

Abstract
The enzyme neuropathy target esterase (NTE) is present in neurons and deacylates the major membrane phospholipid, phosphatidylcholine (PtdCho). Mutation of the NTE gene or poisoning by neuropathic organophosphates—chemical inhibitors of NTE—causes distal degeneration of long spinal axons in humans. However, analogous neuropathological changes have not been reported in nestin-cre:NTEfl/fl mice with NTE-deficient neural tissue. Furthermore, altered PtdCho homeostasis has not been detected in NTE-deficient vertebrates. Here, we describe distal degeneration of the longest spinal axons in ∼3-week-old nestin-cre:NTEfl/fl mice and in adult C57BL/6J mice after acute dosing with a neuropathic organophosphate: in both groups early degenerative lesions were followed by swellings comprising accumulated axoplasmic material. In mice dosed acutely with organophosphate, maximal numbers of lesions, in the longest spinal sensory axon tract, were attained within days and were preceded by a transient rise in neural PtdCho. In nestin-cre:NTEfl/fl mice, sustained elevation of PtdCho over many months was accompanied by progressive degeneration and massive swelling of axons in sensory and motor spinal tracts and by increasing hindlimb dysfunction. Axonal lesion distribution closely resembled that in hereditary spastic paraplegia (HSP). The importance of defective membrane trafficking in HSP and the association of NTE with the endoplasmic reticulum—the starting point for the constitutive secretory pathway and transport of neuronal materials into axons—prompted investigation for a role of NTE in secretion. Cultured NTE-deficient neurons displayed modestly impaired secretion, consistent with neuronal viability and damage in vivo initially restricted to distal parts of the longest axons.
Introduction
Hereditary spastic paraplegias (HSP) comprise a genetically heterogenous group of neurological disorders characterized by degeneration of distal parts of long spinal axons (Deluca et al., 2004). Increasing evidence suggests that defects in membrane trafficking and/or axonal transport are important factors in the pathogenesis of these syndromes (Reid, 2003; Soderblom and Blackstone, 2006; Salinas et al., 2008). Recently, mutations within the region of the human gene encoding the catalytic domain of an enzyme, neuropathy target esterase (NTE), were shown to cause a recessively inherited spastic paraplegia (SPG39) (Rainier et al., 2008). NTE is present in neurons throughout the nervous system (Glynn et al., 1998) and is associated with the endoplasmic reticulum (ER) (Li et al., 2003; Akassoglou et al., 2004). NTE was identified originally in brain tissue homogenates as an enzyme activity (assayed with an artificial ester substrate) which was inhibited by neuropathic organophosphorus (OP) compounds (Johnson, 1969). Accidental ingestion of one particular OP, tri-ortho-cresylphosphate (TOCP), paralyzed thousands of people during the 20th century (Cavanagh, 1964). Like HSP, OP-induced neuropathy preferentially damages the distal ends of long axons. Furthermore, large adult vertebrate species (humans, cats, chickens) are far more susceptible to OP-induced neuropathy, and their brain tissue has a higher specific activity of NTE, than smaller animals (mice, rats) (Glynn, 2000).
The foregoing suggest a relationship between NTE activity and the distance that neuronal materials must be transported to reach the distal ends of long axons and that NTE may play a role in adult axonal maintenance by facilitating export of materials from the neuron to its axon. Suprisingly, however, long spinal axon degeneration has not been described in nestin-cre:NTEfl/fl mice with genetic deletion of NTE from neural tissue (Akassoglou et al., 2004). Vacuolation of the hippocampal neuropil—not a known site of neural damage in OP-induced neuropathy—has been reported in these mice, but spinal cord lesions have not (Akassoglou et al., 2004). NTE has several critical roles in mammalian development: its constitutive deletion in mice causes embryonic death attributable to failed formation both of the placenta and vasculature (Moser et al., 2004). In nestin-cre:NTEfl/fl mice, deletion of NTE is essentially restricted to neural tissue and occurs at embryonic day 11 (Akassoglou et al., 2004); thus, the hippocampal lesions might reflect abnormal neural development rather than lack of maintenance in the adult.
In cultured cells, NTE catalyzes OP-sensitive deacylation of phosphatidylcholine (PtdCho), the major phospholipid of eukaryotic membranes, producing water-soluble glycerophosphocholine and fatty acids (Zaccheo et al., 2004). However, the relationship of NTE activity to PtdCho levels in vivo is unclear at present. Thus, although increased PtdCho has been observed in the brains of NTE-deficient (swiss-cheese) Drosophila (Mühlig-Versen et al., 2005), a recent study failed to detect such a change after dosing chickens with TOCP (Hou et al., 2008). We undertook the present investigation to clarify the relationship of NTE-deficiency to spinal axon degeneration and neural PtdCho homeostasis.
Materials and Methods
Mice: breeding, dosing, behavioral, and histological analyses.
The generation of NTEfl/fl mice has been described previously (Akassoglou et al., 2004). Homozygous NTEfl/fl mice were crossed with nestin-cre mice (Jackson Laboratory). Subsequently, crosses between nestin-cre:NTEfl/fl mice generated the progeny analyzed in this study. Genotyping was by PCR: nestin-cre-:NTEfl/fl and nestin-cre+:NTEfl/fl are abbreviated to wild type (WT) and NTE-conditional knock out (cKO), respectively. Age- and sex-matched pairs of cKO and WT were compared in each experiment. To investigate pharmacological inactivation of NTE, adult male C57BL/6J mice were dosed daily (500 mg/kg; oral; dissolved in arachis oil) for 5 d with a neuropathic organophosphate, mono-ortho-cresyldiphenylphosphate (MOCDPP) (Lotti and Johnson, 1980); control mice received equal volumes of arachis oil only. Hindlimb dysfunction in all mice was evaluated in three tests by observing open-field locomotion, hindlimb response during suspension by the tail, and performance in traversing a 2 cm diameter dowel rod. Dosed mice were assessed daily and knock-out mice monthly. All procedures were performed under license from the UK Home Office in accordance with the Animals (Scientific Procedures) Act 1986. Mice were deeply anesthetized and perfused to fix tissue for semithin sections. Lesions were enumerated at ×100 magnification (full details in supplemental Methods, available at www.jneurosci.org as supplemental material). All histological data are representative of at least two mice.
Biochemical analyses.
PtdCho in mouse brain was determined essentially as described by Murai et al. (2004). In brief, rapidly dissected and frozen individual cerebral hemispheres were homogenized in 0.4 m HClO4, lipids were extracted into CHCl3 and PtdCho was hydrolyzed (90°C; 1 h) in 1 m HClO4/0.5% Triton X-100. After neutralization, choline and cholesterol were determined by enzyme-linked assay using Amplex kits (Invitrogen) (full details in supplemental Methods, available at www.jneurosci.org as supplemental material). Control values ± SE for 10 C57BL/6J mice were as follows: PtdCho, 19.5 ± 0.6; cholesterol, 11.8 ± 0.4 nmol/mg brain tissue.
To assay neuronal secretion of reelin and amyloid precursor protein (APP), cerebellar granule neurons (CGNs) were isolated from NTE–cKO and littermate WT mice at postnatal day 7 and plated at 0.5 × 106 cells per well in 24-well plates in neurobasal medium with B27 and 25 mm KCl. Medium was aspirated from CGN at 7 d in vitro; the cells were washed three times with PBS, then covered with 0.5 ml fresh medium and reincubated. For APP secretion assays, a potent, cell-permeable β-secretase inhibitor (Inhibitor IV; Calbiochem) was added (final concentration, 2 μm) 2–3 h before removing the original medium and added again (same concentration) to the fresh medium. After various times (up to 4 h), medium was again removed and saved, the cells again washed with PBS, lysed, and harvested in 0.5 ml RIPA buffer [50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 0.5% Triton X-100, 0.125% sodium deoxycholate, 1% protease inhibitor mixture (Pierce)]. Aliquots of cell lysates and conditioned medium were analyzed by SDS-PAGE (4–20% Lonza gels), transfer (16 h; 25 V) to nitrocellulose blots, probing with monoclonal antibodies to reelin (G10; Abcam; 1:1000) or APP (MAB348; Millipore Bioscience Research Reagents; 1:4000) followed by peroxidase-labeled anti-mouse IgG (Sigma; 1:1000), enhanced chemiluminescence reagents (Amersham), and finally densitometry (full details in supplemental Methods, available at www.jneurosci.org as supplemental material).
Results
NTE deficiency initially induces distal degeneration followed by swelling of the longest spinal axons
In 18–19-d-old (0.6 month) nestin-cre:NTEfl/fl (NTE–cKO) mice, we found axonal lesions in transverse sections of the gracile nucleus in the medulla oblongata (Fig. 1A,E): this region contains distal parts of the longest sensory spinal axons emanating from neurons in lumbo-sacral dorsal root ganglia. By 3 months of age, gracile nucleus axonal lesions were at least twice as numerous (Fig. 1B,E). We compared these neuropathological changes with those in adult C57BL/6J mice dosed acutely with a neuropathic OP, MOCDPP, a close homolog of TOCP. One week after starting a 5 d MOCDPP dosing regime, axonal lesions were found in similar numbers to those in 18–19 d NTE–cKO mice (Fig. 1C,E) but were not more numerous 6 weeks later (Fig. 1D,E).
Figure 1.
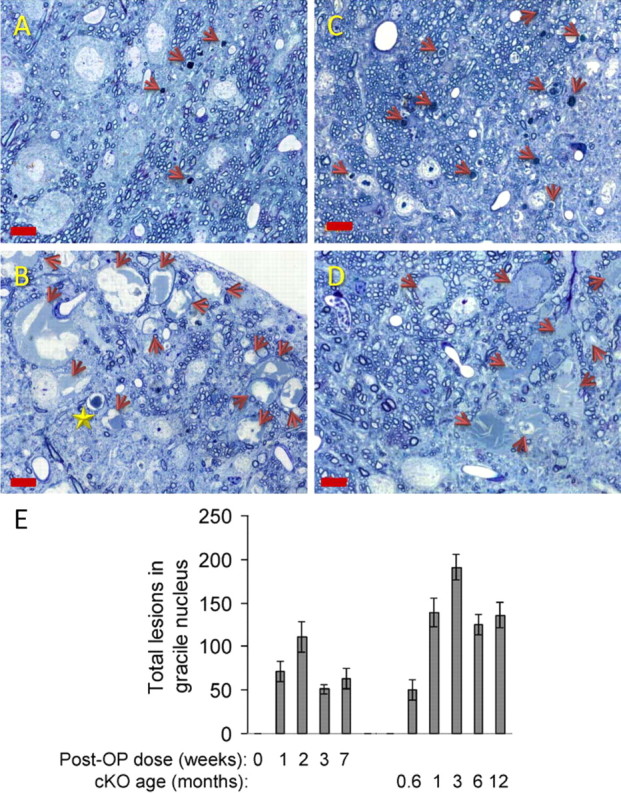
NTE deficiency rapidly induces distal damage to the longest axons in murine spinal sensory tracts. A–D, Axonal lesions in gracile nucleus of the medulla in nestin-cre:NTEfl/fl (NTE–cKO) mice at 18 d (A) and 12 weeks (B) and in adult C57BL/6J mice after MOCDPP-dosing regime at 1 (C) and 7 (D) weeks. Arrows indicate dense-body-type axonal lesions in A and C and swollen lesions in B and D. Star in B indicates dense-body. Scale bar, 10 μm. E, Gracile lesions enumerated at indicated times: data are the mean and range of determinations in two mice per time point (NTE–cKO) and mean ± SEM of determinations in three mice per time point (MOCDPP dosed) except 0 weeks, which represents the number of lesions (0, 0) in two vehicle-dosed mice. There were no significant differences in lesion numbers between 1–7 weeks post-OP, whereas, in NTE–cKO mice, lesion numbers increased significantly (p = 0.028; Student's t test) between 0.6 and 3 months.
In qualitative terms, the axonal lesions in young NTE–cKO mice strongly resembled those induced by MOCDPP dosing of adult C57BL/6J mice. In both groups, three morphologically distinct gracile nucleus lesions were observed: dark-staining axons with dense bodies comprising multilamellar figures or ovoids, translucent intra-axonal vacuoles, swollen axons with accumulations of vesicular and cytoskeletal elements (Fig. 2A). We classed both dense body and vacuolar types as degenerative lesions: these predominated initially but were accompanied progressively by swollen axons (Fig. 2B). The same progression was observed in sections of lumbar spinal cord from NTE–cKO mice. In ventral and lateral lumbar tracts, most of the initial lesions were of the degenerative type, but swollen axons eventually comprised 30–50% of the total (Fig. 2C). In older NTE–cKO mice, distal axons in the medullary gracile nucleus swelled to massive sizes (supplemental Fig. S2, available at www.jneurosci.org as supplemental material) and collections of swollen axons occupied large areas of the tract (Fig. 2D,E). In lumbar lateral tracts of NTE–cKO mice also, cross-sectional areas of swollen axons increased from typically 150–200 μm2 at 1 month (Fig. 2F) to 800–1000 μm2 at 14 months (Fig. 2H).
Figure 2.

Initial distal degeneration is followed by swelling in spinal axons of NTE-deficient mice. A, Three types of axonal lesion in medullary gracile nucleus (examples are from MOCDPP-dosed mice but were observed also in NTE–cKO mice). Scale bar, 2 μm. B, Percentage representation of axonal lesion types in medullary gracile nucleus (degenerating = dense-body and vacuolated types; see A). Data are mean ± SEM of three mice per time point (MOCDPP dosed) and mean and range of two mice per time point (NTE–cKO). C, Percentage representation of lesion types in lumbar spinal tracts (Fig. 3B) of NTE–cKO mice. Data are the mean and range of two mice per time point. D, E, Swollen axons in medullary gracile nucleus of 12-month-old NTE–cKO mouse. Scale bars: D, 10 μm; E, 5 μm. F–H, Swollen axons (arrows) and progressively increasing area of disordered structure in lumbar lateral tract of NTE–cKO mice at 1, 4, and 14 months. Scale bar, 10 μm.
Sustained NTE deficiency induces hindlimb dysfunction and damage to axons in corticospinal motor tracts
In NTE–cKO mice, clinical signs of hindlimb dysfunction progressed over a period of many months. The most obvious behavioral abnormality was the tendency to clench the digits of the hindlimbs: this was apparent both in the suspension reflex test (Fig. 3A) and in observations of open-field movement (supplemental Video, available at www.jneurosci.org as supplemental material). Inability to fully support the lower body in the beam-walking test became apparent by ∼4 months, and by 14 months, open-field movement was noticeably abnormal (although not grossly impaired) (supplemental Video, available at www.jneurosci.org as supplemental material). No clinical signs of hindlimb dysfunction were detected in C57BL/6J mice dosed acutely with MOCDPP at any time during the 7 week observation period.
Figure 3.

Sustained NTE deficiency induces hindlimb dysfunction and damage to axons in corticospinal motor tracts. A, Suspension reflex test: WT mouse (left) splays hindlimb digits on suspension, whereas those in NTE–cKO mouse (right) are clenched. B, Transverse section of mouse lumbar spinal cord showing areas enumerated for axonal lesions: D, dorsal; L, lateral; V, ventral. C, Enumeration of total axonal lesions in lumbar spinal tracts of NTE–cKO mice (mean and range of 2 per time point). D, Wallerian-like degenerating axon (arrow) in lumbar lateral tract of 14-month-old NTE–cKO mouse. Scale bar, 5 μm. E, F, NTE–cKO lumbar spinal cord showing distribution of degenerating (red) and swollen (green) axons at 1 (E) and 14 (F) months.
We examined lumbar spinal cord sections of NTE–cKO mice to detect distal lesions in axons in motor tracts which emanate from neurons in the brain cortex. At this level, we compared the incidence of lesions in lateral (major) and ventral (minor) corticospinal motor tracts with those in the dorsal sensory tract (Fig. 3B). Few lesions were observed in axons of the dorsal tract, which, at this level, are relatively close to their neuronal cell bodies (Fig. 3C). In contrast, lesions were numerous in the corticospinal motor tracts, particularly in the major lateral tracts (Fig. 3C,E,F). Degenerating type axonal lesions were present from early ages in both the lateral and ventral motor tracts (Figs. 2C, 3E,F) and often displayed a Wallerian-like morphology (Fig. 3D). In older NTE–cKO mice, swollen axons were increasingly present in lumbar tracts (Figs. 2C, 3E,F), and the lateral tracts in particular showed progressively larger areas of disordered structure (Fig. 2F–H).
NTE deficiency induces increased neural PtdCho levels and impairs neuronal secretion
We investigated neural PtdCho levels in adult C57BL/6J mice subjected to the same 5 d MOCDPP dosing regime described in Figure 1. One week after the start of dosing brain PtdCho levels were elevated but, a week later, had normalized and remained so for the following 5 weeks (Fig. 4A). We measured PtdCho in brain cerebral hemispheres, a neural area not affected morphologically in OP-induced neuropathy, to detect the primary biochemical consequence of NTE-deficiency rather than a secondary change resulting from tissue damage. This was supported by the lack of change in another lipid, cholesterol, in the same samples (Fig. 4A).
Figure 4.

NTE deficiency induces increased neural PtdCho levels and impairs neuronal secretion. A, B, PtdCho and cholesterol levels in brain cortex of vehicle-dosed (10 mice) and MOCDPP-dosed (4 per time point) adult C57BL/6J mice (A), and WT and NTE–cKO mice (1–3 months, 6 mice; 6–12 months, 12 mice per group, respectively) (B). Data are mean ± SEM. *Significantly different (p < 0.05; Student's t test) from vehicle-dosed control value (A) or respective WT mean value (B). C–F, Secretion from cultured CGN of reelin and APP. C, D, Top, Representative Western blots of paired cell lysates and conditioned media harvested from cultures of WT CGN and probed for reelin (C) or APP (D). Bottom, Data from multiple blots quantified to show secretion index (the ratio of extracellular to intracellular reelin or APP) as a function of incubation time. Data are means ± SEM of 4 (C) or 6–8 (D) individual culture wells from each of three separate WT CGN preparations. E, F, Representative Western blots of paired cell lysates and conditioned media harvested at 2 h from WT and littermate NTE–cKO CGN and probed for reelin (E) or APP (F).
In young (1–3 month) NTE–cKO mice, brain PtdCho levels were elevated, whereas cholesterol was unchanged (Fig. 4B). The magnitude of the PtdCho increase was similar to that detected in C57BL/6J mice 1 week after the first MOCDPP dose. However, in contrast to MOCDPP-dosed C57BL/6J mice, PtdCho remained elevated at the same levels in older (6–12 month) NTE–cKO mice (Fig. 4B).
Finally, we examined the possibility (see Introduction) that NTE facilitates export of materials from the neuronal cell body. This is consistent with NTE's localization to neuronal ER, the starting point for constitutive secretion and axonal transport. Both reelin (Lacor et al., 2000) and APP traverse the constitutive secretory pathway: APP is proteolytically cleaved at the cell surface, generating a large secreted fragment, sAPPα (Thinkaran and Koo, 2008). CGNs from NTE–cKO mice were morphologically indistinguishable from and equally viable in culture as CGN from WT littermates. We established incubation conditions with approximately linear rates of secretion from CGN of both reelin and sAPP (Fig. 4C,D). For both these proteins, the rate of secretion from NTE–cKO CGN was ∼80% of that from littermate WT CGN (Fig. 4E,F) [Reelin: WT = 100 ± 3.2%, NTE–cKO = 82.4 ± 4.1%; n = 20; p = 0.00148. sAPP: WT = 100 ± 3.1%, NTE–cKO = 79.7 ± 3.0%; n = 59; p = 4.936E-07. Means ± SE. Student's t test. For details see supplemental Methods, available at www.jneurosci.org as supplemental material]. These data indicate that the constitutive secretory pathway is moderately impaired in NTE-deficient neurons.
Discussion
We have shown that, in mice, NTE activity is required for neural PtdCho homeostasis and maintenance of adult axons. The transience and modest size of PtdCho elevation in brain cortex after acute OP dosing observed here may explain why others failed to detect a change after OP dosing in chickens (Hou et al., 2008). The small increase (15–20%) in PtdCho in NTE-deficient mouse brain is comparable with that in NTE-deficient Drosophila (Mühlig-Versen et al., 2005). The similar sized increase in OP-dosed mice and young and old NTE–cKO mice indicates that sustained NTE deficiency does not cause progressive PtdCho accumulation, but rapid establishment of a new elevated level which is maintained stably thereafter. Moreover, the rapid normalization of PtdCho levels after cessation of OP-dosing indicates that this PtdCho pool is relatively dynamic. The association of transiently increased neural PtdCho (in OP-dosed mice) with degeneration confined to the longest spinal axons and of sustained PtdCho elevation (in NTE–cKO mice) with increasing areas of axonal damage suggests a linkage between neural PtdCho homeostasis and axonal maintenance. In yeast, PtdCho homeostasis and constitutive secretion are linked via the phospholipid-binding protein, sec14p (Mousley et al., 2007).
Axonal maintenance depends on membrane trafficking and microtubule-based transport. For instance, HSP is caused by mutations in the genes encoding kinesin heavy chain KIF5A (SPG10), a motor protein for anterograde transport, or spastin (SPG4), a protein involved in microtubule turnover (Reid, 2003; Soderblom and Blackstone, 2006; Salinas et al., 2008). In cultured HeLa cells, the 68 kDa isoform of spastin regulates ER to Golgi secretory traffic (Connell et al., 2009). Interestingly, spastin has been shown to bind another HSP gene product, atlastin (SPG3A) (Sanderson et al., 2006), and atlastin-1, the major neural isoform, has a key role in ER and Golgi morphogenesis (Rismanchi et al., 2008).
Mice with a deletion mutation in spastin display swollen spinal axons, whereas neuronal soma appear normal (Tarrade et al., 2006). Preservation of neuronal cell bodies, even when there is extensive axonal degeneration, is a feature of both HSP (Soderblom and Blackstone, 2006) and OP-induced neuropathy (Cavanagh, 1964; Glynn, 2000). As in human HSP, distal parts of the longest spinal axons are the first to be damaged in both spastin- (Tarrade et al., 2006) and NTE-deficient mice. In the latter, we showed that the initial degenerative type of distal axonal lesion is succeeded by progressive swelling. The similar neuropathological changes in the gracile nucleus of young NTE–cKO mice and OP-dosed adult C57BL/6J mice strongly suggest that these lesions result from disruption in the maintenance of mature axons rather than abnormal neural development. With sustained NTE-deficiency over many months, massively swollen axons are present in motor and sensory spinal tracts. The relative distribution of axonal lesions in the motor corticospinal and sensory dorsal tracts in upper and lower spinal cord of NTE–cKO mice closely mirrors that reported in human HSP (Deluca et al., 2004). Despite the numerous forms of human HSP, relatively few animal models of these syndromes have been reported, and so NTE-deficient mice comprise a useful new model of an HSP.
Defective membrane trafficking is emerging as an important theme in the pathogenesis of HSP (Reid, 2003; Soderblom and Blackstone, 2006; Salinas et al., 2008). NTE localizes to neuronal ER, the starting point for constitutive secretion and axonal transport; thus, we hypothesized that these processes might be impaired by NTE deficiency. We measured secretion of reelin and APP from CGN because these proteins traverse the constitutive pathway (see above), and CGNs provide a source of relatively abundant and homogenous neurons which express NTE and can be used for a quantifiable and independent biochemical assay. We do not imply that reelin or APP per se are causally related to either axonal degeneration or HSP, or that CGN axons are particularly vulnerable in NTE deficiency. Using this assay, we found that in cultured NTE-deficient neurons, constitutive secretion is modestly impaired; thus, export of neuronal materials to the associated axon may be similarly affected. The minor degree of impairment observed is consistent with the apparent viability of NTE-deficient neurons and with the observation that, initially, damage in vivo is confined to distal regions of the longest spinal axons.
The neurological and histopathological signs and their gradual appearance over many months in NTE–cKO mice resemble those reported in wild-type mice dosed chronically with TOCP (Lapadula et al., 1985). This contrasts with larger animals (humans, chickens, cats) in which neuropathy appears within 1–2 weeks of a single TOCP dose (Cavanagh, 1964; Glynn, 2000). Moreover, although signs of hindlimb dysfunction were not detected in NTE–cKO mice until early adulthood (4–5 months), in humans with recessively inherited NTE-deficiency spastic paraplegia begins in childhood (Rainier et al., 2008). Similarly, although calcium-independent phospholipase A2 (iPLA2)-deficient mice do not display hindlimb dysfunction and swollen spinal axons until one year of age (Shinzawa et al., 2008), humans with iPLA2-deficiency have neurological signs in infancy and die before puberty (Nardocci et al., 1999). Thus, in two mouse models of deficient phospholipid deacylation, neuropathological changes occur much more slowly than in the respective human syndrome. This may be attributable to the relative lengths of human and mouse spinal axons and the greater need of the former for optimal levels of maintenance.
Footnotes
This work was funded by the Medical Research Council (D.J.R., Y.L., P.G.) and by National Institutes of Health Grant NS051282 (M.V.C.). We thank Pierluigi Nicotera for support; Judy MacWilliam, Maria Guerra Martin, Tim Smith, Lynda Wilkinson, Lynda Langford, and Helen Barbour for excellent assistance; David Dinsdale for advice on electron microscopy; and Toxicology Unit colleagues for helpful discussion.
References
- Akassoglou K, Malester B, Xu J, Tessarollo L, Rosenbluth J, Chao MV. Brain-specific deletion of neuropathy target esterase/swiss cheese results in neurodegeneration. Proc Natl Acad Sci U S A. 2004;101:5075–5080. doi: 10.1073/pnas.0401030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh JB. The significance of the “dying-back” process in experimental and human neurological disease. Int Rev Exp Pathol. 1964;3:219–267. [PubMed] [Google Scholar]
- Connell JW, Lindon C, Luzio JP, Reid E. Spastin couples microtubule severing to membrane traffic in completion of cytokinesis and secretion. Traffic. 2009;10:42–56. doi: 10.1111/j.1600-0854.2008.00847.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deluca GC, Ebers GC, Esiri MM. The extent of axonal loss in the long tracts in hereditary spastic paraplegia. Neuropathol Appl Neurobiol. 2004;30:576–584. doi: 10.1111/j.1365-2990.2004.00587.x. [DOI] [PubMed] [Google Scholar]
- Glynn P. Neural development and neurodegeneration: two faces of neuropathy target esterase. Prog Neurobiol. 2000;61:61–74. doi: 10.1016/s0301-0082(99)00043-x. [DOI] [PubMed] [Google Scholar]
- Glynn P, Holton JL, Nolan CC, Read DJ, Brown L, Hubbard A, Cavanagh JB. Neuropathy target esterase: immunolocalisation to neuronal cell bodies and axons. Neuroscience. 1998;83:295–302. doi: 10.1016/s0306-4522(97)00388-6. [DOI] [PubMed] [Google Scholar]
- Hou WY, Long DX, Wang HP, Wang Q, Wu YJ. The homeostasis of phosphatidylcholine and lysophosphatidylcholine was not disrupted during tri-o-cresyl phosphate-induced delayed neurotoxicity in hens. Toxicology. 2008;252:56–63. doi: 10.1016/j.tox.2008.07.061. [DOI] [PubMed] [Google Scholar]
- Johnson MK. The delayed neurotoxic action of some organophosphorus compounds: identification of the phosphorylation site as an esterase. Biochem J. 1969;114:711–717. doi: 10.1042/bj1140711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Grayson DR, Auta J, Sugaya I, Costa E, Guidotti A. Reelin secretion from glutamatergic neurons in culture is independent from neurotransmitter regulation. Proc Natl Acad Sci U S A. 2000;97:3556–3561. doi: 10.1073/pnas.050589597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapadula DM, Patton SE, Campbell GA, Abou-Donia MB. Characterisation of delayed neurotoxicity in the mouse following chronic oral administration of tri-o-cresyl phosphate. Toxicol Appl Pharmacol. 1985;79:83–90. doi: 10.1016/0041-008x(85)90370-9. [DOI] [PubMed] [Google Scholar]
- Li Y, Dinsdale D, Glynn P. Protein domains, catalytic activity and subcellular distribution of neuropathy target esterase in mammalian cells. J Biol Chem. 2003;278:8820–8825. doi: 10.1074/jbc.M210743200. [DOI] [PubMed] [Google Scholar]
- Lotti M, Johnson MK. Reapeated small doses of a neurotoxic organophosphate: monitoring of neurotoxic esterase in brain and spinal cord. Arch Toxicol. 1980;45:263–271. doi: 10.1007/BF00293807. [DOI] [PubMed] [Google Scholar]
- Moser M, Li Y, Vaupel K, Kretzschmar D, Kluge R, Glynn P, Buettner R. Placental failure and impaired vasculogenesis result in embryonic lethality for neuropathy target esterase-deficient mice. Mol Cell Biol. 2004;24:1667–1679. doi: 10.1128/MCB.24.4.1667-1679.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousley CJ, Tyeryar KR, Vincent-Pope P, Bankaitis VA. The sec14-superfamily and the regulatory interface between phospholipid metabolism and membrane trafficking. Biochim Biophys Acta. 2007;1771:727–736. doi: 10.1016/j.bbalip.2007.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mühlig-Versen M, da Cruz AB, Tschäpe JA, Moser M, Büttner R, Athenstaedt K, Glynn P, Kretzschmar D. Loss of swiss cheese/neuropathy target esterase causes disruption of phosphatidylcholine homeostasis and neuronal and glial death in adult Drosophila. J Neurosci. 2005;25:2865–2873. doi: 10.1523/JNEUROSCI.5097-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai S, Saito H, Shirato R, Tamura H, Yamada A, Kato H. An improved method for assaying phosphatidylcholine in mouse tissue. J Pharmacol Toxicol Methods. 2004;50:223–229. doi: 10.1016/j.vascn.2004.06.001. [DOI] [PubMed] [Google Scholar]
- Nardocci N, Zorzi G, Farina L, Binelli S, Scaioli W, Ciano C, Verga L, Angelini L, Savoiardo M, Bugiani O. Infantile neuroaxonal dystrophy: clinical spectrum and diagnostic criteria. Neurology. 1999;52:1472–1478. doi: 10.1212/wnl.52.7.1472. [DOI] [PubMed] [Google Scholar]
- Rainier S, Bui M, Mark E, Thomas D, Tokarz D, Ming L, Delaney C, Richardson RJ, Albers JW, Matsunami N, Stevens J, Coon H, Leppert M, Fink JK. Neuropathy target esterase mutations cause motor neuron disease. Am J Hum Genet. 2008;82:780–785. doi: 10.1016/j.ajhg.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid E. Science in motion: common molecular pathological themes emerge in hereditary spastic paraplegias. J Med Genet. 2003;40:81–86. doi: 10.1136/jmg.40.2.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rismanchi N, Soderblom C, Stadler J, Zhu PP, Blackstone C. Atlastin GTPases are required for Golgi apparatus and ER morphogenesis. Hum Mol Genet. 2008;17:1591–1604. doi: 10.1093/hmg/ddn046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinas S, Proukakis C, Crosby A, Warner TT. Hereditary spastic paraplegia: clinical features and pathogenic mechanisms. Lancet Neurol. 2008;7:1127–1138. doi: 10.1016/S1474-4422(08)70258-8. [DOI] [PubMed] [Google Scholar]
- Sanderson CM, Connell JW, Edwards TL, Bright NA, Duley S, Thompson A, Luzio JP, Reid E. Spastin and atlastin, two proteins mutated in autosomal-dominant hereditary spastic paraplegia, are binding partners. Hum Mol Genet. 2006;15:307–318. doi: 10.1093/hmg/ddi447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinzawa K, Sumi H, Ikawa M, Matsuoka Y, Okabe M, Sakoda S, Tsujimoto Y. Neuroaxonal dystrophy caused by Group VIA phospholipase A2 deficiency in mice: a model of human neurodegenerative disease. J Neurosci. 2008;28:2212–2220. doi: 10.1523/JNEUROSCI.4354-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderblom C, Blackstone C. Traffic accidents: molecular genetic insights into the pathogenesis of the hereditary spastic paraplegias. Pharmacol Ther. 2006;109:42–56. doi: 10.1016/j.pharmthera.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Tarrade A, Fassier C, Courageot S, Charvin D, Vitte J, Peris L, Thorel A, Mouisel E, Fonknechten N, Roblot N, Seilhean D, Diérich A, Hauw JJ, Melki J. A mutation of spastin is responsible for swellings and impairment in transport in a region of axon characterized by changes in microtubule composition. Hum Mol Genet. 2006;15:3544–3558. doi: 10.1093/hmg/ddl431. [DOI] [PubMed] [Google Scholar]
- Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing and function. J Biol Chem. 2008;283:29615–29619. doi: 10.1074/jbc.R800019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccheo O, Dinsdale D, Meacock PA, Glynn P. Neuropathy target esterase and its yeast homologue degrade phosphatidylcholine to glycerophosphocholine in living cells. J Biol Chem. 2004;279:24024–24033. doi: 10.1074/jbc.M400830200. [DOI] [PubMed] [Google Scholar]