasis, and, in the periphery, its defective response is implicated in insulin resistance or type 2 diabetes (Reaven, 1995; Kruszynska and Olefsky, 1996; Kahn and Flier, 2000; Pessin and Saltiel, 2000). Adult-onset diabetes is a consistent AD risk factor associated with increased accumulation of pathological Aβ and/or hyperphosphorylated tau (Ott et al., 1996, 1999; Leibson et al., 1997; Cole and Frautschy, 2007). When presymptomatic mice expressing mutant human amyloid precursor protein and presenilin are made diabetic, they develop cognitive deficits associated with the loss of the synaptic spine protein drebrin (Burdo et al., 2008), a postsynaptic marker that is lost in APP transgenic mice with DHA depletion and restored by DHA supplements (Calon et al., 2004). The insulin/IR complex is abundantly distributed in cerebral cortical and hippocampal synaptic membranes (Heidenreich et al., 1988; Matsumoto and Rhoads, 1990; Zhao et al., 1999), in which it regulates activities required for learning and memory (Hendricks et al., 1984; Lowe et al., 1986; Biessels et al., 1996; Wan et al., 1997; Zhao and Alkon, 2001). Consistent with our findings, an IR and IRS-1 defect was reported to occur in AD brain (Moloney et al., 2008). In cultured hippocampal neurons, Aβ oligomers appeared to cause loss and redistribution of IR receptors on dendrites via the NMDA receptor (Zhao et al., 2008). An increase in phospho-IRS-1 would impact signaling through other trophic factor receptors. Together with our data, this suggests Aβ oligomers are likely playing a major and direct role in insulin-signaling deficits through effects on IR and IRS-1.
In the current study, Aβ oligomers induced aberrant phosphorylation of IRS-1 at Ser616, a phospho-epitope implicated in the periphery in disrupting insulin–insulin receptor signaling (De Fea and Roth, 1997; Andreozzi et al., 2004a,b). In peripheral tissues, both JNK and ERK phosphorylate IRS-1 at Ser307 (rodent equivalent for human Ser312) (Aguirre et al., 2000) and Ser616 (D'Alessandris et al., 2007). The resulting loss of IRS-1 coupling to PI3-K/Akt is predicted to activate another major tau kinase, GSK3β, which is known to be activated by Aβ (Takashima et al., 1996; Ma et al., 2006; Hu et al., 2009). This process further dysregulates insulin signaling since GSK3 itself directly phosphorylates IRS-1 at Ser332 to help inactivate IRS-1 (Liberman and Eldar-Finkelman, 2005). This suggests a positive-feedback loop in which Aβ activation of JNK initiates an uncoupling of the pathway.
We demonstrated a progression of changes with age in the 3xTg-AD model leading to colocalization of pIRS-1 and ptau that resembles similar colocalization in AD. Similar patterns of staining were observed in the 3xTg AD model on the high-fat, high-n-6 DHA-depleting diet, which increased pJNK, pIRS-1, and ptau and accelerated Y-maze deficits, and these changes were ameliorated by DHA-rich dietary fish oil.
DHA deficiency contributes to the loss of the p85 PI3-K subunit and peripheral insulin resistance in adult-onset diabetes (Liolitsa et al., 2002). In the brain, DHA deficiency can exacerbate insulin signaling deficits, particularly in the presence of an AD mutant APP transgene (Calon et al., 2004; Delarue et al., 2004). DHA, an important dietary n-3 (omega-3) fatty acid was the plasma lipid most predictive of AD risk in the Framingham study in which higher blood levels of DHA correlated with reduced AD assessed 10 years later (Johnson and Schaefer, 2006). Our own and previous work from other groups indicate that dietary DHA significantly improves cognitive deficits, protects from synaptic protein loss, and lowers insoluble Aβ in transgenic AD mouse models (Calon et al., 2004; Lim et al., 2005; Oksman et al., 2006; Ma et al., 2007). Our current data are consistent with a report that algal DHA reduced Aβ accumulation, suggesting this effect may have reduced JNK activation and ptau in 3xTg-AD mice (Green et al., 2007), but the effects we observed need not be secondary to Aβ reduction because we show that DHA reduces the JNK, IRS-1, and tau phosphorylation response to exogenous Aβ oligomers. Both protective activities are likely, but we did not find robust Aβ labeling at 9 months in the line of 3xTg-AD mice on the diets used. This line showed limited intraneuronal labeling but lacked plaques at 9 months (data not shown), consistent with a recent report on the same “low expressing” line (Hirata-Fukae et al., 2008).
Curcumin mediates many of its pleiotropic effects by modulation of several important molecular targets, including transcription factors [NF-κB, AP-1, PPARγ (peroxisome proliferator-activated receptor γ)], enzymes [COX2, 5-LOX, iNOS (inducible nitric oxide synthase), hemeoxygenase-1, JNK], and cytokines (TNF, IL-1β, IL-6) (Shishodia et al., 2005). As a neuroprotective drug, curcumin exerts antiinflammatory, antioxidant, and anti-protein aggregate and neurogenic effects in CNS and AD models (Frautschy et al., 2001; Cole, 2003; Cole et al., 2005; Lim et al., 2005; Yang et al., 2005; Cole and Frautschy, 2007). Curcumin reduced plaque deposition, proinflammatory cytokines, and pJNK in Tg2576 APPsw mice that lack tau pathology (Begum et al., 2008). Here, we found that curcumin significantly suppressed a JNK/IRS-1/tau signaling pathway that leads to AD-like p-IRS-1/insulin signaling defects in 3xTg-AD mice, providing an additional mechanism for efficacy in an AD model with tau pathology.
The combination of fish oil with curcumin had a synergistic effect on suppressing JNK/IRS-1 and tau phosphorylation and limited cognitive deficits at 1 month, providing evidence for the possible combination of fish oil and curcumin for AD treatment or prevention. One explanation may be that DHA and curcumin target some different steps in the Aβ cascade. For example, curcumin has a direct anti-Aβ binding activities, which can directly antagonize Aβ effects on insulin signaling in addition to its inhibition of CNS JNK in vivo (Begum et al., 2008). Furthermore, both influence arachidonic acid metabolites affecting inflammation but at different sites: DHA competitively reduces availability of arachidonic acid substrate in phospholipids, whereas curcumin reduces activity of the phospholipases, cyclooxygenases, and lipoxygenases enzymes, which produce proinflammatory lipid mediators. Although inflammation is limited in 9 month 3xTg-AD mice, anti-inflammatory effects of DHA and curcumin should reduce proinflammatory cytokines like TNFα. Since IRS-1 phosphorylation can also be induced by inflammatory cytokines, notably TNFα (Rui et al., 2001) and possibly via reducing JNK activation (Kallunki et al., 1994), the anti-inflammatory activity of DHA and curcumin provides another mechanism for protecting insulin/trophic factor signaling.
DHA is susceptible to lipid peroxidation in AD and in AD models, arguing that DHA should be combined with an antioxidant-like α-lipoate or curcumin (Calon and Cole, 2007). In AD brain, oxidative stress (Rotstein et al., 2003) causes nonenzymatic oxidation of DHA to F4-isoprostane (neuroprostane) (Montine et al., 2004), depleting it from synaptic membranes and further exacerbating signaling defects (Calon et al., 2004). Potential synergism between fish oil/DHA and curcumin could also accrue through a reduction in oxidative stress since we have previously observed both DHA (Calon et al., 2004) and curcumin (Yang et al., 2005) suppress oxidative damage in APPsw Tg2576 mice. In peripheral models, oxidative damage has been shown to contribute to insulin resistance (Paolisso et al., 1994; Paolisso and Giugliano, 1996), causing enhanced serine/threonine phosphorylation on the IR or IRS because of increased oxidative stress-sensitive kinase activity [e.g., NF-κB-activating kinases, p38 MAPK (mitogen-activated protein kinase), JNK/SAPK (stress-activated protein kinase), PKC, or other serine/threonine kinase(s)] (Evans et al., 2002).
Fish oil, curcumin, and the combination all significantly prevented cognitive decline on SAB in the Y-maze, a task involving working memory that we measured within sessions in 2-month-treated 3xTg-AD mice. The 3xTg-AD mouse line we used here performs poorly in the Y-maze at 5–9 months of age, whereas they show no impairment in MWM, a task involving spatial learning memory. Studies indicate intrasession working memory deficits may not necessarily be associated with delayed spatial learning memory in MWM, and there is lesion evidence suggesting that working and reference memory may depend on different brain systems (Dudchenko, 2004). Although Y-maze may not fully isolate memory from other causes of behavioral defects, deficits in working memory measured with SAB in the Y-maze have been widely used to identify cognitive deficits in different transgenic AD mouse models (Hughes, 2004), including APPsw Tg2576 mice (King and Arendash, 2002; Ognibene et al., 2005), double mutant APP and PS1 transgenic mice (Holcomb et al., 1998), and β-APP751 transgenic mice (Moran et al., 1995). SAB deficits also occur early in the line of 3xTg-AD mice we used, in which they have been linked to hippocampal-dependent SAB impairment (Rosario et al., 2006).
In summary, pleiotropic effects of both drugs on multiple pathways involved in contributing to insulin resistance could contribute to protective effects in AD. We have identified an Aβ oligomer and JNK-dependent pathway that disrupts an insulin signaling adaptor protein in AD models leading to defects found in AD brain. These defects can be potentially improved in the models by safe and inexpensive pleiotropic interventions with fish oil or curcumin, most effectively with the combination.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant R01 AT003008 (G.M.C.); National Institute on Aging–NIH Grants R01 AG16570 (G.M.C., H.V.V.), AG13471 (G.M.C.), U01 AG028583 (S.A.F.), and R01 AG021975 (S.A.F.); and Alzheimer's Association Grant NIRG-07-59659 (Q.-L.M.). H.V.V. was supported in part by the Daljit S. and Elaine Sarkaria Chair in Diagnostic Medicine.
The authors declare no competing financial interests.
References
- Aguirre V, Uchida T, Yenush L, Davis R, White MF. The c-Jun NH2-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307) J Biol Chem. 2000;275:9047–9054. doi: 10.1074/jbc.275.12.9047. [DOI] [PubMed] [Google Scholar]
- Aguirre V, Werner ED, Giraud J, Lee YH, Shoelson SE, White MF. Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J Biol Chem. 2002;277:1531–1537. doi: 10.1074/jbc.M101521200. [DOI] [PubMed] [Google Scholar]
- Aksenov MY, Aksenova MV, Butterfield DA, Geddes JW, Markesbery WR. Protein oxidation in the brain in Alzheimer's disease. Neuroscience. 2001;103:373–383. doi: 10.1016/s0306-4522(00)00580-7. [DOI] [PubMed] [Google Scholar]
- Andreozzi F, Laratta E, Sciacqua A, Perticone F, Sesti G. Angiotensin II impairs the insulin signaling pathway promoting production of nitric oxide by inducing phosphorylation of insulin receptor substrate-1 on Ser312 and Ser616 in human umbilical vein endothelial cells. Circ Res. 2004a;94:1211–1218. doi: 10.1161/01.RES.0000126501.34994.96. [DOI] [PubMed] [Google Scholar]
- Andreozzi F, D'Alessandris C, Federici M, Laratta E, Del Guerra S, Del Prato S, Marchetti P, Lauro R, Perticone F, Sesti G. Activation of the hexosamine pathway leads to phosphorylation of insulin receptor substrate-1 on Ser307 and Ser612 and impairs the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin insulin biosynthetic pathway in RIN pancreatic beta-cells. Endocrinology. 2004b;145:2845–2857. doi: 10.1210/en.2003-0939. [DOI] [PubMed] [Google Scholar]
- Begum AN, Jones MR, Lim GP, Morihara T, Kim P, Heath DD, Rock CL, Pruitt MA, Yang F, Hudspeth B, Hu S, Faull KF, Teter B, Cole GM, Frautschy SA. Curcumin structure-function, bioavailability, and efficacy in models of neuroinflammation and Alzheimer's disease. J Pharmacol Exp Ther. 2008;326:196–208. doi: 10.1124/jpet.108.137455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begum N, Sandu OA, Ito M, Lohmann SM, Smolenski A. Active Rho kinase (ROK-alpha) associates with insulin receptor substrate-1 and inhibits insulin signaling in vascular smooth muscle cells. J Biol Chem. 2002;277:6214–6222. doi: 10.1074/jbc.M110508200. [DOI] [PubMed] [Google Scholar]
- Biessels GJ, Kamal A, Ramakers GM, Urban IJ, Spruijt BM, Erkelens DW, Gispen WH. Place learning and hippocampal synaptic plasticity in streptozotocin-induced diabetic rats. Diabetes. 1996;45:1259–1266. doi: 10.2337/diab.45.9.1259. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Burdo JR, Chen Q, Calcutt NA, Schubert D Advance online publication. The pathological interaction between diabetes and presymptomatic Alzheimer's disease. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2008.02.010. Retrieved June 25, 2009. doi: 10.1016/jneurobiolaging.2008.02.010. [DOI] [PubMed] [Google Scholar]
- Calon F, Cole G. Neuroprotective action of omega-3 polyunsaturated fatty acids against neurodegenerative diseases: evidence from animal studies. Prostaglandins Leukot Essent Fatty Acids. 2007;77:287–293. doi: 10.1016/j.plefa.2007.10.019. [DOI] [PubMed] [Google Scholar]
- Calon F, Lim GP, Yang F, Morihara T, Teter B, Ubeda O, Rostaing P, Triller A, Salem N, Jr, Ashe KH, Frautschy SA, Cole GM. Docosahexaenoic acid protects from dendritic pathology in an Alzheimer's disease mouse model. Neuron. 2004;43:633–645. doi: 10.1016/j.neuron.2004.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calon F, Lim GP, Morihara T, Yang F, Ubeda O, Salem N, Jr, Frautschy SA, Cole GM. Dietary n-3 polyunsaturated fatty acid depletion activates caspases and decreases NMDA receptors in the brain of a transgenic mouse model of Alzheimer's disease. Eur J Neurosci. 2005;22:617–626. doi: 10.1111/j.1460-9568.2005.04253.x. [DOI] [PubMed] [Google Scholar]
- Chen YR, Tan TH. Inhibition of the c-Jun N-terminal kinase (JNK) signaling pathway by curcumin. Oncogene. 1998;17:173–178. doi: 10.1038/sj.onc.1201941. [DOI] [PubMed] [Google Scholar]
- Cole GM. Ironic fate: can a banned drug control metal heavies in neurodegenerative diseases? Neuron. 2003;37:889–890. doi: 10.1016/s0896-6273(03)00160-0. [DOI] [PubMed] [Google Scholar]
- Cole GM, Frautschy SA. The role of insulin and neurotrophic factor signaling in brain aging and Alzheimer's Disease. Exp Gerontol. 2007;42:10–21. doi: 10.1016/j.exger.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Cole GM, Lim GP, Yang F, Teter B, Begum A, Ma Q, Harris-White ME, Frautschy SA. Prevention of Alzheimer's disease: Omega-3 fatty acid and phenolic anti-oxidant interventions. Neurobiol Aging. 2005;26(Suppl 1):133–136. doi: 10.1016/j.neurobiolaging.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Craft S, Peskind E, Schwartz MW, Schellenberg GD, Raskind M, Porte D., Jr Cerebrospinal fluid and plasma insulin levels in Alzheimer's disease: relationship to severity of dementia and apolipoprotein E genotype. Neurology. 1998;50:164–168. doi: 10.1212/wnl.50.1.164. [DOI] [PubMed] [Google Scholar]
- Craft S, Asthana S, Cook DG, Baker LD, Cherrier M, Purganan K, Wait C, Petrova A, Latendresse S, Watson GS, Newcomer JW, Schellenberg GD, Krohn AJ. Insulin dose-response effects on memory and plasma amyloid precursor protein in Alzheimer's disease: interactions with apolipoprotein E genotype. Psychoneuroendocrinology. 2003;28:809–822. doi: 10.1016/s0306-4530(02)00087-2. [DOI] [PubMed] [Google Scholar]
- D'Alessandris C, Lauro R, Presta I, Sesti G. C-reactive protein induces phosphorylation of insulin receptor substrate-1 on Ser307 and Ser612 in L6 myocytes, thereby impairing the insulin signalling pathway that promotes glucose transport. Diabetologia. 2007;50:840–849. doi: 10.1007/s00125-006-0522-y. [DOI] [PubMed] [Google Scholar]
- De Fea K, Roth RA. Protein kinase C modulation of insulin receptor substrate-1 tyrosine phosphorylation requires serine 612. Biochemistry. 1997;36:12939–12947. doi: 10.1021/bi971157f. [DOI] [PubMed] [Google Scholar]
- Delarue J, LeFoll C, Corporeau C, Lucas D. N-3 long chain polyunsaturated fatty acids: a nutritional tool to prevent insulin resistance associated to type 2 diabetes and obesity? Reprod Nutr Dev. 2004;44:289–299. doi: 10.1051/rnd:2004033. [DOI] [PubMed] [Google Scholar]
- Dudchenko PA. An overview of the tasks used to test working memory in rodents. Neurosci Biobehav Rev. 2004;28:699–709. doi: 10.1016/j.neubiorev.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr Rev. 2002;23:599–622. doi: 10.1210/er.2001-0039. [DOI] [PubMed] [Google Scholar]
- Frautschy SA, Hu W, Kim P, Miller SA, Chu T, Harris-White ME, Cole GM. Phenolic anti-inflammatory antioxidant reversal of Abeta-induced cognitive deficits and neuropathology. Neurobiol Aging. 2001;22:993–1005. doi: 10.1016/s0197-4580(01)00300-1. [DOI] [PubMed] [Google Scholar]
- Gao Z, Hwang D, Bataille F, Lefevre M, York D, Quon MJ, Ye J. Serine phosphorylation of insulin receptor substrate 1 by inhibitor kappa B kinase complex. J Biol Chem. 2002;277:48115–48121. doi: 10.1074/jbc.M209459200. [DOI] [PubMed] [Google Scholar]
- Gao Z, Zhang X, Zuberi A, Hwang D, Quon MJ, Lefevre M, Ye J. Inhibition of insulin sensitivity by free fatty acids requires activation of multiple serine kinases in 3T3–L1 adipocytes. Mol Endocrinol. 2004;18:2024–2034. doi: 10.1210/me.2003-0383. [DOI] [PubMed] [Google Scholar]
- Garcia-Alloza M, Borrelli LA, Rozkalne A, Hyman BT, Bacskai BJ. Curcumin labels amyloid pathology in vivo, disrupts existing plaques, and partially restores distorted neurites in an Alzheimer mouse model. J Neurochem. 2007;102:1095–1104. doi: 10.1111/j.1471-4159.2007.04613.x. [DOI] [PubMed] [Google Scholar]
- Green KN, Martinez-Coria H, Khashwji H, Hall EB, Yurko-Mauro KA, Ellis L, LaFerla FM. Dietary docosahexaenoic acid and docosapentaenoic acid ameliorate amyloid-β and tau pathology via a mechanism involving presenilin 1 levels. J Neurosci. 2007;27:4385–4395. doi: 10.1523/JNEUROSCI.0055-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Heidenreich KA, Zahniser NR, Berhanu P, Brandenburg D, Olefsky JM. Structural differences between insulin receptors in the brain and peripheral target tissues. J Biol Chem. 1983;258:8527–8530. [PubMed] [Google Scholar]
- Heidenreich KA, de Vellis G, Gilmore PR. Functional properties of the subtype of insulin receptor found on neurons. J Neurochem. 1988;51:878–887. doi: 10.1111/j.1471-4159.1988.tb01824.x. [DOI] [PubMed] [Google Scholar]
- Hendricks SA, Agardh CD, Taylor SI, Roth J. Unique features of the insulin receptor in rat brain. J Neurochem. 1984;43:1302–1309. doi: 10.1111/j.1471-4159.1984.tb05387.x. [DOI] [PubMed] [Google Scholar]
- Hirata-Fukae C, Li HF, Hoe HS, Gray AJ, Minami SS, Hamada K, Niikura T, Hua F, Tsukagoshi-Nagai H, Horikoshi-Sakuraba Y, Mughal M, Rebeck GW, LaFerla FM, Mattson MP, Iwata N, Saido TC, Klein WL, Duff KE, Aisen PS, Matsuoka Y. Females exhibit more extensive amyloid, but not tau, pathology in an Alzheimer transgenic model. Brain Res. 2008;1216:92–103. doi: 10.1016/j.brainres.2008.03.079. [DOI] [PubMed] [Google Scholar]
- Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O'Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- Hu S, Begum AN, Jones MR, Oh MS, Beech WK, Beech BH, Yang F, Chen P, Ubeda OJ, Kim PC, Davies P, Ma Q, Cole GM, Frautschy SA. GSK3 inhibitors show benefits in an Alzheimer's disease (AD) model of neurodegeneration but adverse effects in control animals. Neurobiol Dis. 2009;33:193–206. doi: 10.1016/j.nbd.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes RN. The value of spontaneous alternation behavior (SAB) as a test of retention in pharmacological investigations of memory. Neurosci Biobehav Rev. 2004;28:497–505. doi: 10.1016/j.neubiorev.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Johnson EJ, Schaefer EJ. Potential role of dietary n-3 fatty acids in the prevention of dementia and macular degeneration. Am J Clin Nutr. 2006;83:1494S–1498S. doi: 10.1093/ajcn/83.6.1494S. [DOI] [PubMed] [Google Scholar]
- Kahn BB, Flier JS. Obesity and insulin resistance. J Clin Invest. 2000;106:473–481. doi: 10.1172/JCI10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallunki T, Su B, Tsigelny I, Sluss HK, Dérijard B, Moore G, Davis R, Karin M. JNK2 contains a specificity-determining region responsible for efficient c-Jun binding and phosphorylation. Genes Dev. 1994;8:2996–3007. doi: 10.1101/gad.8.24.2996. [DOI] [PubMed] [Google Scholar]
- King DL, Arendash GW. Behavioral characterization of the Tg2576 transgenic model of Alzheimer's disease through 19 months. Physiol Behav. 2002;75:627–642. doi: 10.1016/s0031-9384(02)00639-x. [DOI] [PubMed] [Google Scholar]
- Kruszynska YT, Olefsky JM. Cellular and molecular mechanisms of non-insulin dependent diabetes mellitus. J Investig Med. 1996;44:413–428. [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, Klein WL. Synaptic targeting by Alzheimer's-related amyloid-β oligomers. J Neurosci. 2004;24:10191–10200. doi: 10.1523/JNEUROSCI.3432-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YH, Giraud J, Davis RJ, White MF. c-Jun N-terminal kinase (JNK) mediates feedback inhibition of the insulin signaling cascade. J Biol Chem. 2003;278:2896–2902. doi: 10.1074/jbc.M208359200. [DOI] [PubMed] [Google Scholar]
- Leibson CL, Rocca WA, Hanson VA, Cha R, Kokmen E, O'Brien PC, Palumbo PJ. The risk of dementia among persons with diabetes mellitus: a population-based cohort study. Ann N Y Acad Sci. 1997;826:422–427. doi: 10.1111/j.1749-6632.1997.tb48496.x. [DOI] [PubMed] [Google Scholar]
- Liberman Z, Eldar-Finkelman H. Serine 332 phosphorylation of insulin receptor substrate-1 by glycogen synthase kinase-3 attenuates insulin signaling. J Biol Chem. 2005;280:4422–4428. doi: 10.1074/jbc.M410610200. [DOI] [PubMed] [Google Scholar]
- Lim GP, Calon F, Morihara T, Yang F, Teter B, Ubeda O, Salem N, Jr, Frautschy SA, Cole GM. A diet enriched with the omega-3 fatty acid docosahexaenoic acid reduces amyloid burden in an aged Alzheimer mouse model. J Neurosci. 2005;25:3032–3040. doi: 10.1523/JNEUROSCI.4225-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liolitsa D, Powell J, Lovestone S. Genetic variability in the insulin signalling pathway may contribute to the risk of late onset Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2002;73:261–266. doi: 10.1136/jnnp.73.3.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YF, Paz K, Herschkovitz A, Alt A, Tennenbaum T, Sampson SR, Ohba M, Kuroki T, LeRoith D, Zick Y. Insulin stimulates PKCzeta-mediated phosphorylation of insulin receptor substrate-1 (IRS-1). A self-attenuated mechanism to negatively regulate the function of IRS proteins. J Biol Chem. 2001;276:14459–14465. doi: 10.1074/jbc.M007281200. [DOI] [PubMed] [Google Scholar]
- Lowe WL, Jr, Boyd FT, Clarke DW, Raizada MK, Hart C, LeRoith D. Development of brain insulin receptors: structural and functional studies of insulin receptors from whole brain and primary cell cultures. Endocrinology. 1986;119:25–35. doi: 10.1210/endo-119-1-25. [DOI] [PubMed] [Google Scholar]
- Ma QL, Lim GP, Harris-White ME, Yang F, Ambegaokar SS, Ubeda OJ, Glabe CG, Teter B, Frautschy SA, Cole GM. Antibodies against beta-amyloid reduce Abeta oligomers, glycogen synthase kinase-3beta activation and tau phosphorylation in vivo and in vitro. J Neurosci Res. 2006;83:374–384. doi: 10.1002/jnr.20734. [DOI] [PubMed] [Google Scholar]
- Ma QL, Teter B, Ubeda OJ, Morihara T, Dhoot D, Nyby MD, Tuck ML, Frautschy SA, Cole GM. Omega-3 fatty acid docosahexaenoic acid increases SorLA/LR11, a sorting protein with reduced expression in sporadic Alzheimer's disease (AD): relevance to AD prevention. J Neurosci. 2007;27:14299–14307. doi: 10.1523/JNEUROSCI.3593-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma QL, Yang F, Calon F, Ubeda OJ, Hansen JE, Weisbart RH, Beech W, Frautschy SA, Cole GM. P21-activated kinase aberrant activation and translocation in Alzheimer's disease pathogenesis. J Biol Chem. 2008;283:14132–14143. doi: 10.1074/jbc.M708034200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto H, Rhoads DE. Specific binding of insulin to membranes from dendrodendritic synaptosomes of rat olfactory bulb. J Neurochem. 1990;54:347–350. doi: 10.1111/j.1471-4159.1990.tb13321.x. [DOI] [PubMed] [Google Scholar]
- Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- Moloney AM, Griffin RJ, Timmons S, O'Connor R, Ravid R, O'Neill C Advance online publication. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer's disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2008.04.002. Retrieved June 25, 2009. doi: 10.1016/jneurobiolaging.2008.04.002. [DOI] [PubMed] [Google Scholar]
- Montine KS, Quinn JF, Zhang J, Fessel JP, Roberts LJ, 2nd, Morrow JD, Montine TJ. Isoprostanes and related products of lipid peroxidation in neurodegenerative diseases. Chem Phys Lipids. 2004;128:117–124. doi: 10.1016/j.chemphyslip.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Moran PM, Higgins LS, Cordell B, Moser PC. Age-related learning deficits in transgenic mice expressing the 751-amino acid isoform of human beta-amyloid precursor protein. Proc Natl Acad Sci U S A. 1995;92:5341–5345. doi: 10.1073/pnas.92.12.5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer's disease. Neurobiol Aging. 2003;24:1063–1070. doi: 10.1016/j.neurobiolaging.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Ognibene E, Middei S, Daniele S, Adriani W, Ghirardi O, Caprioli A, Laviola G. Aspects of spatial memory and behavioral disinhibition in Tg2576 transgenic mice as a model of Alzheimer's disease. Behav Brain Res. 2005;156:225–232. doi: 10.1016/j.bbr.2004.05.028. [DOI] [PubMed] [Google Scholar]
- Oksman M, Iivonen H, Hogyes E, Amtul Z, Penke B, Leenders I, Broersen L, Lütjohann D, Hartmann T, Tanila H. Impact of different saturated fatty acid, polyunsaturated fatty acid and cholesterol containing diets on beta-amyloid accumulation in APP/PS1 transgenic mice. Neurobiol Dis. 2006;23:563–572. doi: 10.1016/j.nbd.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Ott A, Stolk RP, Hofman A, van Harskamp F, Grobbee DE, Breteler MM. Association of diabetes mellitus and dementia: the Rotterdam Study. Diabetologia. 1996;39:1392–1397. doi: 10.1007/s001250050588. [DOI] [PubMed] [Google Scholar]
- Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology. 1999;53:1937–1942. doi: 10.1212/wnl.53.9.1937. [DOI] [PubMed] [Google Scholar]
- Ozes ON, Akca H, Mayo LD, Gustin JA, Maehama T, Dixon JE, Donner DB. A phosphatidylinositol 3-kinase/Akt/mTOR pathway mediates and PTEN antagonizes tumor necrosis factor inhibition of insulin signaling through insulin receptor substrate-1. Proc Natl Acad Sci U S A. 2001;98:4640–4645. doi: 10.1073/pnas.051042298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolisso G, Giugliano D. Oxidative stress and insulin action: is there a relationship? Diabetologia. 1996;39:357–363. doi: 10.1007/BF00418354. [DOI] [PubMed] [Google Scholar]
- Paolisso G, D'Amore A, Volpe C, Balbi V, Saccomanno F, Galzerano D, Giugliano D, Varricchio M, D'Onofrio F. Evidence for a relationship between oxidative stress and insulin action in non-insulin-dependent (type II) diabetic patients. Metabolism. 1994;43:1426–1429. doi: 10.1016/0026-0495(94)90039-6. [DOI] [PubMed] [Google Scholar]
- Pedersen WA, Flynn ER. Insulin resistance contributes to aberrant stress responses in the Tg2576 mouse model of Alzheimer's disease. Neurobiol Dis. 2004;17:500–506. doi: 10.1016/j.nbd.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Pederson TM, Kramer DL, Rondinone CM. Serine/threonine phosphorylation of IRS-1 triggers its degradation: possible regulation by tyrosine phosphorylation. Diabetes. 2001;50:24–31. doi: 10.2337/diabetes.50.1.24. [DOI] [PubMed] [Google Scholar]
- Pessin JE, Saltiel AR. Signaling pathways in insulin action: molecular targets of insulin resistance. J Clin Invest. 2000;106:165–169. doi: 10.1172/JCI10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praticò D, Uryu K, Leight S, Trojanoswki JQ, Lee VM. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J Neurosci. 2001;21:4183–4187. doi: 10.1523/JNEUROSCI.21-12-04183.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig B, Gómez-Isla T, Ribé E, Cuadrado M, Torrejón-Escribano B, Dalfó E, Ferrer I. Expression of stress-activated kinases c-Jun N-terminal kinase (SAPK/JNK-P) and p38 kinase (p38-P), and tau hyperphosphorylation in neurites surrounding betaA plaques in APP Tg2576 mice. Neuropathol Appl Neurobiol. 2004;30:491–502. doi: 10.1111/j.1365-2990.2004.00569.x. [DOI] [PubMed] [Google Scholar]
- Reaven GM. Pathophysiology of insulin resistance in human disease. Physiol Rev. 1995;75:473–486. doi: 10.1152/physrev.1995.75.3.473. [DOI] [PubMed] [Google Scholar]
- Rivera EJ, Goldin A, Fulmer N, Tavares R, Wands JR, de la Monte SM. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer's disease: link to brain reductions in acetylcholine. J Alzheimers Dis. 2005;8:247–268. doi: 10.3233/jad-2005-8304. [DOI] [PubMed] [Google Scholar]
- Rondinone CM, Wang LM, Lonnroth P, Wesslau C, Pierce JH, Smith U. Insulin receptor substrate (IRS) 1 is reduced and IRS-2 is the main docking protein for phosphatidylinositol 3-kinase in adipocytes from subjects with non-insulin-dependent diabetes mellitus. Proc Natl Acad Sci U S A. 1997;94:4171–4175. doi: 10.1073/pnas.94.8.4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario ER, Carroll JC, Oddo S, LaFerla FM, Pike CJ. Androgens regulate the development of neuropathology in a triple transgenic mouse model of Alzheimer's disease. J Neurosci. 2006;26:13384–13389. doi: 10.1523/JNEUROSCI.2514-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotstein NP, Politi LE, German OL, Girotti R. Protective effect of docosahexaenoic acid on oxidative stress-induced apoptosis of retina photoreceptors. Invest Ophthalmol Vis Sci. 2003;44:2252–2259. doi: 10.1167/iovs.02-0901. [DOI] [PubMed] [Google Scholar]
- Ruby AJ, Kuttan G, Babu KD, Rajasekharan KN, Kuttan R. Anti-tumour and antioxidant activity of natural curcuminoids. Cancer Lett. 1995;94:79–83. doi: 10.1016/0304-3835(95)03827-j. [DOI] [PubMed] [Google Scholar]
- Rui L, Aguirre V, Kim JK, Shulman GI, Lee A, Corbould A, Dunaif A, White MF. Insulin/IGF-1 and TNF-alpha stimulate phosphorylation of IRS-1 at inhibitory Ser307 via distinct pathways. J Clin Invest. 2001;107:181–189. doi: 10.1172/JCI10934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saad MJ, Araki E, Miralpeix M, Rothenberg PL, White MF, Kahn CR. Regulation of insulin receptor substrate-1 in liver and muscle of animal models of insulin resistance. J Clin Invest. 1992;90:1839–1849. doi: 10.1172/JCI116060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage MJ, Lin YG, Ciallella JR, Flood DG, Scott RW. Activation of c-Jun N-terminal kinase and p38 in an Alzheimer's disease model is associated with amyloid deposition. J Neurosci. 2002;22:3376–3385. doi: 10.1523/JNEUROSCI.22-09-03376.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HM, Liu ZG. JNK signaling pathway is a key modulator in cell death mediated by reactive oxygen and nitrogen species. Free Radic Biol Med. 2006;40:928–939. doi: 10.1016/j.freeradbiomed.2005.10.056. [DOI] [PubMed] [Google Scholar]
- Shishodia S, Sethi G, Aggarwal BB. Curcumin: getting back to the roots. Ann N Y Acad Sci. 2005;1056:206–217. doi: 10.1196/annals.1352.010. [DOI] [PubMed] [Google Scholar]
- Shoji M, Iwakami N, Takeuchi S, Waragai M, Suzuki M, Kanazawa I, Lippa CF, Ono S, Okazawa H. JNK activation is associated with intracellular beta-amyloid accumulation. Brain Res Mol Brain Res. 2000;85:221–233. doi: 10.1016/s0169-328x(00)00245-x. [DOI] [PubMed] [Google Scholar]
- Singh S, Aggarwal BB. Activation of transcription factor NF-kappa B is suppressed by curcumin (diferuloylmethane) [corrected] J Biol Chem. 1995;270:24995–25000. doi: 10.1074/jbc.270.42.24995. [DOI] [PubMed] [Google Scholar]
- Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, Xu XJ, Wands JR, de la Monte SM. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer's disease—is this type 3 diabetes? J Alzheimers Dis. 2005;7:63–80. doi: 10.3233/jad-2005-7107. [DOI] [PubMed] [Google Scholar]
- Sun XJ, Wang LM, Zhang Y, Yenush L, Myers MG, Jr, Glasheen E, Lane WS, Pierce JH, White MF. Role of IRS-2 in insulin and cytokine signalling. Nature. 1995;377:173–177. doi: 10.1038/377173a0. [DOI] [PubMed] [Google Scholar]
- Sun XJ, Goldberg JL, Qiao LY, Mitchell JJ. Insulin-induced insulin receptor substrate-1 degradation is mediated by the proteasome degradation pathway. Diabetes. 1999;48:1359–1364. doi: 10.2337/diabetes.48.7.1359. [DOI] [PubMed] [Google Scholar]
- Takashima A, Noguchi K, Michel G, Mercken M, Hoshi M, Ishiguro K, Imahori K. Exposure of rat hippocampal neurons to amyloid beta peptide (25–35) induces the inactivation of phosphatidyl inositol-3 kinase and the activation of tau protein kinase I/glycogen synthase kinase-3 beta. Neurosci Lett. 1996;203:33–36. doi: 10.1016/0304-3940(95)12257-5. [DOI] [PubMed] [Google Scholar]
- Talbot K, Han LY, Schneider SA, Wilson RS, Bennett DA, Arnold SE. Expression of pIRS-1 (S312 and S616) is elevated in MCI and AD and correlates with cognitive impairment and neurofibrillary pathology. Alzheimers Dement. 2006;2(Suppl 3):A54. [Google Scholar]
- Troy CM, Rabacchi SA, Xu Z, Maroney AC, Connors TJ, Shelanski ML, Greene LA. beta-Amyloid-induced neuronal apoptosis requires c-Jun N-terminal kinase activation. J Neurochem. 2001;77:157–164. doi: 10.1046/j.1471-4159.2001.t01-1-00218.x. [DOI] [PubMed] [Google Scholar]
- Virkamäki A, Ueki K, Kahn CR. Protein-protein interaction in insulin signaling and the molecular mechanisms of insulin resistance. J Clin Invest. 1999;103:931–943. doi: 10.1172/JCI6609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Q, Xiong ZG, Man HY, Ackerley CA, Braunton J, Lu WY, Becker LE, MacDonald JF, Wang YT. Recruitment of functional GABAA receptors to postsynaptic domains by insulin. Nature. 1997;388:686–690. doi: 10.1038/41792. [DOI] [PubMed] [Google Scholar]
- White MF. IRS proteins and the common path to diabetes. Am J Physiol Endocrinol Metab. 2002;283:E413–E422. doi: 10.1152/ajpendo.00514.2001. [DOI] [PubMed] [Google Scholar]
- Yang F, Mak K, Vinters HV, Frautschy SA, Cole GM. Monoclonal antibody to the C-terminus of beta-amyloid. Neuroreport. 1994;5:2117–2120. doi: 10.1097/00001756-199410270-00032. [DOI] [PubMed] [Google Scholar]
- Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen PP, Kayed R, Glabe CG, Frautschy SA, Cole GM. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem. 2005;280:5892–5901. doi: 10.1074/jbc.M404751200. [DOI] [PubMed] [Google Scholar]
- Zhao W, Chen H, Xu H, Moore E, Meiri N, Quon MJ, Alkon DL. Brain insulin receptors and spatial memory. Correlated changes in gene expression, tyrosine phosphorylation, and signaling molecules in the hippocampus of water maze trained rats. J Biol Chem. 1999;274:34893–34902. doi: 10.1074/jbc.274.49.34893. [DOI] [PubMed] [Google Scholar]
- Zhao WQ, Alkon DL. Role of insulin and insulin receptor in learning and memory. Mol Cell Endocrinol. 2001;177:125–134. doi: 10.1016/s0303-7207(01)00455-5. [DOI] [PubMed] [Google Scholar]
- Zhao WQ, De Felice FG, Fernandez S, Chen H, Lambert MP, Quon MJ, Krafft GA, Klein WL. Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J. 2008;22:246–260. doi: 10.1096/fj.06-7703com. [DOI] [PubMed] [Google Scholar]
- Zhu X, Raina AK, Rottkamp CA, Aliev G, Perry G, Boux H, Smith MA. Activation and redistribution of c-Jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer's disease. J Neurochem. 2001;76:435–441. doi: 10.1046/j.1471-4159.2001.00046.x. [DOI] [PubMed] [Google Scholar]