

Abstract
Adenylosuccinate lyase (ADSL; also called “adenylosuccinase”) catalyzes two steps in the synthesis of purine nucleotides: (1) the conversion of succinylaminoimidazolecarboxamide ribotide into aminoimidazolecarboxamide ribotide and (2) the conversion of adenylosuccinate into adenosine monophosphate. ADSL deficiency, a recessively inherited disorder, causes variable—but most often severe—mental retardation, frequently accompanied by epilepsy and/or autism. It is characterized by the accumulation, in body fluids, of succinylaminoimidazolecarboxamide riboside and succinyladenosine, the dephosphorylated derivatives of the two substrates of the enzyme. Analysis of the ADSL gene of three unrelated patients with ADSL deficiency, in whom one of the ADSL alleles displayed a normal coding sequence, revealed a −49T→C mutation in the 5′ untranslated region of this allele. Measurements of the amount of mRNA transcribed from the latter allele showed that it was reduced to ∼33% of that transcribed from the alleles mutated in their coding sequence. Further investigations showed that the −49T→C mutation provokes a reduction to 25% of wild-type control of promoter function, as evaluated by luciferase activity and mRNA level in transfection experiments. The mutation also affects the binding of nuclear respiratory factor 2 (NRF-2), a known activator of transcription, as assessed by gel-shift studies. Our findings indicate that a mutation of a regulatory region of the ADSL gene might be an unusually frequent cause of ADSL deficiency, and they suggest a role for NRF-2 in the gene regulation of the purine biosynthetic pathway.
Introduction
Adenylosuccinate lyase (ADSL; also called “adenylosuccinase” [Enzyme Commission number 4.3.2.2]) catalyzes two steps in the synthesis of purine nucleotides: (1) the conversion of succinylaminoimidazolecarboxamide ribotide (SAICAR) into aminoimidazolecarboxamide ribotide (AICAR), the eighth step in the de novo synthesis of IMP, and (2) the conversion of adenylosuccinate (S-AMP) into adenosine monophosphate (AMP), the second step in the conversion of inosine monophosphate into AMP. ADSL deficiency (MIM 103050), first described by Jaeken and Van den Berghe (1984), is an autosomal recessive disorder characterized by the accumulation, in body fluids, of succinylaminoimidazolecarboxamide riboside (SAICAriboside) and succinyladenosine (S-Ado), the dephosphorylated derivatives of the two substrates of ADSL. To date, ∼60 patients with ADSL deficiency have been diagnosed. Most of them display severe mental retardation, often accompanied by epilepsy and/or autism (Van den Berghe and Jaeken 2001). The ADSL gene has been mapped to chromosome region 22q13.1-13.2 in humans (Van Keuren et al. 1987; Delattre et al. 1991; Fon et al. 1993). It is 23 kb long and was entirely sequenced by Kmoch et al. (2000) (GenBank accession number AF106656). The human coding sequence comprises 1,452 nucleotides, encoding a protein of 484 amino acids (European Molecular Biology Laboratory [EMBL] Database accession number X65867).
Molecular analysis of 31 affected families has revealed the existence of 30 different mutations (Adenylosuccinate Lyase Mutations Database Home Page). Most of them are found in compound heterozygous form. The large majority are missense mutations, among which a predominant mutation, R426H, is found in 13 families. A splicing error (Marie et al. 1999) and a nonsense mutation (Kmoch et al. 2000) have also been identified. The present article describes the identification, in three unrelated patients, of the first mutation found in the 5′ UTR of the ADSL gene, as well as its functional evaluation as a disease-causing gene.
Patients, Material, and Methods
Patients
The three patients were from unrelated families, and they presented with profound psychomotor delay and convulsions. Diagnosis was based on the detection of SAICAriboside and S-Ado by HPLC analysis of cerebrospinal fluid and/or urine. The clinical features of patient 1, an Italian American girl, have been published elsewhere (Nassogne et al. 2000). Patients 2 and 3 are from Norway and Australia, respectively. In all three patients, one of the ADSL alleles displayed a normal coding sequence. On the other allele, a missense mutation was found: R426H in patient 1, S447P in patient 2, and P318L (D. M. Cowley and I. McGown, personal communication) in patient 3. In accordance with findings in other patients (Race et al. 2000), measurements of the activity of ADSL in cultured skin fibroblasts showed 33%, 50%, and 28% of control activity (2.51±0.17 nmol/min per mg of protein) with S-AMP, and 33%, 46%, and 10% of control activity (1.95±0.24 nmol/min per mg of protein) with SAICAR, in patients 1, 2, and 3, respectively.
Cloning and Sequencing
Total RNA was extracted from two 75-cm2 flasks of cultured fibroblasts from the patients through use of TRIZOL reagent (Life-Technologies). M-MLV-Reverse transcriptase (Life-Technologies) was used to obtain the cDNA. PCR was performed at 64°C through use of Pwo polymerase (Roche) and two ADSL-specific primers: S1, starting 47 bp upstream of the ATG codon (−47 CCGCTCTTCCCTGGTCCAGTC −27); and AS1, starting 26 bp downstream of the stop codon (+1483 GATTTTCGTTTAATTCTCTTCCAACT +1458) (coding sequence positions are given with respect to EMBL sequence X65867).
Genomic DNA was extracted from one flask of cultured fibroblasts, using the QIAamp DNA kit from QIAgen. Two primers were used to amplify ∼400 bp of the 5′ flanking region of the gene: S2 (−398 GAGGGTCTCCCTGTGTTGCCCAG −376) and AS2 (+34 TCGGGCGAACCATGATCGCCTC +13)
PCR products were either cloned in the pBluescript plasmid (pBS; Stratagene) and sequenced using the Thermosequenase fluorescent labeled primer cycle sequencing kit (Amersham) with a 4000L automatic sequencer (Li-Cor), or they were directly sequenced using the CEQ 200 Dye Terminator cycle sequencing kit with a capillary automatic sequencer from Beckman Coulter.
Measurements of Relative Amounts of ADSL mRNA Transcribed from the Patients’ Two Alleles
Quantification of ADSL mRNA harboring the 5′ UTR mutation, as compared with ADSL mRNA carrying the patients’ missense mutations, was performed by PCR amplification of specific allele (PASA), as described by Bottema and Sommer (1998). For each of the three patients, two different pairs of oligonucleotides (numbered “1” and “2” below) were used at the following annealing temperatures: for patient 1, (1) WT-426 (+1300 AAGTAGGCATCAACCTGGATAC +1279) and P1-2, and (2) Mut-426 (+1300 AAGTAGGCATCAACCTGGATAT +1279) and P1-2, at 68°C; for patient 2, (1) WT-447 (+1361 GGCACGACCAGTGAAAGAAGA +1341) and P1-2, and (2) Mut-447 (+1361 GGCACGACCAGTGAAAGAAGG +1341) and P1-2, at 70°C; for patient 3, (1) WT-318 (+975 GGACAGATGCTGTCTGTAGCG +955) and P3, and (2) Mut-318 (+975 GGACAGATGCTGTCTGTAGCA +955) and P3, at 66°C. The sequence of P1-2, used in the analysis of patients 1 and 2, is +558 TTGAAGCGTGTCCGAGATGACC +579, and the sequence of P3, used in the analysis of patient 3, is +370 TTCTTAGAAATGCACTTGACCTGCT +394. Amplification of ribosomal protein S26 was used as an internal control, with the following primers (Vincent et al. 1993): +50 TCGTGCCAAAAAGGGCCGCG +69 (sense strand) and +376 GCTCCTTACATGGGCTTTGGTGG +354 (antisense strand).
Samples were withdrawn after 18, 21, 24, 27, and 30 cycles of PCR. Reaction products were resolved on 1.5% agarose gels. Pictures of the gels were scanned, and PCR products were quantified by densitometry with the NIH Image program.
Cell Transfection and Luciferase Assays
A series of DNA constructs were prepared in the pGL2-basic plasmid from Promega, containing the firefly luciferase gene (F-luc) in the absence of any promoter. Various lengths of the promoter region of the ADSL gene, including the 5′ UTR, were inserted in front of the firefly luciferase reporter gene. Site-directed mutagenesis was performed by PCR amplification. The 5′ flanking region cloned into pBS was amplified using Pwo DNA polymerase and two back-to-back primers, one of them containing a single modified base to generate one of three mutants, −49T→C, −46C→T, or −40T→C. The PCR-amplified plasmids were purified on 0.8% agarose gels and were ligated after phosphorylation of the 5′ ends with ATP and polynucleotide kinase. Clones were checked by sequencing, and the mutated sequences were subcloned into the pGL2-basic plasmid.
HEK-293 cells were seeded at 3×105 cells per well in six-well plates. After 24 h, through use of calcium phosphate precipitation, cells were cotransfected with 2 μg of the pGL2 plasmid, containing the F-luc constructs, and with 20 ng of the pRL-CMV reference plasmid, containing the Renilla luciferase gene (R-luc) under the control of the cytomegalovirus promoter. F-luc and R-luc activities were assayed 48 h later, through use of the dual-luciferase reporter assay system from Promega and a DLR Turner Design luminometer. Luciferase activity was expressed as the ratio of F-luc and R-luc values. Similar experiments were performed with NIH-3T3 cells that were seeded at 5×104 cells per well in 24-well plates and transfected with 2 μg of F-luc constructs and 20 ng of pRL-CMV.
Gel-Shift Experiments
Nuclear extracts from HEK-293 cells were prepared as described by Ausubel et al. (2000, pp. 12.1.1–12.1.3). HeLa cell nuclear extracts were purchased from Promega. Double strand oligonucleotides were labeled using T4 polynucleotide kinase and γ[32P]-ATP. Protein-DNA binding reactions were performed in a total volume of 20 μl containing 3 μg of nuclear extract, 1 μg poly(dI-dC), 10 mM Hepes (pH 7.6), 1 mM dithiothreitol, 5 mM MgCl2, 0.1 mM EDTA, 50 mM KCl, 10% glycerol (v/v), and 5×104 cpm of 32P-labeled oligonucleotide. Incubation was for 20 min on ice. For competition experiments, 20 or 50 ng of cold oligonucleotide were added at the same time. Protein-DNA complexes were resolved by electrophoresis on 6% native polyacrylamide gels for 3 h at 200 V. Gels were dried and subjected to autoradiography.
Results
Sequence and Mutation Analysis of the 5′ UTR of the Human ADSL Gene
The putative transcription initiation start point of the human ADSL gene was localized, 55 nucleotides upstream of the ATG codon, by Kmoch et al. (2000). When we analyzed the 5′ UTR, we found an additional G in position −51 (fig. 1), bringing its length to 56 bp. Analysis of 400 bp of the 5′ flanking region of the DNA of patient 1 revealed a T→C substitution that was 49 bp upstream of the ATG codon and thus was located in the 5′ UTR. As shown in figure 1, patient 1 is a T/C heterozygote at position −49, which contains T in the control DNA. The −49T→C mutation was also found in the heterozygous state in the mother's DNA. The same mutation was found in patient 2 from Norway and in patient 3 from Australia, as well as in her father. It was not found in a series of 121 control DNAs.
Figure 1.

Sequence analysis of patient 1 DNA (A) and control DNA (B).
Measurements of the Relative Amounts of mRNA Transcribed from the Two Alleles of the Patients
Since the −49T→C mutation is located too close to the 5′ end of mRNA, oligonucleotides that would be specific for the mutated allele (−49C) or the normal allele (−49T) could not be used. Three different pairs of oligonucleotides, one for each missense mutation—R426H, S447P, or P318L—were therefore designed, as described in the “Patients, Material, and Methods” section, so as to match perfectly with the normal sequence (WT-oligo) or the mutant sequence (Mut-oligo) of ADSL. In all cases, WT-oligo amplified the mRNA with mutation −49T→C.
Figure 2 shows the results obtained in the study of patient 1, who carries mutations −49T→C and R426H, with the oligonucleotides specifically amplifying her two alleles. As shown in panel A, distinctly less mRNA was transcribed from the patient’s −49T→C allele (amplified with WT-426) than from her R426H allele (amplified with Mut-426). Quantification after 27 and 30 cycles showed that the amount of mRNA transcribed from the −49T→C allele reached 30% and 35%, respectively, of that transcribed from the R426H allele. In contrast (fig. 2B), in an individual heterozygous for the R426H mutation, equal amounts of mRNA were transcribed from the WT and R426H alleles. The specificity of the two oligonucleotides at 68°C is shown in the bottom panels of figure 2: the cDNA from control cells (C) was amplified only with the WT-426 oligonucleotide, and the cDNA from a patient homozygous for mutation R426H (D) was amplified only with the Mut-426 oligonucleotide. Results similar to those depicted in panel A were obtained for patients 2 and 3, with WT- and Mut-447 and with WT- and Mut-318 oligonucleotides, respectively (not illustrated).
Figure 2.

PASA in patient 1. cDNAs from patient 1 (−49T→C/R426H) (A), a heterozygote (WT/R426H) (B), a control (WT/WT) (C), and a homozygote (R426H/R426H) (D) were amplified by PCR with one oligonucleotide specific for the WT allele (WT-426) and one oligonucleotide specific for the mutated allele (Mut-426), as described in the “Patients, Material, and Methods” section. PCR products were analyzed at several numbers of cell cycles, as indicated at the top of the figure. Ribosomal protein S26 was used as an internal control.
Mapping of the Promoter Region of the Human ADSL Gene
Analysis of the 5′ UTR sequence in the transcription factor database TRANSFAC showed that mutation −49T→C is located within the consensus sequence CNCTTCCGGT (core sequence underlined) of a potential binding site for nuclear respiratory factor 2 (NRF-2), a stimulatory regulator of transcription (Virbasius et al. 1993). To test the effect that a mutation of this potential binding site has on promoter function, we first analyzed the basal promoter activity of the 5′ flanking region of the ADSL gene.
As shown in figure 3, several potential binding sites for nuclear factors are found in the 5′ flanking region of the ADSL gene, including the NRF-2 site at position −52/−44. Of note, close to this site, at position −42/−38, a second potential binding site for NRF-2 is found with the essential core sequence CTTCC. To map promoter activity of the 5′ flanking region of the ADSL gene, four DNA constructs were used. Fragments starting at positions −1113, −398, −270, or −68 and ending at position −3, with respect to the ATG codon (fig. 3), were cloned into the pGL2-basic plasmid for transient expression in HEK-293 cells. Figure 4 shows promoter activities obtained with these constructs, measured as relative light units (firefly luciferase activity/Renilla luciferase activity). Maximal promoter activity was obtained with the longest (−1113 bp) fragment, which displayed 400-fold more luciferase activity than the background activity measured with the empty pGL2-basic vector. Promoter activity decreased to 66% and 43%, respectively, when the 5′ construct was shortened from −1113 bp to −398 bp and −270 bp. It was reduced to 1.7% when the construct was shortened to −68 bp, indicating that the minimal promoter is contained in the 202 bp deleted between the two last constructs. Strikingly, constructs in which promoter regions were inserted in reverse orientation (Rev-398 and Rev-270) showed even higher promoter activities than their equivalent in forward orientation.
Figure 3.

Sequence of the 5′ flanking region of the human ADSL gene. Bases are numbered by reference to the ATG codon. The bold arrow indicates the start point of the initiation of transcription. Thin arrows indicate the start points of the different constructs used for luciferase assay. Potential binding sites for nuclear factors are underlined. The core sequence of the tandem NRF-2 sites are double-underlined. Base −49T within the −52/−44 NRF-2 site is indicated in boldface.
Figure 4.

Promoter activity of 5′ flanking region of the human ADSL gene. Lengths of constructions are indicated on the left, and activities of firefly luciferase are given on the right as a percentage of the 1,113-bp construct.
Site-Directed Mutagenesis of the Human ADSL Promoter
To assess the functional significance of the −49T→C mutation in the 5′ UTR, we performed site-directed mutagenesis of the −398-bp construct. Figure 5 shows that the presence of the mutation provoked a reduction to 25% of promoter activity as compared with WT in HEK-293 cells. Similar results were obtained in NIH-373 cells, with a reduction to 31% of promoter activity (not illustrated). Mutation of another base located in the core sequence of the position −52/−44 NRF-2 site—namely, −46C→T—provoked an even more marked reduction, to 12%, of promoter activity in HEK-293 cells (fig. 5). In contrast, mutation −40T→C, located within the CTTCC core sequence of the second position −42/−38 potential NRF-2 binding site, had no significant effect. The −49T→C mutation did not affect the activity of the promoter in reverse orientation (results not shown). Northern blots showed that, in the cells transfected with the −49T→C mutated construct, the luciferase mRNA level was reduced three- to fourfold (results not shown).
Figure 5.

Effect of site-directed mutagenesis of the ADSL 5′ UTR. Location of mutations, within or outside of the NRF-2 binding site (underlined), are shown on top. Luciferase activities are expressed as percentages of the activity of the 398-bp construct without mutation.
Gel-Shift Experiments
To further evaluate the implication of the NRF-2 transcription factor in the expression of the ADSL gene, we performed gel-shift experiments. Synthetic oligonucleotides used in the present study, given in figure 6, were derived from the ADSL 5′ flanking region, without (WT-ADSL) or with (Mut-ADSL) the −49T→C mutation, as well as from the rat cytochrome c oxidase gene (RCO4-NRF-2). The latter oligonucleotide, which also contains a functional binding site for NRF-2 (underlined in the sequence shown in fig. 6) (Virbasius and Scarpulla 1991), serves as a positive control for NRF-2 in the gel-shift assays.
Figure 6.

Synthetic oligonucleotides used in gel-shift analysis of the NRF-2 binding site of ADSL. The binding site for NRF-2 is underlined.
Figure 7 shows that the labeled WT-ADSL oligonucleotide probe was able to form a specific complex with a nuclear extract of HEK-293 cells, which was completely displaced by 20–50 ng of the cold WT-ADSL oligonucleotide. In contrast, the presence of the −49T→C mutation abolished the capacity of the Mut-ADSL oligonucleotide to compete with the WT-ADSL probe, even in the presence of 50 ng of DNA. As expected, the RCO4-NRF-2 oligonucleotide was a very good competitor of the WT-ADSL oligonucleotide probe.
Figure 7.

Gel-shift analysis of NRF-2 binding site of ADSL. Binding of nuclear proteins to labeled WT oligonucleotide is shown in the absence (0 ng) or in the presence of 20 and 50 ng of WT-ADSL, −49T→C mutated ADSL (Mut-ADSL), and cytochrome c oxidase (RCO4-NRF-2) cold competitor oligonucleotides.
Upon incubation of the three oligonucleotides in the labeled form with HEK-293 nuclear extracts, WT-ADSL and RCO4-NRF-2 formed a complex of the same apparent size, whereas Mut-ADSL formed a complex of larger size (fig. 8). The same results were obtained with nuclear extracts from HeLa cells (data not shown).
Figure 8.
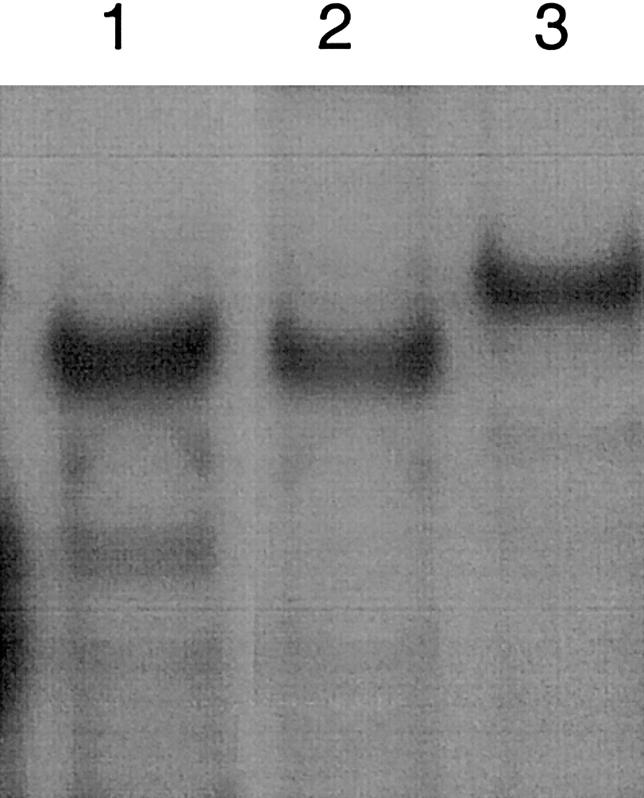
Comparison of protein-DNA complexes formed with WT and mutated ADSL, as well as with RCO4-NRF-2. Apparent sizes of protein-DNA complexes formed after incubation of nuclear extracts with labeled RCO4-NRF-2 (lane 1), WT-ADSL (lane 2), and −49T→C mutated ADSL (lane 3) oligonucleotides are shown.
Discussion
In the present study, we show that analysis of the ADSL gene of three unrelated patients with ADSL deficiency in whom one of the ADSL alleles displayed a normal coding sequence revealed a −49T→C mutation in the 5′ UTR of this allele. Further investigation showed that the mutation plays a causal role in their disease, most likely by affecting the binding of NRF-2, a known activator of transcription. The studies also suggest a role for NRF-2 in the gene regulation of the purine biosynthetic pathway.
The observations that the −49T→C mutation decreased the amount of mRNA transcribed from this allele, to ∼33% of that transcribed from the other allele (fig. 2A), and reduced luciferase activity to 25% of WT in transfection experiments (fig. 5) provide a strong indication that, in the patients' cells, the allele carrying this mutation produces a lower amount of the ADSL enzyme protein. Together with the ADSL lesion provoked by the mutation on the other allele, this would explain the ADSL deficiency.
The finding of reduced amounts of mRNA in both the studies of transcription and the transfection experiments performed with the 5′ UTR mutation does not allow us to differentiate between a transcriptional or translational effect of the mutation. Indeed, decreased translation of the mRNA might render it less stable. However, the finding that the −49T→C mutation is located within a potential binding site for the activating transcription factor NRF-2 is an argument in favor of a transcriptional effect. NRF-2 is a member of the Ets factor family (LaMarco et al. 1991; Thompson et al. 1991; Wasylyk et al. 1993) and is identical to the previously characterized GABP and E4TF1 (Virbasius et al. 1993; Suzuki et al. 1998). NRF-2 is known to activate the transcription of several nuclear and mitochondrial genes encoding mitochondrial respiratory proteins, such as cytochrome c oxidase subunits (Virbasius and Scarpula 1991; Seelan et al. 1996; Wong-Riley et al. 2000) and succinate dehydrogenase iron protein (Au and Scheffler 1998). Components of the mtDNA transcription and replication machinery, such as mtTFA, are also controlled by NRF-2 (Virbasius and Scarpulla 1994). Of note, in all of these promoters, the NRF-2 binding site is frequently located within the 5′ UTR, as in ADSL, or only a few nucleotides upstream of the transcription initiation site.
Our gel-shift experiments provide a good indication that NRF-2 is the factor that binds to the ADSL promoter. Indeed, the RCO4-NRF-2 oligonucleotide competes very well with the WT-ADSL oligonucleotide (fig. 7), indicating that they probably bind the same nuclear factor. The finding that the ADSL oligonucleotide with the −49T→C mutation is not able to compete with the wild-type sequence corroborates the functional importance of the mutation and indicates that the lack of NRF-2 binding may play a role in the decrease in transcription. The observation that the protein-DNA complex formed with WT-ADSL DNA comigrates with the complex formed with the RCO4-NRF-2 oligonucleotide (fig. 8), which was used to purify NRF-2 from HeLa cells (Virbasius et al. 1993), also indicates binding of NRF-2 to the ADSL promoter. The observation that, in binding assays with nuclear proteins (fig. 8), the oligonucleotide probe with the −49T→C mutation revealed a complex of higher molecular weight suggests that the mutation might also allow the binding of an unknown repressing factor.
It has been shown that NRF-2 binds as a dimer to its site, with one subunit containing the DNA binding domain and the other subunit modulating its affinity for DNA (Thompson et al. 1991; Virbasius et al. 1993). Moreover, the presence of two very proximal NRF-2 sites is a rather frequent situation in genes controlled by NRF-2 (Seelan et al. 1996; Wong-Riley et al. 2000). The presence of a tandem site allows the binding of NRF-2 as a tetramer, resulting in a stronger interaction between the factor and DNA. In the ADSL 5′ UTR, we found a second core sequence of the NRF-2 binding site, CTTCC, at position −42/−38, very close to the −52/−44 site. This tandem site could thus bind NRF-2 as a tetramer. However, the observation in the luciferase experiments that mutation −40T→C within this second core sequence did not significantly affect transcription (fig. 5) leads us to suppose that this second site is not implicated in the binding of the factor.
In the course of our studies, we also observed that the ADSL promoter displays bidirectional activity. Divergent transcription has been reported for other genes, like Surf and collagen genes (Lennard and Fried 1991; Fischer et al. 1993). Two closely linked genes of the de novo pathway of purine synthesis—namely, GPAT and AIRC—are also divergently transcribed in chicken (Gavalas et al. 1993; Gavalas and Zalkin 1995), human (Brayton et al. 1994), and rat (Iwahana et al. 1995). GPAT encodes glutamine 5′-phosphoribosyl-pyrophosphate amidotransferase, catalyzing step 1 of the de novo purine synthesis, and AIRC encodes a bifunctional enzyme, 5′-phosphoribosyl-aminoimidazole carboxylase/5′-phosphoribosyl 4-(N-succininocarboxamide)-5-aminoimidazole synthetase, catalyzing steps 6 and 7. Interestingly, another nuclear respiratory factor, NRF-1, implicated in the expression of several genes whose products function in the mitochondria (Evans and Scarpulla 1990; Chau et al. 1992), has been shown to play a key role in the bidirectional transcription of the human GPAT-AIRC genes (Chen et al. 1997). A potential NRF-1 binding site was also found in the rat GPAT-AIRC promoter (Iwahana et al. 1995) but not in the chicken promoter (Gavalas et al. 1993). Whereas the NRF-1 site of the GPAT-AIRC promoter was found to be implicated in the transcription in both orientations, this was not the case for the NRF-2 site in the ADSL promoter, since we found that mutations in the latter site did not affect the transcription in the reverse direction. Also of note, the identity of the neighbor gene of the ADSL gene is not known, even though the entire sequence of human chromosome 22 is available.
Taken together, our studies suggest that a mutation of a regulatory region of the ADSL gene might be an unusually frequent cause of ADSL deficiency. Indeed, regulatory mutations account for <1% of all mutation types (Human Gene Mutation Database), whereas a disease-causing mutation of the 5′ UTR has now been identified in 3 of 31 families with the ADSL enzyme defect. On the other hand, in vertebrates, little is known about gene regulation in purine nucleotide synthesis. Our observation that NRF-2 plays a role in the transcription of ADSL lends further support to the proposal that nuclear respiratory factors may play a role in the coordination of respiratory metabolism with biosynthetic and degradative pathways (Seelan et al. 1996; Scarpulla 1997).
Acknowledgments
We thank Dr. Eli Anne Kvittingen (Institute of Clinical Biochemistry, University of Oslo, Norway), as well as Dr. David M. Cowley and Mr. Ivan McGown (Mater Misericordiae Hospitals, Brisbane, Australia), for providing patient information and material. This work was supported by grants from the Fund for Medical Scientific Research (Belgium), the Actions de Recherche Concertées, and the Interuniversity Network for Basic Research of the Belgian Federal Service for Scientific, Technical and Cultural Affairs, as well as by European Community Concerted Action grant BMH4-CT98-3079. G.V.D.B. is Director of Research of the Belgian National Fund for Scientific Research.
Electronic-Database Information
Accession numbers and URLs for data presented herein are as follows:
- Adenylosuccinate Lyase Mutations Database Home Page, http://www.icp.ucl.ac.be/adsldb/
- European Molecular Biology Laboratory (EMBL) Nucleotide Sequence Database, http://www.ebi.ac.uk/embl/index.html (for the ADSL gene [accession number X65867])
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for the ADSL coding sequence [accession number AF106656])
- Human Gene Mutation Database, The, http://www.uwcm.ac.uk/uwcm/mg/hgmd0.html
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for ADSL [MIM 103050])
References
- Au HC, Scheffler IE (1998) Promoter analysis of the human succinate dehydrogenase iron-protein gene. Both nuclear respiratory factors NRF-1 and NRF-2 are required. Eur J Biochem 251:164–74 [DOI] [PubMed] [Google Scholar]
- Ausubel FM, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K (eds) (2000) Current protocols in molecular biology. John Wiley & Sons, New York [Google Scholar]
- Bottema CDK, Sommer SS (1998) Selective amplification of specific alleles. In: Cotton RGH, Edkins E, Forrest S (eds) Mutation detection: a practical approach. IRL Press, Oxford, pp 161–187 [Google Scholar]
- Brayton KA, Chen Z, Zhou G, Nagy PL, Gavalas A, Trent JM, Deaven LL, Dixon JE, Zalkin H (1994) Two genes for de novo purine nucleotide synthesis on human chromosome 4 are closely linked and divergently transcribed. J Biol Chem 269:5313–5321 [PubMed] [Google Scholar]
- Chau CA, Evans MJ, Scarpulla RC (1992) Nuclear respiratory factor 1 activation sites in genes encoding the γ-subunit of ATP synthase, eukaryotic initiation factor 2a, and tyrosine aminotransferase. J Biol Chem 267:6999–7006 [PubMed] [Google Scholar]
- Chen S, Nagy PL, Zalkin H (1997) Role of NRF-1 in bidirectional transcription of the human GPAT-AIRC purine biosynthesis locus. Nucleic Acids Res 25:1809–1816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delattre O, Azambuja CJ, Aurias A, Zucman J, Peter M, Zhang F, Hors-Cayla MC, Rouleau G, Thomas G (1991) Mapping of human chromosome 22 with a panel of somatic cell hybrids. Genomics 9:721–727 [DOI] [PubMed] [Google Scholar]
- Evans MJ, Scarpulla RC (1990) NRF-1: a trans-activator of nuclear-encoded respiratory genes in animal cells. Genes Dev 4:1023–1034 [DOI] [PubMed] [Google Scholar]
- Fischer G, Schmidt C, Opitz J, Cully Z, Kuhn K, Poschl E (1993) Identification of a novel sequence element in the common promoter region of human collagen type IV genes, involved in the regulation of divergent transcription. Biochem J 292:687–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fon EA, Demczuk S, Delattre O, Thomas G, Rouleau GA (1993) Mapping of human adenylosuccinate lyase (ADSL) gene to chromosome 22q13.1→q13.2. Cytogenet Cell Genet 64:201–203 [DOI] [PubMed] [Google Scholar]
- Gavalas A, Dixon JE, Brayton KA, Zalkin H (1993) Coexpression of two closely linked avian genes for purine nucleotide synthesis from a bidirectional promoter. Mol Cell Biol 13:4784–4792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavalas A, Zalkin H (1995) Analysis of the chicken GPAT/AIRC bidirectional promoter for de novo purine nucleotide synthesis. J Biol Chem 270:2403–2410 [DOI] [PubMed] [Google Scholar]
- Iwahana H, Honda S, Tsujisawa T, Takahashi Y, Adzuma K, Katashima R, Yamaoka T, Moritani M, Yoshimoto K, Itakura M (1995) Rat genomic structure of amidophosphoribosyltransferase, cDNA sequence of aminoimidazole ribonucleotide carboxylase, and cell cycle-dependent expression of these two physically linked genes. Biochim Biophys Acta 1261:369–380 [DOI] [PubMed] [Google Scholar]
- Jaeken J, Van den Berghe G (1984) An infantile autistic syndrome characterised by the presence of succinylpurines in body fluids. Lancet 2:1058–1061 [PubMed] [Google Scholar]
- Kmoch S, Hartmannová H, Stiburková B, Krijt J, Zikanova M, Sebesta I (2000) Human adenylosuccinate lyase (ADSL), cloning and characterization of full-length cDNA and its isoform, gene structure and molecular basis for ADSL deficiency in six patients. Hum Mol Genet 9:1501–1513 [DOI] [PubMed] [Google Scholar]
- LaMarco K, Thompson CC, Byers BP, Walton EM, McKnight SL (1991) Identification of Ets- and Notch-related subunits in GA binding protein. Science 253:789–792 [DOI] [PubMed] [Google Scholar]
- Lennard AC, Fried M (1991) The bidirectional promoter of the divergently transcribed mouse Surf-1 and Surf-2 genes. Mol Cell Biol 11:1281–1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie S, Cuppens H, Heuterspreute M, Jaspers M, Zambrano Tola E, Gu XX, Legius E, Vincent MF, Jaeken J, Cassiman JJ, Van den Berghe G (1999) Mutation analysis in adenylosuccinate lyase deficiency: eight novel mutations in the re-evaluated full ADSL coding sequence. Hum Mutat 13:197–202 [DOI] [PubMed] [Google Scholar]
- Nassogne MC, Henrot B, Aubert G, Bonnier C, Marie S, Saint-Martin C, Van den Berghe G, Sébire G, Vincent MF (2000) Adenylosuccinase deficiency: an unusual cause of early-onset epilepsy associated with acquired microcephaly. Brain Dev 22:383–386 [DOI] [PubMed] [Google Scholar]
- Race V, Marie S, Vincent MF, Van den Berghe G (2000) Clinical, biochemical and molecular genetic correlations in adenylosuccinate lyase deficiency. Hum Mol Genet 9:2159–2165 [DOI] [PubMed] [Google Scholar]
- Scarpulla RC (1997) Nuclear control of respiratory chain expression in mammalian cells. J Bioenerg Biomembr 29:109–119 [DOI] [PubMed] [Google Scholar]
- Seelan RS, Gopalakrishnan L, Scarpulla RC, Grossman LI (1996) Cytochrome c oxidase subunit VIIa liver isoform: characterization and identification of promoter elements in the bovine gene. J Biol Chem 271:2112–2120 [DOI] [PubMed] [Google Scholar]
- Suzuki F, Goto M, Sawa C, Ito S, Watanabe H, Sawada J, Handa H (1998) Functional interactions of transcription factor human GA-binding protein subunits. J Biol Chem 273:29302–29308 [DOI] [PubMed] [Google Scholar]
- Thompson CC, Brown TA, McKnight SL (1991) Convergence of Ets- and Notch-related structural motifs in a heterodimeric DNA binding complex. Science 253:762–768 [DOI] [PubMed] [Google Scholar]
- Van den Berghe G, Jaeken J (2001) Adenylosuccinate lyase deficiency. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic and molecular bases of inherited disease. McGraw Hill, New York, pp 2653–2662 [Google Scholar]
- Van Keuren ML, Hart IM, Kao FT, Neve RL, Bruns GAP, Kurnit DM, Patterson D (1987) A somatic cell hybrid with a single human chromosome 22 corrects the defect in the CHO mutant (Ade-1) lacking adenylosuccinase activity. Cytogenet Cell Genet 44:142–147 [DOI] [PubMed] [Google Scholar]
- Vincent S, Marty L, Fort P (1993) S26 ribosomal protein RNA: an invariant control for gene regulation experiments in eukaryotic cells and tissues. Nucleic Acids Res 21:1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virbasius JV, Scarpulla RC (1991) Transcriptional activation through ETS domain binding sites in the cytochrome c oxidase subunit IV gene. Mol Cell Biol 11:5631–5638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— (1994) Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: a potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc Natl Acad Sci USA 91:1309–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virbasius JV, Virbasius CA, Scarpulla RC (1993) Identity of GABP with NRF-2, a multisubunit activator of cytochrome oxidase expression, reveals a cellular role for an ETS domain activator of viral promoters. Genes Dev 7:380–392 [DOI] [PubMed] [Google Scholar]
- Wasylyk B, Hahn SL, Giovane A (1993) The Ets family of transcription factors. Eur J Biochem 211:7–18 [DOI] [PubMed] [Google Scholar]
- Wong-Riley M, Guo A, Bachman NJ, Lomax MI (2000) Human COX6A1 gene: promoter analysis, cDNA isolation and expression in the monkey brain. Gene 247:63–75 [DOI] [PubMed] [Google Scholar]