

Abstract
Newly synthesized synaptic proteins and mitochondria are transported along lengthy neuronal processes to assist in the proper assembly of developing synapses and activity-dependent remodeling of mature synapses. Neuronal transport is mediated by motor proteins that associate with their cargoes via adaptors and travel along the cytoskeleton within neuronal processes. Our previous studies in developing hippocampal neurons revealed that syntabulin acts as a KIF5B motor adaptor and mediates anterograde transport of presynaptic cargoes and mitochondria, presynaptic assembly, and activity-induced plasticity. Here, using cultured superior cervical ganglion neurons combined with manipulation of syntabulin expression or interference with its interaction with KIF5B, we uncover a crucial role for syntabulin in the maintenance of presynaptic function. Syntabulin loss-of-function delayed the appearance of synaptic activity in developing neurons and impaired synaptic transmission in mature neurons, including reduced basal activity, accelerated synaptic depression under high-frequency firing, slowed recovery rates after synaptic vesicle depletion, and impaired presynaptic short-term plasticity. These defects correlated with reduced mitochondrial distribution along neuronal processes and were rescued by the application of ATP within presynaptic neurons. These results suggest that syntabulin supports the axonal transport of mitochondria and concomitant ATP production at presynaptic terminals. ATP supply from locally stationed mitochondria is in turn necessary for the efficient mobilization of synaptic vesicles into the readily releasable pool. These findings emphasize the critical role of KIF5B-syntabulin-mediated axonal transport in the maintenance of presynaptic function and regulation of synaptic plasticity.
Introduction
Synaptic terminals undergo activity-dependent maturation and remodeling, and these dynamic changes require the targeted delivery of synaptic components to axo-dendritic contact sites, a process that begins with transport of synaptic carrier vesicles along the secretory pathway (Horton and Ehlers, 2004). Mitochondria and presynaptic proteins are incorporated or attached to cargo vesicles at the soma and transported along microtubules to synaptic terminals (Nakata et al., 1998; Ahmari et al., 2000; Zhai et al., 2001). Microtubule-based motor proteins of the kinesin family are responsible for anterograde transport of mitochondria and presynaptic components to synaptic destinations (Tanaka et al., 1998; Martin et al., 1999; Ligon and Steward, 2000; Diefenbach et al., 2002; Stowers et al., 2002; Górska-Andrzejak et al., 2003; Cai et al., 2005; Guo et al., 2005; Glater et al., 2006; Pilling et al., 2006). KIF5B is the first identified kinesin-1 family motor for anterograde transport of mitochondria (Tanaka et al., 1998; Pilling et al., 2006). Our previous studies identified syntabulin as a KIF5B adaptor protein for transport of presynaptic cargo (Su et al., 2004) and functional depletion of syntabulin disrupts presynaptic assembly and activity-dependent plasticity in developing hippocampal neurons (Cai et al., 2007). Syntabulin also associates with mitochondria to mediate their KIF5B-dependent anterograde transport along neuronal processes (Cai et al., 2005). However, the role of the KIF5B-syntabulin transport machinery in the presynaptic function of mature neurons, in particular the maintenance of synaptic transmission and plasticity, remains unclear.
The primary function of presynaptic terminals is the activity-dependent release of neurotransmitters and subsequent recycling of their carrier synaptic vesicles, processes which critically depend on ATP and calcium. To accomplish this, neurons require mechanisms to localize mitochondria in the vicinity of synaptic terminals (Shepherd and Harris, 1998), where energy production and calcium homeostasis are in high demand (Hollenbeck and Saxton, 2005). Accordingly, the loss of mitochondria and ATP biogenesis from nerve terminals results in impaired synaptic transmission (Stowers et al., 2002; Guo et al., 2005; Verstreken et al., 2005; Kang et al., 2008). Basal and sustained synaptic transmission and presynaptic short term plasticity are also regulated by residual Ca2+, which enters and accumulates in the presynaptic terminal during trains of action potentials (Zucker and Regehr, 2002). Residual Ca2+ is sensed by Ca2+ binding proteins which, among other potential effectors, have been shown to mediate synaptic facilitation and augmentation via effects on Ca2+ channel gating (Mochida et al., 2008). Mitochondria are also associated with short-term synaptic plasticity due to an intrinsic Ca2+ buffering capacity (Tang and Zucker, 1997; Billups and Forsythe, 2002; Levy et al., 2003; Yang et al., 2003; Kang et al., 2008). However, it is not known how ongoing transport of mitochondria to mature synapses affects the steady-state maintenance of synaptic transmission, including efficient replenishment of the readily releasable pool (RRP) of synaptic vesicles, the generation of short term plasticity, and the relative balance of mitochondrial ATP biogenesis and Ca2+ homeostasis to the maintenance of synaptic function.
In the present study, we investigate the functional role of syntabulin and the KIF5B-syntabulin interaction in sympathetic SCG neurons, which form a well characterized cholinergic synapse in long-term culture (Mochida, 1995; Ma and Mochida, 2007). This system is an ideal cell model to address the function of syntabulin for several reasons. First, the axon of this neuron forms synaptic contacts with the soma of neighboring neurons after 1–2 weeks in culture which increases in synaptic strength with further maturation (Mochida et al., 1994a,b). This synaptic maturation can be monitored by recording basal and evoked EPSPs for up to 10 weeks or more. Second, the SCG neuron has a large cell body and nucleus, which allows the manipulation of gene expression and function in mature neurons via acute microinjection of cDNA, small interfering RNA (siRNA), dominant-negative transgenes, peptides, antibodies, and metabolites (Mochida et al., 2003, 2008; Baba et al., 2005; Krapivinsky et al., 2006; Ma and Mochida, 2007), an approach technically not feasible for cultured neurons from the CNS. Third, synaptic activity and short term plasticity, as it relates to the size and replenishment of functional synaptic vesicle pools, can be accurately monitored by recording EPSPs evoked by paired or repetitive action potentials in presynaptic neurons. With this approach, we uncovered a crucial role for syntabulin in the maintenance of presynaptic function in mature neurons, thus highlighting a molecular mechanism through which presynaptic function and plasticity is sustained by KIF5B-syntabulin-mediated mitochondrial transport in the nervous system.
Materials and Methods
Culture of SCG neurons.
Postnatal day 7 Wistar ST rats were decapitated under diethyl ether anesthesia according to the Guidelines of the Physiological Society of Japan. Isolated SCG neurons were maintained in culture as described previously (Mochida et al., 1994a; Mochida, 1995). In brief, SCGs were dissected, desheathed, and incubated with 0.5 mg/ml collagenase (Worthington Biochemical) in L-15 (Invitrogen) at 37°C for 10 min. After enzyme digestion, the semidissociated ganglion was triturated gently through a small pore glass pipette until a cloudy suspension was observed. After washing by low-speed centrifugation at 1300 rpm for 3 min, the collected cells were plated onto coverslips in plastic dishes (Corning; 35 mm diameter; approximately one ganglion per dish) containing a growth medium of 84% MEM (Invitrogen), 10% FCS (Invitrogen), 5% horse serum (Invitrogen), 1% penicillin/streptomycin (Invitrogen), and 25 ng/ml nerve growth factor (2.5 S; grade II; Alomone Labs). The cells were maintained up to 10 weeks at 37°C in a 95% air/5% CO2 humidified incubator; cell medium was changed twice per week.
Manipulation of syntabulin in SCG neurons.
To suppress syntabulin expression, the expression vector pRNAT-H1.1/neo (GenScript) was used to generate siRNA. A 21 nt sequence corresponding to amino acid residues 101–107 of syntabulin [syntabulin-targeted siRNA (STB-siRNA)] and cGFP marker were co-encoded in the vector (Su et al., 2004). Scrambled control siRNA (con-siRNA), not homologous to any sequence in GenBank, was used as a negative control. To rescue the RNA interference (RNAi) phenotype, a syntabulin mutant (STB*) was generated (Cai et al., 2007), in which a single nucleotide was altered within the sequence target of STB-siRNA without changing the amino acid sequence of syntabulin. To interfere with the function of syntabulin, the KIF5B-binding domain (KBD) of syntabulin was encoded in the EGFP-C1 vector (Millipore Bioscience Research Reagents).
The above expression vectors were dissolved in 150 mm KAc, 5 mm Mg2-ATP, and 10 mm HEPES, pH 7.3, and microinjected into the nuclei of SCG neurons through a microglass pipette as described previously (Baba et al., 2005; Krapivinsky et al., 2006). Fast Green FCF (5%) (Sigma-Aldrich) was included in the injection solution to confirm entry into the SCG neuron. Injections were performed at days 10, 19, 24, or 8 weeks in culture to study synapse coupling (Fig. 1), and 7–9 weeks in culture for other studies (Figs. 2–8). Cells were maintained at 37°C in a 95% air/5% CO2humidified incubator. Electrophysiological analysis was performed 4 d after siRNA injection or 2 d after KBD transfection. The injected neurons were identified by green fluorescent protein (GFP) expression via an inverted microscope (TE 300; Nikon) equipped with an epifluorescence unit.
Figure 1.

Syntabulin is required for synaptic maturation in SCG neurons. A, B, SCG neurons at 10, 17, and 24 d and 8 weeks in culture were transfected with STB-siRNA or con-siRNA (A), or SCG neurons at 12, 19, and 26 d and 8 weeks in culture were transfected with GFP-KBD or GFP (B). Four days after siRNA transfection or 2 d after GFP-KBD transfection, EPSPs were recorded in nontransfected neurons in response to action potentials elicited in neighboring paired transfected neurons. The percentage of synaptic coupling was calculated from randomly chosen 20 pairs in 4–9 different cultures (marked above bars) and expressed as mean ± SEM (*p < 0.05, **p < 0.01, ***p < 0.001; unpaired Student's t test).
Figure 2.

Syntabulin maintains basal synaptic transmission in mature neurons. A, Synaptic transmission was compared in long-term (7–9 weeks) cultured SCG neuron pairs, in which presynaptic neurons were transfected with STB-siRNA, cotransfected with STB-siRNA and STB*, or transfected with GFP-KBD. EPSPs were recorded 4 d after siRNA transfection or 2 d after GFP-KBD transfection in neighboring nontransfected neurons. EPSPs from one representative experiment are shown in the traces. B, Mean amplitudes of three EPSPs recorded at 0.1 Hz were averaged and expressed as bar graphs. The number of experiments is indicated above bars. *p < 0.001; unpaired Student's t test. amp, Amplitude.
Figure 3.
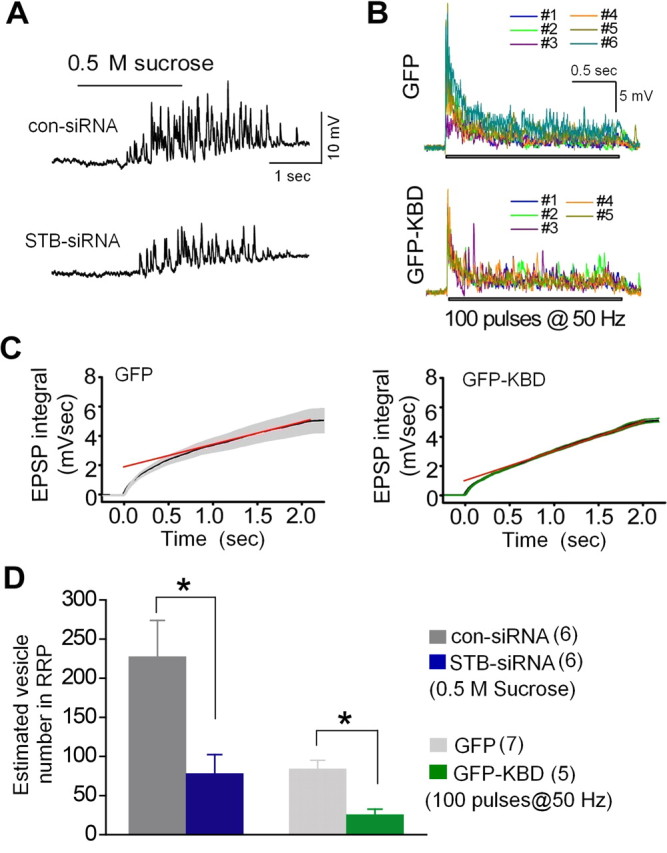
Syntabulin maintains the RRP size. A, Sucrose (0.5 m) was puff-applied with N2 pressure for 2 s to presynaptic neurons transfected with STB-siRNA or con-siRNA. Response traces to the sucrose application are shown from one representative experiment. B, EPSPs were elicited by a train of 100 action potentials at 50 Hz applied to presynaptic neurons expressing GFP or GFP-KBD. C, Averaged cumulative EPSP integral values with SEM (gray bars, GFP; green bars, GFP-KBD) were calculated from the EPSP traces shown in B. The red line represents a linear regression fit to data points from 1 to 2 s to estimate the integral value at time 0, indicating the RRP size. D, Number of synaptic vesicles in the RRP was estimated. The cumulative integral value of sucrose responses as shown in A and the value at time 0 shown in C were divided by the mean cumulative integral value of the quantal EPSP (0.019 mVsec). Number of experiments is indicated in parentheses. *p < 0.05; unpaired Student's t test.
Figure 4.

Syntabulin prevents synaptic depression during high-frequency firing. A–E, Normalized EPSP amplitudes (amp) recorded from paired SCG neurons in which presynaptic neurons were transfected with STB-siRNA or con-siRNA (A–C), cotransfected with STB-siRNA and STB* or con-siRNA and STB* (D), or transfected with GFP-KBD or GFP (E). Four days (A–D) or 2 d (E) after transfection, EPSP amplitudes were recorded at 0.1 Hz (A), 0.3 Hz (B), and 1 Hz (C–E), and averaged values are plotted (filled circles) with error bars (SEM). Mean values are fitted with a single exponential decay curve (red lines, STB-siRNA or GFP-KBD; yellow lines, con-siRNA or GFP) to calculate the decay time constant τ. F, The mean decay time constant, at 1 Hz recordings, calculated from individual recording of various transfection conditions (C–E). The number of experiments is indicated in parentheses. *p < 0.05; unpaired Student's t test.
Figure 5.

Syntabulin promotes recovery from synaptic vesicle depletion. Presynaptic neurons were transfected with STB-siRNA or con-siRNA (A, B), and GFP-KBD or GFP (C, D). A, C, EPSPs were recorded at 1 Hz. A 4 min train of action potentials at 5 Hz was applied, as indicated, to deplete releasable synaptic vesicles at presynaptic terminals. EPSP amplitudes (amp) were normalized to the averaged EPSP amplitude measured for 1 min before application of the train. Normalized values (circles) were plotted with the smoothed lines (green, gray, and blue) using an 8 point-moving average algorithm. EPSP amplitude recovered with two different rates: a first fast phase (red arrow) and then a slow phase (orange arrow), after the cessation of synaptic vesicle depletion. The slow recovery phase can be fit with a linear line (cyan and pink). B, D, Mean EPSP amplitudes shown in A, C are plotted with SEM at 1 min intervals. The number of experiments is indicated in parentheses. *p < 0.05; unpaired Student's t test.
Figure 6.

Syntabulin maintains presynaptic short-term plasticity. Presynaptic neurons were transfected with STB-siRNA or GFP-KBD. A–C, Changes in EPSP amplitude after an evoked transmitter release. EPSPs were elicited by two action potentials with various interstimulus intervals. The ratio of the peak amplitude of the second EPSP to the first EPSP is plotted against the paired-action potential interval (A, C), and the normalized paired-pulse ratio with an interval of 50 ms (upper) and 120 ms (lower) is shown in B. *p < 0.05; unpaired Student's t test. D, E, Changes in EPSP amplitude after high-frequency stimulations. Normalized EPSP amplitudes recorded in STB-siRNA (blue) or con-siRNA (black) synapses (D). Conditioning stimuli at 10 Hz for 10 s to generate augmentation and at 10 Hz for 60 s to generate PTP were applied at the indicated time. EPSP amplitudes shown in D at 5 s after conditioning stimuli at 10 Hz for 10 s (left), and at 10 s after conditioning stimuli at 10 Hz for 60 s (right) are divided by the averaged EPSP amplitude measured for 1 min before the conditioning pulse (E). The number of experiments is indicated in parentheses.
Figure 7.
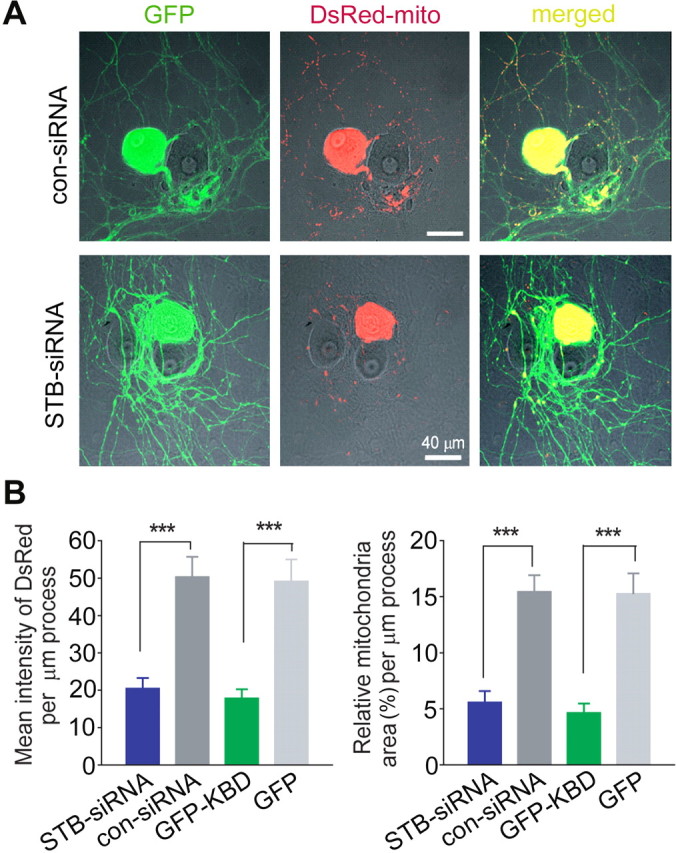
Syntabulin normalizes mitochondrial distribution along neuronal processes. A, Representative images of mitochondrial distribution in long-term cultured SCG neurons. A neuron (8 weeks in culture) was cotransfected with DsRed-mito and STB-siRNA or con-siRNA, followed by differential interference contrast and fluorescence images 6 d after transfection. GFP encoded by the siRNA expression vector serves as a marker for transfected neurons (left column). Scale bars, 40 μm. B, Mean intensity of DsRed-mito (left) and relative mitochondria area (right) per μm process. Note, reduced mitochondrial distribution in SCG processes expressing STB-siRNA or EGFP-KBD. Data were analyzed with DsRed images taken 6 d after transfection with STB-siRNA, or 2 d after transfection with GFP-KBD (from at least 12 cells and 5000 μm of processes).
Figure 8.
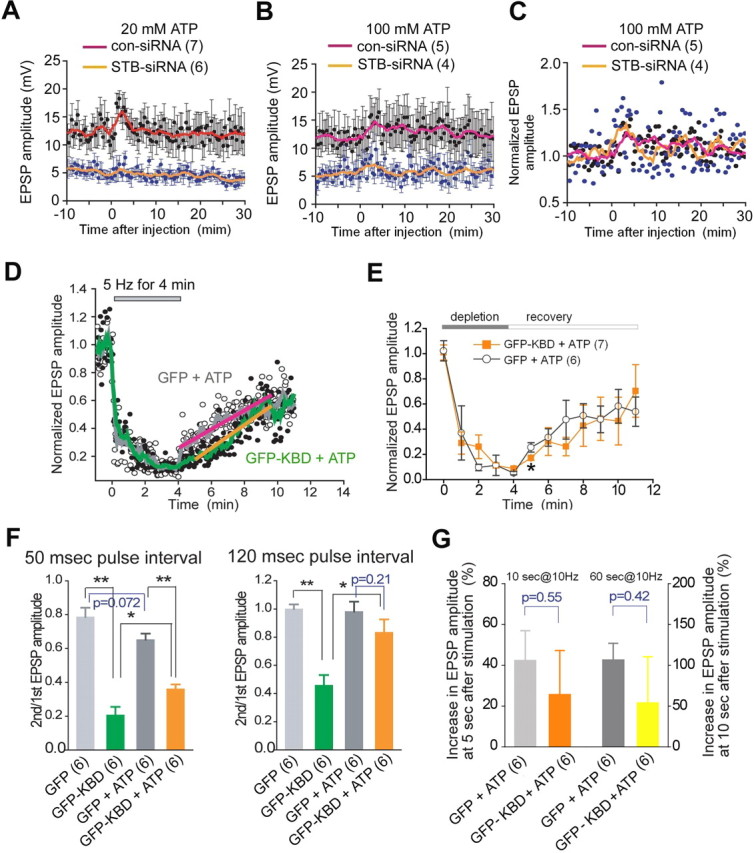
ATP partially rescues defective synaptic transmission in syntabulin-deficient synapses. A–C, Inability of ATP to restore basal transmitter release. Presynaptic neurons were transfected with STB-siRNA or con-siRNA. EPSP was recorded at 0.1 Hz. At time 0, ATP at 20 (A) or 100 mm (B) in the pipette was injected into presynaptic neurons with the outward current passage for 3 min. Averaged EPSP amplitudes were plotted against time with SEM (• ± bar, black for con-siRNA and blue for STB-siRNA). The smoothed value with a moving average algorithm was plotted with line. Normalized EPSP amplitudes are shown in C. D, E, ATP efficiently rescued the slow recovery from vesicle depletion in the synapses expressing GFP-KBD. ATP at 20 mm in the pipette was injected into the presynaptic neuron and EPSPs were recorded 5 min later with the same procedure shown in Figure 5. D, Normalized and averaged EPSP amplitudes are plotted (circles) with the smoothed values (green and gray lines). The slow recovery phase is fit with a linear line (orange line for GFP-KBD + ATP; pink line for GFP + ATP). E, Mean EPSP amplitudes shown in D are plotted with SEM at 1 min intervals. *p < 0.05; unpaired Student's t test. F, ATP rescues paired-EPSP decrease. Normalized paired-pulse ratio with the interval of 50 ms (left) and 120 ms (right) at 5 min after injection of ATP at 20 mm in the pipette. *p < 0.05, **p < 0.01, unpaired Student's t test. G, ATP rescues augmentation and PTP. EPSP amplitude at 5 s after conditioning stimuli at 10 Hz for 10 s (left two bars), and at 10 s after conditioning stimuli at 10 Hz for 60 s (right two bars) were divided by the averaged EPSP amplitude measured for 1 min before the conditioning. EPSP recording with the same procedure shown in Figure 6D started at 5 min after injection of ATP at 20 mm in the pipette. Unpaired Student's t test. The number of experiments is indicated in parentheses.
Synaptic transmission between SCG neurons.
EPSPs were recorded as described previously (Mochida et al., 2003; Baba et al., 2005). Conventional intracellular recordings were made from two neighboring neurons using microelectrodes filled with 1 m KAc (70–90 MΩ). EPSPs were recorded from a nontransfected neuron, whereas action potentials were generated in the transfected neuron by passing current through an intracellular recording electrode. Synaptic couples with subthreshold EPSPs that did not produce postsynaptic action potentials were studied. Neurons were superfused with a modified Krebs' solution consisting of 136 mm NaCl, 5.9 mm KCl, 2.5 mm CaCl2, 1.2 mm MgCl2, 11 mm glucose, and 3 mm Na-HEPES, pH 7.4. Electrophysiological data were collected and analyzed using software written by the late L. Tauc (Centre National de la Recherche Scientifique, Gif-sur-Yvette, France) (Mochida et al., 1994b), and analyzed with Origin 7.5 or 8 software (Microcal Software). To measure the RRP size, 0.5 m sucrose was puff-applied to presynaptic neurons every 3 min (Mochida et al., 1998; Baba et al., 2005). Sucrose responses (minimum 3 responses) were recorded with Clampex; integral values measured by Clampfit (pClamp 10; Molecular Devices) were averaged and normalized. Quantal integral values calculated from EPSP recordings in 0.2 mm Ca2+ and 5 mm Mg2+ were used to estimate the number of synaptic vesicles (SVs) in the RRP (Krapivinsky et al., 2006).
Imaging mitochondria.
DsRed-mito along with the KBD or the siRNA vector expressing GFP as a marker was microinjected into the nuclei of SCG neurons. Two days after the KBD transfection or 6 d after the siRNA transfection, images of living neurons were taken with a confocal laser microscope (Nikon EZ-C1) using a 40× objective lens. Images were acquired using the same settings below saturation at a resolution of 1024 × 1024 pixels (12 bit) and mitochondrial density was quantified by measuring DsRed-mito fluorescence along a major process with the MetaMorph software. To quantify the change of mitochondrial distribution and density, we first set the image threshold by subtracting the averaged background from neighboring regions of untransfected cells under the same image. To avoid under-estimation of mitochondrial density due to overlaid mitochondria in a neuronal process, the mean intensity and area of DsRed-mito per 1 μm length of process from a total process length >5000 μm and 12–20 cells were measured (Ruthel and Hollenbeck, 2003; Miller and Sheetz, 2004; Cai et al., 2005). These data were calculated and scored automatically by the MetaMorph software based on the fluorescence intensity profile.
Statistics.
Experiments were performed with a minimum of 4 synaptically coupled SCG neuronal pairs. Data values with associated error are shown in the text and figures as the mean ± SEM. A two-tailed t test was applied to compare significant effects.
Results
Syntabulin is required for synaptic maturation in SCG neurons
Our previous study showed that syntabulin loss-of-function impaired synaptic formation in developing hippocampal neurons (Cai et al., 2007). To address whether syntabulin is also required for synaptic maturation of SCG neurons, we suppressed syntabulin expression in presynaptic neurons with syntabulin-targeted siRNA (STB-siRNA), which was rigorously and extensively tested for its specificity and efficiency in our previous studies (Su et al., 2004; Cai et al., 2005, 2007). A con-siRNA that was not homologous to any sequence in GenBank was used as a control. We previously reported that synaptic coupling in SCG neurons, detected by recording EPSPs, was observed after 1 week in culture and increased up to 2 weeks (Mochida et al., 1994b). To determine whether syntabulin-mediated axonal transport is required to support synaptic activity in long-term cultured SCG neurons (2–8 weeks), we introduced siRNA-expressing vectors into presynaptic neurons by microinjection 4 d before electrophysiological recording. EPSPs were recorded from a nontransfected neuron in response to action potentials elicited in a neighboring transfected neuron. The percentage of synaptic coupling by neurons expressing STB-siRNA in ≤4 weeks culture was significantly lower than that for neurons expressing con-siRNA (p < 0.05, unpaired Student's t test) but increased up to 8 weeks (Fig. 1A), indicating a critical role for syntabulin in supporting synapse maturation in peripheral SCG neurons. Suppressing syntabulin expression delays synapse formation by ∼2 weeks when compared with nontransfected (Mochida et al., 1994b) or con-siRNA-transfected SCG neurons.
To address whether syntabulin supports synapse formation through its role as a KIF5B adaptor, we interrupted syntabulin-KIF5B interaction by expressing the KIF5B-binding domain (KBD) transgene in presynaptic neurons. KBD was derived from the syntabulin sequence (81–230) and significantly inhibited the KIF5B-syntabulin-mediated transport in hippocampal neurons by competitively blocking endogenous syntabulin binding to KIF5B (Cai et al., 2005, 2007). Expressing GFP-KBD significantly reduced the incidence of synaptic coupling in ≤4 weeks culture (Fig. 1B), in contrast to the neuron pairs transfected with GFP control. Consistent with the siRNA effect, this deficiency was rescued when neurons matured (up to 8 weeks). The delayed synaptic coupling in SCG neurons with syntabulin loss-of-function supports our proposal that KIF5B-syntabulin-mediated axonal transport is required for proper synapse maturation not only in the CNS but also in sympathetic neurons.
Syntabulin maintains basal synaptic transmission in mature neurons
To explore the significance of the KIF5B-syntabulin (motor-adaptor) complex in neurotransmission at mature sympathetic synapses, we examined basal synaptic transmission in long-term (7–9 week) cultured SCG neurons following expression of STB-siRNA or GFP-KBD (Fig. 2A). The mean EPSP amplitude recorded from STB-siRNA synapses was significantly smaller than that from con-siRNA synapses (Fig. 2B). To rescue the RNAi phenotype, we applied a syntabulin mutant (STB*) by altering the nucleotide sequence targeted by STB-siRNA without changing the amino acid sequence of syntabulin. Expressing STB*, but not wild-type syntabulin, was resistant to STB-siRNA in both COS cells and hippocampal neurons (Cai et al., 2007). In synapses coexpressing STB* and STB-siRNA, the mean EPSP amplitude was similar to that in STB*/con-siRNA synapses or that in con-siRNA synapses (Fig. 2B). Thus, coexpressing siRNA-resistant STB* rescued the phenotype, further indicating an essential role for syntabulin in synaptic activity. Furthermore, in GFP-KBD synapses, mean EPSP amplitudes were significantly reduced in contrast to those in GFP synapses (Fig. 2B). A marked reduction in mean EPSP amplitudes in response to low-frequency stimulation when syntabulin expression was suppressed or the KIF5B-syntabulin interaction was disrupted highlights the physiological importance of KIF5B-syntabulin-mediated axonal transport in maintaining basal synaptic function of mature synapses.
A decreased synaptic release probability or smaller size of the RRP may contribute to reduced EPSP amplitudes (Katz, 1969; Johnson and Wernig, 1971; Zucker, 1973; Quastel, 1997; Schneggenburger et al., 2002). To compare RRP sizes between paired SCG neurons transfected with STB-siRNA and con-siRNA, we estimated the number of SVs in the RRP by applying 0.5 m sucrose to presynaptic terminals, followed by dividing the mean of the integral value of the sucrose response with the mean value of the quantal EPSP integral (Rosenmund and Stevens, 1996) (Fig. 3A,D). The estimated RRP size for STB-siRNA synapses is significantly smaller (34%, p < 0.05) than that for con-siRNA synapses. A significant discrepancy in estimating the RRP size based on measurements made via the application of hypertonic solution versus action potential trains has been reported (Moulder and Mennerick, 2005). Therefore, to further validate our findings, we alternatively applied a train of action potentials to estimate the RRP size from synapses expressing GFP-KBD, a protocol widely used in the field (Murthy and Stevens, 1998; Schneggenburger et al., 1999; Schikorski and Stevens, 2001; Inchauspe et al., 2007). One hundred action potentials at 50 Hz produced multipeak EPSPs, which depressed and then reached an apparent steady state over the course of 100 stimuli during the train (Fig. 3B). This depression typically represents RRP depletion, with the steady state representing ongoing replenishment of the depleted RRP (Schneggenburger et al., 1999; Hagler and Goda, 2001). Excluding the replenishment component, the cumulative EPSP integral was plotted and back-extrapolated to time 0 to determine the cumulative EPSP integral representing the RRP (Schneggenburger et al., 1999; Inchauspe et al., 2007) (Fig. 3C). To estimate the number of SVs in the RRP, we divided the cumulative EPSP integral by the mean of the quantal EPSP integral. GFP synapses had 84 ± 11 vesicles (n = 7) compared with 26 ± 7 vesicles (n = 5) for GFP-KBD synapses (p < 0.05) (Fig. 3D), reflecting a 69% reduction in the RRP size when KIF5B-syntabulin coupling was disrupted. This result corresponds well to the 66% reduction in the RRP size measured in presynaptic neurons when syntabulin expression was suppressed with RNAi, although the RRP size in control synapses measured by hypertonic solution was 2.7-fold larger (Fig. 3D). To estimate a release probability for GFP control and GFP-KBD synapses we calculated the ratio of SV number producing the first EPSP (shown in Fig. 2B) to that in the RRP. The estimated release probability for GFP synapses was 0.61 ± 0.08 (n = 7) and for GFP-KBD synapses was 1.06 ± 0.15 (n = 5). The release probability appears higher at GFP-KBD synapses because the RRP is already reduced by 69%. Thus, theoretically, at GFP control synapses an action potential can release 51 of 84 vesicles of the RRP, but at GFP-KBD synapses can release essentially all available vesicles (∼28). These results suggest that disruption of the syntabulin-KIF5B interaction (and presumably syntabulin RNAi knockdown) reduces the RRP size resulting in a more profound fractional depletion of vesicles from the RRP in response to action potentials, but importantly does not appear to alter the overall release kinetics of individual vesicles. Altogether, these results provide evidence that ongoing KIF5B-syntabulin-mediated axonal transport in mature synapses is critical to maintain basal synaptic transmission.
Syntabulin prevents synaptic depression during high-frequency firing
To further characterize the role of syntabulin in synaptic transmission, we monitored EPSP amplitudes under three different presynaptic firing frequencies 0.1, 0.3, and 1 Hz for 60 min after transfection of long-term (7–9 weeks) cultured SCG neurons. With the low-frequency (0.1 Hz) stimuli, EPSPs recorded from STB-siRNA synapses decreased at a similar rate to those in con-siRNA synapses (Fig. 4A). The decay time constant τ, fitted with single exponential curve, was 7.8 ± 0.6 min for STB-siRNA and 8.1 ± 0.8 min for con-siRNA. Similarly, EPSPs recorded at 0.3 Hz showed no significant difference in the decay time constant τ: 15.4 ± 2.9 min for STB-siRNA and 16.8 ± 2.1 min for con-siRNA (Fig. 4B). In contrast, EPSP amplitudes at 1 Hz were gradually reduced during the 60 min recording; the reduction rate in STB-siRNA synapses was significantly faster than that in con-siRNA synapses (11.9 ± 0.7 min vs 38.9 ± 4.9 min, respectively, p < 0.05) (Fig. 4C). Coexpressing STB* rescued the RNAi phenotype at 1 Hz (20.9 ± 1.3 min for STB-siRNA + STB*; 20.1 ± 1.7 min for con-siRNA + STB*) (Fig. 4D). Furthermore, we confirmed the phenotype by expressing KBD, followed by repetitive presynaptic firing at 1 Hz. Again, normalized EPSP amplitudes, although gradually reduced in GFP synapses (τ = 13.5 ± 0.5 min), decreased more rapidly in GFP-KBD synapses (τ = 5.5 ± 0.2 min, p < 0.05) (Fig. 4E). The decay time constant calculated from individual recording at 1 Hz was significantly shorter in either the synaptic pairs with RNAi-suppressed syntabulin or with disruption of KIF5B-syntabulin interaction (Fig. 4F). Altogether, the accelerated synaptic depression under high-frequency stimulation suggests that KIF5B-syntabulin-mediated axonal transport sustains high levels of synaptic excitability in mature sympathetic neurons.
Syntabulin promotes recovery from synaptic vesicle depletion and short-term plasticity
We next examined various properties of presynaptic vesicle pools in mature SCG neurons. Presynaptic activity-dependent reduction in EPSP amplitudes suggests that replenishment of the RRP with SVs might be impaired in synapses with functional depletion of syntabulin. To examine RRP replenishment in those synapses, we monitored the recovery rate of EPSP amplitudes after depleting all releasable SVs in presynaptic terminals (Fig. 5). Our previous study showed that in mature SCG neurons, the recovery consists of two phases, a fast and a slow recovery, corresponding to a rapid RRP refilling with SVs from the reserve pool (RP) and a gradual RRP refilling with SVs through endocytic pathways (Lu et al., 2009). In con-siRNA synapses, the rates of both fast and slow recovery were 55%/min and 3.9%/min, respectively. In contrast, in synapses with syntabulin depletion, the fast recovery phase disappeared and the slow recovery rate decreased to 2.4%/min (Fig. 5A,B). Consistently, in synapses expressing GFP-KBD, the slow recovery rate decreased to 2.9%/min. In contrast, EPSP amplitude recovered with a rapid rate of 80%/min followed by a slow rate of 9.2%/min in GFP synapses (Fig. 5C,D). The significantly slower recovery in syntabulin-dysfunctional synapses suggests that KIF5B-syntabulin-mediated axonal transport supports efficient RRP refilling after prolonged presynaptic activity.
To further confirm whether KIFB-syntabulin-dependent transport is required for refilling RRPs after evoked transmitter release, a paired-pulse protocol was applied. Our previous study revealed that synaptic responses induced by two consecutive action potentials showed depression of the second response [paired-pulse depression (PPD)] when interstimulus intervals (ISIs) were between 20 and 100 ms, whereas PPD was not observed when ISIs were in the range of 200–2000 ms (Lu et al., 2009). In the STB-siRNA synapses, the paired-pulse ratio (second EPSP/first EPSP) with ISIs from 80 to 500 ms was significantly smaller than that in con-siRNA synapses (Fig. 6A,B). Coexpressing STB* partially rescued the phenotype (Fig. 6B), further indicating a critical role for syntabulin in maintaining the efficient refilling of the RRP. Consistently, expressing GFP-KBD also enhanced PPD (Fig. 6C), suggesting that syntabulin loss-of-function decreased the RRP size after evoked transmitter release.
Rapid replenishment of the RRP is critical for short-term synaptic plasticity which is important for encoding information in the nervous system (Abbott and Regehr, 2004). Next, we examined two forms of presynaptic short-term plasticity, termed augmentation and post-tetanic potentiation (PTP) which last for seconds to minutes (Zucker and Regehr, 2002). Augmentation and PTP were monitored by measuring EPSP amplitudes before and after applying a 10 s conditioning train at 10 Hz (Stevens and Wesseling, 1998), and a 60 s train at 10 Hz (Magleby and Zengel, 1975). In syntabulin dysfunctional synapses, augmentation and PTP were almost abolished (Fig. 6D,E). Coexpressing STB* with STB-siRNA rescued the phenotype (Fig. 6E). Similarly, expressing GFP-KBD in presynaptic neurons inhibited both augmentation and PTP (Fig. 6E). These results indicate that presynaptic short-term plasticity was efficiently supported by KIF5B-syntabulin-mediated axonal transport.
Syntabulin normalizes mitochondrial distribution along neuronal processes
The KIF5B-syntabulin transport machinery drives anterograde movement of mitochondria along neuronal processes of hippocampal neurons and syntabulin loss-of-function reduces mitochondrial density in neuronal processes (Cai et al., 2005). Furthermore, the loss of mitochondria at neuromuscular junctions in the drp1 mutant Drosophila resulted in faster depression of synaptic responses during prolonged pulse train stimulation (Verstreken et al., 2005), a phenotype similar to that observed in syntabulin-defective synapses (Figs. 4, 5). To provide a mechanistic link for the observed phenotypes, we compared the DsRed-mito-labeled mitochondria in the processes of mature SCG neurons (Fig. 7). Expressing STB-siRNA for 6 d significantly reduced the mean fluorescence intensity of DsRed-mito in neuronal processes and relative mitochondrial area per μm length of process compared with that from con-siRNA-transfected neurons (Fig. 7B). Mitochondria predominantly remain in the soma and proximal processes (within 60 μm from the soma) on expression of STB-siRNA, but spread along processes (>120 μm from the soma) on con-siRNA expression, reflecting defective anterograde transport of mitochondria in syntabulin-deficient SCG neurons (Fig. 7A). Consistently, the striking alteration in mitochondrial distribution with syntabulin knockdown was recapitulated after the disruption of syntabulin-KIF5B interaction by GFP-KBD (Fig. 7B). These results suggest that KIF5B-syntabulin loss-of-function appears to impair mitochondrial anterograde transport resulting in a reduction in overall density within neuronal processes of long-term cultured mature SCG neurons, where axons typically form synaptic contacts on the soma of adjacent neurons (Mochida, 1994b).
ATP partially rescues defective synaptic transmission in syntabulin-deficient synapses
Our imaging analysis provides direct evidence that KIF5B-syntabulin-mediated anterograde transport is essential for proper density of axonal mitochondria in mature SCG neurons, thus highlighting the possibility that insufficient ATP supply at nerve terminals fails to support basal and sustained presynaptic activity. To test this possibility, we injected ATP into presynaptic neurons for 3 min (at 20 or 100 mm in the injection pipette) and recorded EPSP elicited by low-frequency stimuli. EPSP amplitude was increased slightly and then returned rapidly to the preinjection level (Fig. 8A–C). The transient increase in EPSP amplitude in STB-siRNA synapses (1.34- and 1.36-fold for 20 mm and 100 mm ATP, respectively) was not significantly greater than that from con-siRNA synapses (1.25- and 1.35-fold). The inability of ATP to restore basal transmitter release in syntabulin-suppressed synapses suggests impaired assembly of the release machinery, which is consistent with our previous findings that KIF5B-syntabulin transports presynaptic components to nerve terminals in hippocampal synapses (Cai et al., 2007). Since it is technically difficult to rescue basal synaptic transmission by bypassing the defective axonal transport of presynaptic proteins, this hypothesis remains untested in the current study.
We next asked whether exogenous ATP introduced into the cell via micropipette rescues affected recovery of the RRP after depletion in synapses with syntabulin loss-of-function. Interestingly, ATP rescued the slow phase of the recovery in GFP-KBD synapses. Injecting ATP (20 mm) accelerated the slow phase of the recovery rate to 8.3%/min (Fig. 8D), compared with 2.9%/min without ATP injection (Fig. 5C). This result suggests that syntabulin loss-of-function may reduce the ATP supply in presynaptic terminals, thus slowing SV mobilization into the RRP via a myosin motor-driven and ATP-dependent transport mechanism (Mochida et al., 1994a; Takagishi et al., 2005; Verstreken et al., 2005). To further explain this phenomenon for PPD, ATP was injected into presynaptic neurons expressing GFP-KBD. Five minutes later, paired-pulse ratios for ISI = 50 and 120 ms were significantly increased in contrast to those without ATP supply (Fig. 8F). These results indicate that the slower RRP replenishment rate in SCG neurons with impaired mitochondrial transport is efficiently rescued by exogenous ATP supplement.
Since the generation of augmentation and PTP is also supported by syntabulin function (Fig. 6), we next examined whether impaired short-term plasticity is attributed to reduced ATP production at presynaptic terminals. By supplying ATP to presynaptic neurons expressing GFP-KBD, we compared augmentation and PTP with those synapses without ATP injection (Fig. 6E). EPSP amplitudes after a conditioning stimulus for augmentation and PTP were greatly increased in the presence of ATP. The increases were not statistically different from that in control GFP-expressing synapses supplemented with ATP (Fig. 8G). These results further support our hypothesis that syntabulin dysfunction impairs axonal transport of mitochondria, leading to ATP depletion in presynaptic terminals. Thus, these data highlight the importance of ATP, produced from locally stationed mitochondria in the presynaptic terminal, for the efficient mobilization of SVs to the RRP and the maintainence of normal presynaptic function.
Discussion
Our study demonstrates that KIF5B-syntabulin-mediated axonal transport is critical not only for synaptic maturation (Fig. 1) but also for maintaining basal and sustained neurotransmitter release (Figs. 2–5) and short-term presynaptic plasticity (Fig. 6) in mature SCG neurons. Syntabulin loss-of-function accelerates synaptic depression under high-frequency firing, slows the recovery rate from SV depletion, and impairs presynaptic short-term plasticity. These defects correlate with reduced mitochondrial distribution along processes (Fig. 7) and can be efficiently rescued by applying ATP to presynaptic neurons (Fig. 8). The results suggest that syntabulin dysfunction results in impaired anterograde mitochondria transport in neuronal processes and subsequently an insufficient ATP supply in presynaptic terminals with adverse consequences for SV mobilization to the RRP. Together, these findings suggest a molecular mechanism linking the KIF5B motor adaptor syntabulin and the regulation and maintenance of synaptic vesicle recycling in developing and mature presynaptic terminals.
Molecular regulation of mitochondrial movement
The efficient control of mitochondrial transport is essential for neuronal development and synaptic function. Approximately one-third of axonal mitochondria are mobile at instantaneous velocities of 0.2–2.0 μm/s in mature neurons, suggesting that mitochondrial distribution is efficiently controlled by the axonal transport machinery (Ligon and Steward, 2000; Kang et al., 2008). Recent advances in identifying the motor-adaptor complexes (Cai et al., 2005; Glater et al., 2006) and docking machinery (Kang et al., 2008) specific for neuronal mitochondria provide molecular targets for the regulation of mitochondrial distribution along axonal and dendritic processes. In addition to the KIF5B-syntabulin complex, Miro serves as a Ca2+ sensor in regulation of motor-adaptor coupling, thus controlling mitochondrial mobility in response to elevated intracellular Ca2+ and synaptic activity (Saotome et al., 2008; Wang and Schwarz, 2009; MacAskill et al., 2009; for review, see Cai and Sheng, 2009). Molecular regulation of mitochondrial movement can also be achieved by altering dynamic mitochondrial fusion/fission events. Mutation of the mitochondrial protein Drp1, a dynamin-like GTPase, impaired axonal transport and synaptic targeting of mitochondria by interfering with the mitochondrial fission process. At Drosophila drp1 mutant neuromuscular junctions, mitochondria were largely absent from synapses (Verstreken et al., 2005). Our previous study using time-lapse imaging in live hippocampal neurons (Cai et al., 2005) demonstrated that syntabulin mediates anterograde transport of mitochondria along neuronal processes and syntabulin loss-of-function selectively impaired anterograde but not retrograde transport of mitochondria. Consistently, live cell imaging analysis in current study demonstrates that syntabulin loss-of-function reduces mitochondrial density in those neuronal processes that wrap a neighboring untransfected SCG neuron (Fig. 7), which might be attributed to impaired presynaptic function under intense stimulation (Figs. 4–6). Thus, KIF5B-syntabulin loss-of-function appears to impair mitochondrial anterograde transport resulting in a reduction in overall mitochondrial density within neuronal processes of SCG neurons.
Mitochondrial function at presynaptic terminals
Mitochondria also regulate Ca2+ homeostasis at presynaptic terminals (Billups and Forsythe, 2002; Levy et al., 2003; Jacobson and Duchen, 2004), thus affecting presynaptic short-term plasticity (Tang and Zucker, 1997; Billups and Forsythe, 2002; Zucker and Regehr, 2002; Levy et al., 2003; Yang et al., 2003; Jacobson and Duchen, 2004). In contrast to the temporal facilitation of synaptic transmission under reduced mitochondrial calcium buffering capacity in various neurons (Zucker and Regehr, 2002; Levy et al., 2003; Jonas, 2006; Kang et al., 2008), no facilitation was seen in EPSPs with repetitive action potentials (Figs. 3, 4, 6) or paired-action potentials (Fig. 6). In addition, exogenous ATP efficiently rescued impaired short-term plasticity, suggesting that lack of mitochondrial energy supply in nerve terminals may lead to defective synaptic transmission. Furthermore, supplying ATP accelerated recovery from depletion of readily releasable SVs in syntabulin-dysfunctional synapses (Fig. 8), while increasing calcium buffering by injecting EGTA into presynaptic SCG neurons failed to recover the depletion of SVs (data not shown). Although we cannot exclude the possibility that mitochondria may act as a calcium buffer to maintain calcium homeostasis in presynaptic terminals and thereby contribute to effects on short term plasticity, our data suggest that the prominent effect on the replenishment of the RRP size during intense synaptic stimulation in syntabulin-impaired neurons may reflect the role of mitochondria as a source of ATP and as a requirement to refill the RRP, rather than as a function of their calcium buffering. However, further studies will be necessary to clarify this issue since ATP is also required to maintain membrane ionic gradients, which may have secondary effects on calcium levels.
Mitochondrial ATP supply and synaptic vesicle mobilization
The impairment of synaptic transmission under intense stimulation (Figs. 4–6) was partially rescued by exogenous ATP (Fig. 8), which also accelerated the recovery of readily releasable SVs from depletion in syntabulin-dysfunctional synapses. These results suggest that mitochondrial ATP supply is required for SV mobilization in presynaptic terminals. At Drosophila drp1 mutant neuromuscular junctions, in which mitochondrial distribution and function were altered, basal synaptic transmission was only slightly affected, and mutant synapses failed to maintain normal neurotransmission during intense stimulation, likely due to defects in mobilizing SVs into the releasable pool. This phenotype was partially rescued by exogenous ATP (Verstreken et al., 2005). The mobilization of SVs within SCG nerve terminals is ATP-dependent and driven by myosin II motors along actin filaments (Mochida et al., 1994a; Takagishi et al., 2005). Efficient SV recruitment from the RP depends on mitochondrial ATP production (Jonas, 2006); insufficient ATP at nerve terminals might limit myosin-propelled mobilization of SVs from the RP (Verstreken et al., 2005). Thus, our study provides a mechanistic link between KIF5B-syntabulin-mediated mitochondrial transport, presynaptic energy homeostasis, and regulation of RRP replenishment during and after intense presynaptic activity in mature SCG neurons. Given that ATP is coreleased with ACh from sympathetic neurons (for review, see Burnstock, 2009), the possibility that insufficient ATP production affects ATP- and adenosine-based signaling remains an issue for future investigation.
Syntabulin and the rate of RRP replenishment
The rate of RRP replenishment can be investigated by measuring the recovery rate from depletion of the RRP under different conditions, after an evoked action potential (Fig. 6C), during a train of action potentials (Fig. 3C) and after the depletion of releasable SVs in nerve terminals by sustained firing of action potentials (Fig. 5C). In our experiments, the rate constants from these three independent approaches were comparable under control conditions but slower when syntabulin or its interaction with KIF5B was deficient. The RRP replenishment rate for the GFP-control, 200 ms, calculated from the values shown in Figure 3C (red line), corresponds well to the recovery rate of the second paired pulse response shown in Figure 6C, whereas the recovery is incomplete for GFP-KBD, in which the syntabulin-motor association is disrupted, with a calculated RRP replenishment rate of 300 ms. The estimated rates suggest that the RRP is replenished during repetitive stimuli at 1 Hz, whereas the EPSP size in syntabulin-deficient synapses declines more rapidly than the control (Fig. 4E), suggesting that syntabulin dysfunction causes a severe and rapid loss of synaptic transmission with repetitive firing. Furthermore, the calculated rate of slow RRP replenishment after depletion of SVs in presynaptic terminals is 4.8 vesicles/min for the GFP-control whereas it is 1 vesicle/min for the GFP-KBD (during EPSP monitoring at 1 Hz) (Fig. 5C), suggesting that loss of syntabulin interaction with KIF1B severely slows the rate of RRP replenishment via a depleted RP.
The mechanism underlying paired pulse depression (PPD) in central synapses is likely depletion of the SV pool available for release in response to the second stimulus (Liley and North, 1953; Betz, 1970; Zucker and Regehr, 2002; Sullivan, 2007). We recently showed that the second EPSP response to paired-action potentials evoked at <200 ms interval was partially reduced by blocking the endocytic pathway (Lu et al., 2009), suggesting that the RRP is partially refilled by recycled SVs after the first EPSPs. Thus, our present study suggests that the recovery of the RRP via a recycling SV pathway may be supported, at least partially, by ATP produced by mitochondria stationed in the vicinity of the presynaptic active zone.
Role of nonmitochondrial cargoes linked to KIF5B-syntabulin transport
We analyzed synaptic depression in neurons with altered syntabulin-KIF5B interactions and the estimated RRP replenishment rate suggests that loss of syntabulin interaction with KIF5B delayed the rate of RRP replenishment more than that of the values calculated from the experimental data. In addition, our results reveal that, in syntabulin dysfunctional synapses, basal synaptic transmission was reduced due to a reduction in the RRP size, in contrast to the Drosophila drp1 mutant neuromuscular junctions where it was slightly affected (Verstreken et al., 2005). Furthermore, the reduced basal synaptic transmission we observed was not rescued by ATP supply. These results suggest additional defects apart from ATP availability in the KIF5B-syntabulin deficient presynaptic terminal. The KIF5B-syntabulin complex mediates axonal transport not only of mitochondria, but also of presynaptic cargoes containing syntaxin and Bassoon in developing hippocampal neurons (Su et al., 2004; Cai et al., 2005, 2007), suggesting that nonmitochondrial cargoes might be responsible for SV priming and fusion in SCG neurons. To partially address this possibility, overexpression of KIF5B-syntabulin cargoes Bassoon or syntaxin-1 in syntabulin dysfunctional presynaptic neurons did not rescue the reduced RRP size or the rapid EPSP decline under repetitive stimuli at 1 Hz (data not shown). Thus, the possibility that nonmitochondrial cargoes may mediate some of the effects of syntabulin on synaptic maintenance will require future investigation with an alternative approach bypassing defective axonal transport, which is not technically feasible under current experimental conditions.
In summary, our study demonstrates that syntabulin-KIF5B-mediated transport is critical for two presynaptic phenomena: replenishment of the RRP and determination of the RRP size. This is the first demonstration of synaptic transmission defects due to a kinesin motor adaptor dysfunction in mature neurons. There is emerging evidence that defective axonal transport is an early and important event in several major human neurodegenerative diseases, including Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis (De Vos et al., 2008). Defective trafficking and dysfunction of axonal mitochondria are also implicated in the pathogenesis of axonal degeneration (Stamer et al., 2002; Pigino et al., 2003; Chan, 2006; Chevalier-Larsen and Holzbaur, 2006). However, adaptor-mediated axonal transport has been mostly explored in developing neurons. Our present study provides evidence that disruption of the KIF5B-syntabulin transport machinery impairs synaptic transmission not only in developing but also mature SCG neurons and emphasizes the tight linkage between ongoing axonal transport and synaptic assembly, function, and energy homeostasis. Future studies using genetic mouse models combined with physiology and live cell imaging of mitochondrial mobility will provide molecular and cellular details on how altered mitochondrial motility and distribution impact synaptic transmission and neuronal homeostasis.
Footnotes
This work was supported by grants-in-aid for Scientific Research B (S.M.) and the Intramural Research Program of the National Institute of Neurological Disorders and Stroke–National Institutes of Health (Z-H.S.).
The authors declare no competing financial interests.
References
- Abbott LF, Regehr WG. Synaptic computation. Nature. 2004;431:796–803. doi: 10.1038/nature03010. [DOI] [PubMed] [Google Scholar]
- Ahmari SE, Buchanan J, Smith SJ. Assembly of presynaptic active zones from cytoplasmic transport packets. Nat Neurosci. 2000;3:445–451. doi: 10.1038/74814. [DOI] [PubMed] [Google Scholar]
- Baba T, Sakisaka T, Mochida S, Takai Y. PKA-catalyzed phosphorylation of tomosyn and its implication in Ca2+-dependent exocytosis of neurotransmitter. J Cell Biol. 2005;170:1113–1125. doi: 10.1083/jcb.200504055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz WJ. Depression of transmitter release at the neuromuscular junction of the frog. J Physiol. 1970;206:629–644. doi: 10.1113/jphysiol.1970.sp009034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billups B, Forsythe ID. Presynaptic mitochondrial calcium sequestration influences transmission at mammalian central synapses. J Neurosci. 2002;22:5840–5847. doi: 10.1523/JNEUROSCI.22-14-05840.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnstock G. Purinergic cotransmission. Exp Physiol. 2009;94:20–24. doi: 10.1113/expphysiol.2008.043620. [DOI] [PubMed] [Google Scholar]
- Cai Q, Sheng ZH. Moving or stopping mitochondria: Miro as a traffic cop by sensing calcium. Neuron. 2009;61:493–496. doi: 10.1016/j.neuron.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q, Gerwin C, Sheng ZH. Syntabulin-mediated anterograde transport of mitochondria along neuronal processes. J Cell Biol. 2005;170:959–969. doi: 10.1083/jcb.200506042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q, Pan PY, Sheng ZH. Syntabulin-kinesin-1 family member 5B-mediated axonal transport contributes to activity-dependent presynaptic assembly. J Neurosci. 2007;27:7284–7296. doi: 10.1523/JNEUROSCI.0731-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125:1241–1252. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Chevalier-Larsen E, Holzbaur EL. Axonal transport and neurodegenerative disease. Biochim Biophys Acta. 2006;1762:1094–1108. doi: 10.1016/j.bbadis.2006.04.002. [DOI] [PubMed] [Google Scholar]
- De Vos KJ, Grierson AJ, Ackerley S, Miller CC. Role of Axonal Transport in Neurodegenerative Diseases. Annu Rev Neurosci. 2008;31:151–173. doi: 10.1146/annurev.neuro.31.061307.090711. [DOI] [PubMed] [Google Scholar]
- Diefenbach RJ, Diefenbach E, Douglas MW, Cunningham AL. The heavy chain of conventional kinesin interacts with the SNARE proteins SNAP25 and SNAP23. Biochemistry. 2002;41:14906–14915. doi: 10.1021/bi026417u. [DOI] [PubMed] [Google Scholar]
- Glater EE, Megeath LJ, Stowers RS, Schwarz TL. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J Cell Biol. 2006;173:545–557. doi: 10.1083/jcb.200601067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Górska-Andrzejak J, Stowers RS, Borycz J, Kostyleva R, Schwarz TL, Meinertzhagen IA. Mitochondria are redistributed in Drosophila photoreceptors lacking milton, a kinesin-associated protein. J Comp Neurol. 2003;463:372–388. doi: 10.1002/cne.10750. [DOI] [PubMed] [Google Scholar]
- Guo X, Macleod GT, Wellington A, Hu F, Panchumarthi S, Schoenfield M, Marin L, Charlton MP, Atwood HL, Zinsmaier KE. The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron. 2005;47:379–393. doi: 10.1016/j.neuron.2005.06.027. [DOI] [PubMed] [Google Scholar]
- Hagler DJ, Jr, Goda Y. Properties of synchronous and asynchronous release during pulse train depression in cultured hippocampal neurons. J Neurophysiol. 2001;85:2324–2334. doi: 10.1152/jn.2001.85.6.2324. [DOI] [PubMed] [Google Scholar]
- Hollenbeck PJ, Saxton WM. The axonal transport of mitochondria. J Cell Sci. 2005;118:5411–5419. doi: 10.1242/jcs.02745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton AC, Ehlers MD. Secretory trafficking in neuronal dendrites. Nat Cell Biol. 2004;6:585–591. doi: 10.1038/ncb0704-585. [DOI] [PubMed] [Google Scholar]
- Inchauspe CG, Forsythe ID, Uchitel OD. Changes in synaptic transmission properties due to the expression of N-type calcium channels at the calyx of Held synapse of mice lacking P/Q-type calcium channels. J Physiol. 2007;584:835–851. doi: 10.1113/jphysiol.2007.139683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson J, Duchen MR. Interplay between mitochondria and cellular calcium signalling. Mol Cell Biochem. 2004;256–257:209–218. doi: 10.1023/b:mcbi.0000009869.29827.df. [DOI] [PubMed] [Google Scholar]
- Johnson EW, Wernig A. The binomial nature of transmitter release at the crayfish neuromuscular junction. J Physiol. 1971;218:757–767. doi: 10.1113/jphysiol.1971.sp009642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas E. BCL-xL regulates synaptic plasticity. Mol Interv. 2006;6:208–222. doi: 10.1124/mi.6.4.7. [DOI] [PubMed] [Google Scholar]
- Kang JS, Tian JH, Pan PY, Zald P, Li C, Deng C, Sheng ZH. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell. 2008;132:137–148. doi: 10.1016/j.cell.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz B. The release of neural transmitter substances. Liverpool, UK: Liverpool UP; 1969. [Google Scholar]
- Krapivinsky G, Mochida S, Krapivinsky L, Cibulsky SM, Clapham DE. The TRPM7 ion channel functions in cholinergic synaptic vesicles and affects transmitter release. Neuron. 2006;52:485–496. doi: 10.1016/j.neuron.2006.09.033. [DOI] [PubMed] [Google Scholar]
- Levy M, Faas GC, Saggau P, Craigen WJ, Sweatt JD. Mitochondrial regulation of synaptic plasticity in the hippocampus. J Biol Chem. 2003;278:17727–17734. doi: 10.1074/jbc.M212878200. [DOI] [PubMed] [Google Scholar]
- Ligon LA, Steward O. Movement of mitochondria in the axons and dendrites of cultured hippocampal neurons. J Comp Neurol. 2000;427:340–350. doi: 10.1002/1096-9861(20001120)427:3<340::aid-cne2>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Liley AW, North KA. An electrical investigation of effects of repetitive stimulation on mammalian neuromuscular junction. J Neurophysiol. 1953;16:509–527. doi: 10.1152/jn.1953.16.5.509. [DOI] [PubMed] [Google Scholar]
- Lu W, Ma H, Sheng ZH, Mochida S. Dynamin and activity regulate synaptic vesicle recycling in sympathetic neurons. J Biol Chem. 2009;284:1930–1937. doi: 10.1074/jbc.M803691200. [DOI] [PubMed] [Google Scholar]
- Ma H, Mochida S. A cholinergic model synapse to elucidate protein function at presynaptic terminals. Neurosci Res. 2007;57:491–498. doi: 10.1016/j.neures.2006.12.015. [DOI] [PubMed] [Google Scholar]
- MacAskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A, Aspenstrom P, Attwell D, Kittler JT. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron. 2009;61:541–555. doi: 10.1016/j.neuron.2009.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magleby KL, Zengel JE. A dual effect of repetitive stimulation on post-tetanic potentiation of transmitter release at the frog neuromuscular junction. J Physiol. 1975;245:163–182. doi: 10.1113/jphysiol.1975.sp010839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M, Iyadurai SJ, Gassman A, Gindhart JG, Jr, Hays TS, Saxton WM. Cytoplasmic dynein, the dynactin complex, and kinesin are interdependent and essential for fast axonal transport. Mol Biol Cell. 1999;10:3717–3728. doi: 10.1091/mbc.10.11.3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KE, Sheetz MP. Axonal mitochondrial transport and potential are correlated. J Cell Sci. 2004;117:2791–2804. doi: 10.1242/jcs.01130. [DOI] [PubMed] [Google Scholar]
- Mochida S. Role of myosin in neurotransmitter release: functional studies at synapses formed in culture. J Physiol Paris. 1995;89:83–94. doi: 10.1016/0928-4257(96)80555-9. [DOI] [PubMed] [Google Scholar]
- Mochida S, Kobayashi H, Matsuda Y, Yuda Y, Muramoto K, Nonomura Y. Myosin II is involved in transmitter release at synapses formed between rat sympathetic neurons in culture. Neuron. 1994a;13:1131–1142. doi: 10.1016/0896-6273(94)90051-5. [DOI] [PubMed] [Google Scholar]
- Mochida S, Nonomura Y, Kobayashi H. Analysis of the mechanism for acetylcholine release at the synapse formed between rat sympathetic neurons in culture. Microsc Res Tech. 1994b;29:94–102. doi: 10.1002/jemt.1070290206. [DOI] [PubMed] [Google Scholar]
- Mochida S, Yokoyama CT, Kim DK, Itoh K, Catterall WA. Evidence for a voltage-dependent enhancement of neurotransmitter release mediated via the synaptic protein interaction site of N-type Ca2+ channels. Proc Natl Acad Sci U S A. 1998;95:14523–14528. doi: 10.1073/pnas.95.24.14523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochida S, Westenbroek RE, Yokoyama CT, Itoh K, Catterall WA. Subtype-selective reconstitution of synaptic transmission in sympathetic ganglion neurons by expression of exogenous calcium channels. Proc Natl Acad Sci U S A. 2003;100:2813–2818. doi: 10.1073/pnas.262787299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochida S, Few AP, Scheuer T, Catterall WA. Regulation of presynaptic Ca(V)2.1 channels by Ca2+ sensor proteins mediates short-term synaptic plasticity. Neuron. 2008;57:210–216. doi: 10.1016/j.neuron.2007.11.036. [DOI] [PubMed] [Google Scholar]
- Moulder KL, Mennerick S. Reluctant vesicles contribute to the total readily releasable pool in glutamatergic hippocampal neurons. J Neurosci. 2005;25:3842–3850. doi: 10.1523/JNEUROSCI.5231-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy VN, Stevens CF. Synaptic vesicles retain their identity through the endocytic cycle. Nature. 1998;392:497–501. doi: 10.1038/33152. [DOI] [PubMed] [Google Scholar]
- Nakata T, Terada S, Hirokawa N. Visualization of the dynamics of synaptic vesicle and plasma membrane proteins in living axons. J Cell Biol. 1998;140:659–674. doi: 10.1083/jcb.140.3.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigino G, Morfini G, Pelsman A, Mattson MP, Brady ST, Busciglio J. Alzheimer's presenilin 1 mutations impair kinesin-based axonal transport. J Neurosci. 2003;23:4499–4508. doi: 10.1523/JNEUROSCI.23-11-04499.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilling AD, Horiuchi D, Lively CM, Saxton WM. Kinesin-1 and Dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol Biol Cell. 2006;17:2057–2068. doi: 10.1091/mbc.E05-06-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quastel DM. The binomial model in fluctuation analysis of quantal neurotransmitter release. Biophys J. 1997;72:728–753. doi: 10.1016/s0006-3495(97)78709-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- Ruthel G, Hollenbeck PJ. Response of mitochondrial traffic to axon determination and differential branch growth. J Neurosci. 2003;23:8618–8624. doi: 10.1523/JNEUROSCI.23-24-08618.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, Rizzuto R, Hajnóczky G. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci U S A. 2008;105:20728–20733. doi: 10.1073/pnas.0808953105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schikorski T, Stevens CF. Morphological correlates of functionally defined synaptic vesicle populations. Nat Neurosci. 2001;4:391–395. doi: 10.1038/86042. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Meyer AC, Neher E. Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron. 1999;23:399–409. doi: 10.1016/s0896-6273(00)80789-8. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Sakaba T, Neher E. Vesicle pools and short-term synaptic depression: lessons from a large synapse. Trends Neurosci. 2002;25:206–212. doi: 10.1016/s0166-2236(02)02139-2. [DOI] [PubMed] [Google Scholar]
- Shepherd GM, Harris KM. Three-dimensional structure and composition of CA3-CA1 axons in rat hippocampal slices: implications for presynaptic connectivity and compartmentalization. J Neurosci. 1998;18:8300–8310. doi: 10.1523/JNEUROSCI.18-20-08300.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow EM. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol. 2002;156:1051–1063. doi: 10.1083/jcb.200108057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF, Wesseling JF. Activity-dependent modulation of the rate at which synaptic vesicles become available to undergo exocytosis. Neuron. 1998;21:415–424. doi: 10.1016/s0896-6273(00)80550-4. [DOI] [PubMed] [Google Scholar]
- Stowers RS, Megeath LJ, Górska-Andrzejak J, Meinertzhagen IA, Schwarz TL. Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron. 2002;36:1063–1077. doi: 10.1016/s0896-6273(02)01094-2. [DOI] [PubMed] [Google Scholar]
- Su Q, Cai Q, Gerwin C, Smith CL, Sheng ZH. Syntabulin is a microtubule-associated protein implicated in syntaxin transport in neurons. Nat Cell Biol. 2004;6:941–953. doi: 10.1038/ncb1169. [DOI] [PubMed] [Google Scholar]
- Sullivan JM. A simple depletion model of the readily releasable pool of synaptic vesicles cannot account for paired-pulse depression. J Neurophysiol. 2007;97:948–950. doi: 10.1152/jn.00554.2006. [DOI] [PubMed] [Google Scholar]
- Takagishi Y, Futaki S, Itoh K, Espreafico EM, Murakami N, Murata Y, Mochida S. Localization of myosin II and V isoforms in cultured rat sympathetic neurones and their potential involvement in presynaptic function. J Physiol. 2005;569:195–208. doi: 10.1113/jphysiol.2005.095943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Kanai Y, Okada Y, Nonaka S, Takeda S, Harada A, Hirokawa N. Targeted disruption of mouse conventional kinesin heavy chain, kif5B, results in abnormal perinuclear clustering of mitochondria. Cell. 1998;93:1147–1158. doi: 10.1016/s0092-8674(00)81459-2. [DOI] [PubMed] [Google Scholar]
- Tang Y, Zucker RS. Mitochondrial involvement in post-tetanic potentiation of synaptic transmission. Neuron. 1997;18:483–491. doi: 10.1016/s0896-6273(00)81248-9. [DOI] [PubMed] [Google Scholar]
- Verstreken P, Ly CV, Venken KJ, Koh TW, Zhou Y, Bellen HJ. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron. 2005;47:365–378. doi: 10.1016/j.neuron.2005.06.018. [DOI] [PubMed] [Google Scholar]
- Wang X, Schwarz TL. The mechanism of Ca2+-dependent regulation of kinesin-mediated mitochondrial motility. Cell. 2009;136:163–174. doi: 10.1016/j.cell.2008.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, He XP, Russell J, Lu B. Ca2+ influx-independent synaptic potentiation mediated by mitochondrial Na(+)-Ca2+ exchanger and protein kinase C. J Cell Biol. 2003;163:511–523. doi: 10.1083/jcb.200307027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai RG, Vardinon-Friedman H, Cases-Langhoff C, Becker B, Gundelfinger ED, Ziv NE, Garner CC. Assembling the presynaptic active zone: a characterization of an active one precursor vesicle. Neuron. 2001;29:131–143. doi: 10.1016/s0896-6273(01)00185-4. [DOI] [PubMed] [Google Scholar]
- Zucker RS. Changes in the statistics of transmitter release during facilitation. J Physiol. 1973;229:787–810. doi: 10.1113/jphysiol.1973.sp010167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]