

Abstract
We used a covariate-based linkage method to reanalyze genome scan data from affected sibships collected by the Alzheimer Disease (AD) Genetics Initiative of the National Institute of Mental Health. As reported in an earlier article, the amyloid-β precursor protein (APP) region is strongly linked to affected sib pairs of the oldest current age (i.e., age either at last exam or at death) who lack E4 alleles at the apolipoprotein E (ApoE) locus. We now report that a region on 20p shows the same pattern. A model that includes current age and the number of E2 alleles as covariates gives a LOD score of 4.1. The signal on 20p is near the location of the gene coding for cystatin-C, previously shown to be associated with late-onset AD and to codeposit with APP in the brains of patients with AD. Two-locus analysis provides evidence of strong epistasis between 20p and the APP region, limited to the oldest age group and to those lacking ApoE4 alleles. We speculate that high-risk polymorphisms in both regions produce a biological interaction between these two proteins that increases susceptibility to a very-late-onset form of AD.
Alzheimer disease (AD) is the most common form of dementia in the elderly and is believed to be genetically complex. To date, three genetic loci have been identified that contribute to early-onset autosomal dominant AD: presenilin 1 (PSEN1 [MIM 104311]), presenilin 2 (PSEN2 [MIM 633044]), and amyloid-β precursor protein (APP [MIM 104760]). However, only apolipoprotein E (ApoE [MIM 107741]) has been well-established as contributing to late-onset AD (see, e.g., Corder et al. 1993; Saunders et al. 1993). Daw et al. (2000) found evidence for multiple genetic loci contributing to the age at onset of AD with effect sizes similar to or larger than the effect of the ApoE locus, suggesting a role of additional genetic-risk factors in the age at onset of AD. However, clear evidence of additional loci that contribute to risk or age at onset has remained elusive, although substantial linkage evidence exists for regions on chromosomes 9 (Pericak-Vance et al. 1997), 10 (Bertram et al. 2000; Ertekin-Taner et al. 2000; Myers et al. 2000), and 12 (Pericak-Vance et al. 1997).
Locus heterogeneity, such as may be present for late-onset AD, presents a challenge in the analysis and interpretation of linkage results. Recently, we developed a model-free linkage method that allows covariates to be included in the analysis (Goddard et al. 2001). This method accounts for locus heterogeneity that is measured by covariates, thereby allowing the discovery of evidence for linkage that might otherwise be obscured. We have demonstrated the potential effectiveness of this method in a recent analysis of the APP region, using affected sib pairs (ASPs) collected as part of the National Institute of Mental Health (NIMH) AD Genetics Initiative (Olson et al. 2001). In the case of the APP region, we observed an increase in the LOD score from .04 to 5.54 after we included current age as a covariate in the linkage model. Our results indicated that the families with AD that have the oldest current age and no ApoE4 alleles exhibit linkage to APP.
In the present study, we revisit the data collected by the NIMH AD Genetics Initiative and analyze the remaining autosomes through use of the same covariate-based method of ASP linkage analysis. We chose, as covariates, variables that we believed were likely to be related to locus heterogeneity. Specifically, we examine the roles that age at onset, current age (age at last exam or age at death), and ApoE genotype play in linkage heterogeneity to late-onset AD. As we shall see, we find substantial evidence for linkage on 20p, in addition to the signal in the APP region that we reported previously. We also present an extension of our linkage analysis method to include two-locus models, to examine a possible epistatic interaction between the signal in the APP region and the new signal on 20p.
The collection, characterization, and genotyping of AD-affected sibships by the NIMH AD Genetics Initiative has been previously described (Blacker et al. 1997). We restricted our analysis to 272 ASPs that had onset at age ⩾60 and also had complete data on the following variables: age at onset (as measured by age at first symptoms), current age (as measured by either age at last exam [33%] or age at death [67%]), and ApoE genotype. The lower bound on age at onset was imposed to eliminate the possible effect of outliers on the linkage analysis; only four ASPs were removed for this reason. For age at onset and current age, the covariate included in the linkage analysis was the sum of the values of the two individuals. For ApoE genotype, we considered two covariates: (1) the total number of E4 alleles in the ASP and (2) the total number of E2 alleles in the ASP. Multipoint identity-by-descent values were computed at 2-cM intervals throughout the genome, by use of all markers available from the genome scan.
To allow for covariate-related locus heterogeneity, we applied a covariate-based ASP LOD-score method (Goddard et al. 2001) to the genotype data. The model is a one-parameter modification of the conditional logistic parameterization of the ASP LOD score introduced by Olson (1999). An optimal mode of inheritance parameter (Whittemore and Tu 1998) is specified that allows one to fit only a single additional parameter per covariate. The model is parameterized in terms of offspring recurrence-risk ratio (λ1), conditional on K covariates xk, as
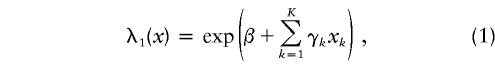 |
where β is a parameter that measures the “average” linkage in the sample and the γk are covariate-specific parameters that measures the change in linkage as a function of the covariates, and in terms of the recurrence-risk ratio for monozygotic twins (λ2), conditional on K covariates xk, as
This model has been implemented in the S.A.G.E. program LODPAL.
To simplify specification of constraints on parameter estimates, to improve numerical stability, and so that β reflects average allele-sharing, all covariates are centered around their sample mean before they are included in the linkage model. Because covariates are centered around their mean, inclusion of a covariate in the model is not expected to give a different value of β than that estimated in the model without covariates, unless the distribution of the covariate is asymmetric, as is the case with the number of E4 alleles. In general, the values of β and the γk depend on the choice of “coding scheme” for the covariates; a linear transformation of the covariate does not change either the LOD score or the estimates of covariate-specific recurrence-risk ratios. More importantly, conclusions about the existence of locus heterogeneity and the extent or nature of locus heterogeneity do not depend on the estimated value of β (which may equal zero).
Asymptotic distributions of the resulting likelihood-ratio tests were used to obtain P values (see Goddard et al. 2001). In the present study, we report as “LOD” scores the likelihood-ratio statistics (LRSs) divided by 4.605 (i.e., 2loge10). Critical values for the LRSs were obtained as follows. The distribution of the LRS for the basic one-parameter model is a 50:50 mixture of a point mass at zero and a χ2 distribution with 1 df. Addition of K covariates gives an LRS with a distribution that is a 50:50 mixture of a χ2 with K df and a χ2 with K+1 df. The difference in LRS between nested models that differ by J covariates has a χ2 distribution with J df. One can therefore test both the significance of the contribution of a covariate and the overall evidence for linkage. The overall evidence for linkage includes information about both the “average” linkage for the sample and the change in linkage as a function of the covariate.
Two-locus models were used to examine regions demonstrating linkage for possible interaction, conditional on one or more covariates. First, let λij(x) be the recurrence-risk ratio corresponding to the sharing of i alleles at the first locus and j alleles at the second locus, for i,j=0,1,2, conditional on covariates xk as in equation (1)—that is,
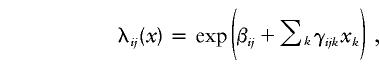 |
where βij and γijk are the corresponding “average” and covariate-specific linkage parameters, respectively. An extension of equation (2) to two loci gives the constraints λi2(x)=3.634λi1(x)-2.634λi0(x) and λ2j(x)=3.634λ1j(x)-2.634λ0j(x), subject to λ00(x)≡1. The model without covariates has three free parameters: two “single-locus” parameters, 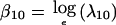 and
and 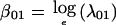 , and the joint (two-locus) parameter,
, and the joint (two-locus) parameter, 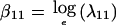 . As many as three parameters for each covariate may be added, one for each risk ratio. Likelihood-ratio statistics then can be used to compare nested models. In addition, a model additive in penetrance (to approximate a heterogeneity model) was fitted by putting λij(x)=λ(1)i(x)+λ(2)j(x)-1, and a model multiplicative in penetrance was fitted by putting λij(x)=λ(1)i(x)λ(2)j(x), for i,j=0,1,2 (see Risch 1990; Olson 1999). In these expressions, λ(l)i(x) represents a risk-ratio–like quantity specific to locus l, for l=1,2. In the absence of covariates, these two models each contain two free parameters, one for each of the two loci; as many as two parameters per covariate may be added, in a manner similar to equation (1).
. As many as three parameters for each covariate may be added, one for each risk ratio. Likelihood-ratio statistics then can be used to compare nested models. In addition, a model additive in penetrance (to approximate a heterogeneity model) was fitted by putting λij(x)=λ(1)i(x)+λ(2)j(x)-1, and a model multiplicative in penetrance was fitted by putting λij(x)=λ(1)i(x)λ(2)j(x), for i,j=0,1,2 (see Risch 1990; Olson 1999). In these expressions, λ(l)i(x) represents a risk-ratio–like quantity specific to locus l, for l=1,2. In the absence of covariates, these two models each contain two free parameters, one for each of the two loci; as many as two parameters per covariate may be added, in a manner similar to equation (1).
Table 1 contains descriptive statistics for the variables used to construct covariates for the linkage analysis. The covariates were included in the model as the sum of the values for the two individuals comprising each ASP; table 1 also includes the mean and SD for these covariates as well as for the original variables. Ages at onset for individuals in this study ranged from 61 to 93 years, with a mean of 74.8 years. As expected, there was a higher frequency of E4 alleles (0.395) than in the general population. We also noted a monotonic increase in current age as the number of E4 alleles decreased.
Table 1.
Descriptive Statistics of Variables and Covariates
| Variable/Covariate | Mean | SD | Range | Within-PairCorrelation |
| Original Variable:a | ||||
| Age at onset (years) | 74.77 | 5.84 | 61–93 | |
| Current age (years) | 83.33 | 6.72 | 63–105 | |
| ApoE allele frequencies: | ||||
| E2 | .024 | |||
| E3 | .581 | |||
| E4 | .395 | |||
| Covariate:b | ||||
| Age at onset (years) | 149.18 | 9.88 | 127–181 | .37 |
| Current age (years) | 166.37 | 11.81 | 134–208 | .55 |
| No. of E4 alleles | 1.62 | 1.13 | 0–4 | .58 |
| No. of E2 alleles | .08 | .32 | 0–2 | .25 |
| Current age (years): | ||||
| 0 E4 alleles (N=56) | 175.02 | 11.50 | 154–208 | |
| 1 E4 allele (N=56) | 167.13 | 11.55 | 143–192 | |
| 2 E4 alleles (N=114) | 165.12 | 9.97 | 141–190 | |
| 3 E4 alleles (N=27) | 159.19 | 10.71 | 134–179 | |
| 4 E4 alleles (N=19) | 156.32 | 8.42 | 144–169 |
N=461 sibs.
N=272 ASPs. Statistics are for the sum of the pair’s values before centering. Covariates were centered before inclusion in linkage models.
Figure 1 shows genome-scan results for the baseline model and single-covariate models with age at onset, current age, and E4 as covariates. Regions in which the total LOD score was significant at the .01 level are given in table 2. The largest single-covariate signal is on chromosome 21; a more detailed analysis of this region is presented by Olson et al. (2001) and will not be repeated here. For the baseline model (i.e., without covariates), five regions are significant at the .01 level (table 2), including the ApoE region; the highest LOD score is at the last marker on chromosome 6. Of the five regions with significant baseline effects, four (chromosomes 1, 6, 10, and 11) show no significant effect of any covariate at the .05 level. The remaining region (chromosome 19) shows evidence that ASPs with more E4 alleles are more likely to be linked. Three additional regions (chromosomes 5, 9, and 14) show little or no linkage when the baseline model is used but do show some effect of E4; for these three regions, ASPs with more E4 alleles are more likely to be linked.
Figure 1.

Genome scan results for baseline (without covariates) and single-covariate models. A LOD score of 2.1 correponds to the α=.001 significance level for the baseline model; a LOD score of 2.8 corresponds to the the α=.001 significance level for models with a single covariate.
Table 2.
LOD Scores, Parameter Estimates, and Significance Levels for Baseline and Single Covariate Models
| Nearest Marker(Distance fromFirst Marker,in cM) | BaselineLODScorea | Covariate | Overall LODScore (P)b | β | γ | LOD-ScoreDifference (P)c |
| D1S1675 (172) | 1.21 | … | 1.21 (.0091) | .16 | … | … |
| D5S1470 (46) | 1.01 | E4 | 2.03 (.0058) | .18 | .15 | 1.02 (.0302) |
| D6S1007 (188) | 3.88 | … | 3.88 (.000012) | .36 | … | … |
| D9S176 (106) | 1.06 | E4 | 2.58 (.0016) | .17 | .16 | 1.52 (.0082) |
| D10S1211 (62) | 2.13 | … | 2.13 (.00087) | .28 | … | … |
| D11S2002 (78) | 1.18 | … | 1.18 (.0099) | .16 | … | … |
| D14S72 (0) | .30 | E4 | 1.92 (.0075) | .09 | .16 | 1.62 (.0063) |
| D14S1015 (90) | .00 | Age at onset | 1.89 (.0080) | .00 | −.02 | 1.89 (.0032) |
| D19S571 (80) | 1.95 | E4 | 3.10 (.0005) | .26 | .17 | 1.15 (.0214) |
| D20S186 (24) | .00 | Current age | 2.69 (.0012) | .00 | .01 | 2.69 (.0004) |
| D21S1435 (30) | .03 | Current age | 5.91 (.0000007) | .11 | .03 | 5.88 (.0000002) |
LOD score for the model without covariates.
LOD score and associated P value for the model with one covariate.
Difference in LOD scores and associated P value between the baseline and overall models, giving the LOD score for the effect on linkage of the covariate.
Of the regions significant at the .01 level that also show an E4 effect, only one is in a region containing a strong candidate locus: the region on chromosome 19 that contains the ApoE locus itself. It has long been known that E4 is a risk factor for late-onset AD, and these results are consistent with previous findings. The addition of E2 to the E4 model on chromosome 19 gives a significant increase in the LOD score, to 4.03 (P=.0385 for E2 effect). Therefore, the best, most parsimonious model contains E2 and E4 (β=.304, γE2=-.323, γE4=.222), indicating increasing evidence for linkage with decreasing numbers of E2 alleles and increasing numbers of E4 alleles.
Three chromosomal regions showed significant evidence for linkage at the .01 level when either current age or age at onset was included in the model (table 2). We followed up the age-at-onset peak (LOD score = 1.89) on chromosome 14, which is ∼15 cM distal to PSEN1, one of the genes linked to early-onset AD (Sherrington et al. 1995). Multiple regression analysis failed to increase the LOD score significantly, and no substantial evidence of linkage for any model was found at 74 cM, the approximate location of PSEN1. In the process of fitting these models, however, we did observe a LOD score of 4.14 (P=.00051) at 60 cM (between D14S63 and D14S43, β=.056) when E2 (γ=-.511), E4 (γ=.038), and age at onset (γ=-.011) were used as covariates. ASPs with more E4 alleles, fewer E2 alleles, and earlier age at onset are most strongly linked.
Excluding chromosome 21, the largest effect for a single covariate was for current age, on chromosome 20 (LOD score = 2.69). Since smaller effects were observed at this location for E2 and E4, we followed up with multiple regression analyses (table 3). The best, most-parsimonious model includes both current age and E2; ASPs most likely to be linked are those with the oldest current age who have E2 alleles. The large difference in LOD score between the model with age at onset (LOD score = 0.85) and the model with current age (LOD score = 2.69) suggests that an analysis with only age at onset would not have detected this region. Although the correlation between current age and age at onset is expected to be high, we note that there is a higher within-pair correlation for current age (0.55) than for age at onset (0.37). The meaning of the current age covariate is unclear, although the difference in the LOD scores suggests that this locus more likely influences disease progression, since current age equals age at onset plus disease duration (Olson et al. 2001).
Table 3.
LOD Scores, Parameter Estimates, and Significance Levels for Chromosome 20 at 24 cM[Note]
| Model | LOD Score (Overall P) | β | γ1 | γ2 | γ3 | LOD-Score Difference from Nearest Model (P) |
| Baseline | .00 (1.0000) | .00 | … | |||
| +ApoE4 (E4) | 1.01 (.0644) | .00 | −.11 | 1.01 (.0310) | ||
| +ApoE2 (E2) | 1.59 (.0163) | .00 | .77 | 1.59 (.0068) | ||
| +E4+E2 | 2.19 (.0122) | .00 | −.08 | .69 | .60 (.0965) | |
| +Age at onset | .85 (.0946) | .00 | .01 | .85 (.0479) | ||
| +Current age (C) | 2.69 (.0012) | .00 | .01 | 2.69 (.0004) | ||
| +C+E2 | 4.09 (.0002) | .00 | .01 | .58 | 1.41 (.0108) | |
| +C+E2+E4 | 4.11 (.0005) | .00 | .01 | .58 | .01 | .02 (.7615) |
Note.— Boldface italics indicate the best, most-parsimonious model.
Our final model for chromosome 20 closely resembles the model previously reported for the APP region on chromosome 21 (Olson et al. 2001). Both locations show that the likelihood of linkage increases with increasing current age and with increasing number of E2 alleles and decreasing number of E4 alleles. Although the final models differ somewhat in their details (for example, the APP region shows no effect of ApoE once current age is accounted for), the striking similarities suggest the possibility that the same subset of families segregates for disease alleles in both locations. For that reason, we fit two-locus models to determine whether epistasis could be detected statistically.
Table 4 gives some results of this analysis. Because of computational difficulties in fitting a full model with both current age and E2, we begin with a full model with only current age as a covariate. This full model gives a LOD score of 12.74, which differs significantly from the two single-locus models, the multiplicative model, and the heterogeneity model. A more parsimonious model is obtained by allowing only the joint recurrence-risk ratio λ11 to depend on current age (LOD-score difference = 1.08; P=.083, 2 df). The addition of E2 to λ11 increases the LOD score by .96 (P=.036, 1 df) to 12.62, but the further addition of E2 to the “single-locus” recurrence-risk ratios λ01 and λ10 gives no further improvement (P=.339, 2 df). We conclude that the best, most parsimonious model includes the covariates only in the joint recurrence-risk ratio and further note that, in this model, the “single-locus” estimates of β equal zero. According this model, ASPs who share no alleles IBD at one or both loci contribute no linkage evidence (i.e., λij(x)=1 for i,j=0,1,2 and all values of x); only ASPs who share at least one allele IBD at both loci contribute linkage evidence, with the most strongly linked pairs being those of the oldest age and, to a lesser extent, those with the most E2 alleles.
Table 4.
Two-Locus Models for 20p and 21p (the APP Region)[Note]
|
Parameter Estimate |
|||||||||||
| 20p |
21p |
Interaction |
|||||||||
| Model | LOD Score | P | β | γage | γE2 | β | γage | γE2 | β | γage | γE2 |
| With current age: | |||||||||||
| 20p | 2.69 | <.00001a | .00 | .01 | … | … | … | … | … | … | … |
| 21p | 5.91 | <.00001a | … | … | … | .11 | .03 | … | … | … | … |
| Multiplicative | 8.60 | .00004a | .00 | .01 | … | .11 | .03 | … | … | … | … |
| Heterogeneity | 8.37 | .00003a | .00 | .01 | … | .04 | .02 | … | … | … | … |
| No single-locus age | 11.66 | .083a | .06 | … | … | .12 | … | … | .04 | .03 | … |
| Full | 12.74 | … | .00 | −.01 | … | .09 | .01 | … | .12 | .02 | … |
| With age and E2: | |||||||||||
| No single-locus E2 | 12.62 | .0355b | .00 | … | … | .00 | … | … | .10 | .02 | .29 |
| Full +E2 | 13.09 | .3389c | .02 | … | .46 | .00 | … | .00 | .10 | .02 | .38 |
Note.— Boldface italics indicate the best, most-parsimonious model.
Compared with full model.
Compared with no single-locus age.
Compared with no single-locus E2.
We thus obtained strong evidence for linkage to chromosome 20p (a location different from the signal in 20q13 detected by Pericak-Vance et al. [1997]). One candidate gene, located at 20p11.2, is cystatin C (CST3 [MIM 119817]), a cysteine protease inhibitor involved in hereditary cerebral hemorrhage with amyloidosis–Icelandic type (HCHWA-I) (Levy et al. 1989). The CST3 locus is ∼15 cM proximal to our peak location. Evidence of genetic association between late-onset AD and a CST3 polymorphism has been described in three case-control studies (Crawford et al. 2000; Finckh et al. 2000; Beyer et al. 2001); all three showed that the strength of this association increased with increasing age. Their results are consistent with our finding that linkage is strongest in the oldest ASPs and, to a lesser extent, in those with E2 alleles. As discussed by Olson et al. (2001), it may be that the effect of ApoE is the result of attrition due to the detrimental effects of E4 at earlier ages. Beyer et al. (2001) also observed an association in the earlier-onset subset and argued that CST3 interacts with ApoE at earlier ages. This conclusion was based on results from a very small portion of their data and is not supported by our finding of lack of linkage to 20p at earlier ages. No association with CST3 was found in a Japanese sample (Maruyama et al. 2001), and no linkage to CST3 was found in eight extended families with mean ages at onset of 36–65 years (Parfitt et al. 1993).
We also report evidence for strong epistasis between the 20p region and the region of the APP locus. Our results indicate that a subset of families with AD segregates for susceptibility loci at both locations and suggest that susceptibility alleles at both loci must be present for AD to occur. In patients with AD, CST3 has been shown to be codeposited with amyloid-β in amyloid plaques in the brain (Levy et al. 2001), as well as colocalized with amyloid-β in brain arteriolar walls (Vinters et al. 1990). In the rare autosomal dominant disease HCHWA-I, a point mutation in CST3 leads to the deposition of amyloid fibrils in the blood vessels of the brain and subsequently to multiple hemorrhages. HCHWA-I is very similar to the Dutch type of HCHWA (HCHWA-D), in which APP, rather than CST3, is the mutated locus (Prelli et al. 1988). Both diseases, however, involve the colocalization of both CST3 and amyloid-β in the cortical blood vessels (Bornebroek et al. 1996; Maat-Schieman et al. 1996; Wei et al. 1998).
Although it has been suggested that CST3 might be involved in APP processing and/or generation of amyloid-β, the exact mechanism of biological interaction remains to be determined. A high-affinity substrate of CST3, cathepsin S, is known to cleave APP into derivatives containing amyloid-β in vitro (Munger et al. 1995) and to increase amyloid-β production in tissue culture (Lemere et al. 1995). A separate line of investigation, using transgenic mice, suggests that amyloid plaque initiation is independent of CST3 levels but also that, once plaques are formed, elevated CST3 levels are associated with deposition of CST3 layers onto the amyloid plaque cores (Steinhoff et al. 2001). Whatever the mechanism, our results support the hypothesis that these two proteins interact to affect susceptibility to or progression of AD.
With the covariates we used, we were unable to detect substantial linkage in two candidate regions: the PSEN2 region on chromosome 1 (Levy-Lehad et al. 1995) and the LRP1 region on chromosome 12 (Pericak-Vance et al. 1997). Covariates failed to significantly improve the modest evidence for linkage to the HLA/TNF region on chromosome 6 (Collins et al. 2000), to 10q21-22 (Myers et al. 2000, Ertekin-Taner et al. 2000), or to the α2-macroglobulin region on 12q13 (Blacker et al. 1998), previously detected in part using these data.
More generally, it is of interest to compare our findings with earlier genome results using the same data. Kehoe et al. (1999) analyzed the whole sample (292 ASPs), a subset without E4 alleles (63 ASPs), and a subset in which both members had at least one E4 allele (162 ASPs). Their results were consistent with the analyses shown in figure 1 and table 2, with some differences in the relative magnitudes of linkage evidence at different locations. In addition, because we formally tested for the effect of E4 on linkage, we did not report as significant some of the subgroup signals reported by Kehoe et al. (1999). If we had used a more liberal significance criterion, such as .01, we would have included some of these signals, as well as additional E4 signals not reported by Kehoe et al. (1999). Interestingly, Kehoe et al. used exclusion mapping to eliminate regions at or near the locations of our signal on chromosomes 20, using a sibling recurrence-risk ratio of 1.4.
To summarize our key findings, we have described evidence for the existence of a genetically identifiable subtype of “late-onset” AD limited to the most elderly and characterized, in these data, by joint linkage to 21p and 20p. Our analysis suggests that the development and/or rate of progression of AD in such families requires the presence of high-risk alleles at both genes, which likely interact biologically to increase disease risk. We believe that this subtype of AD is biologically independent of ApoE and that its association with E2 in this data set is the result of E4-related attrition at earlier ages. In addition, we have found some evidence that a gene in the PSEN1 region contributes to a subtype of AD found in families of earlier onset and in which E4 may play a more direct role.
Because of issues relating to multiple comparisons, we view our analyses as exploratory; our results require confirmation in an independent sample. Like Goddard et al. (2001) and Olson et al. (2001), we note that the ability to include covariates increases the overall probability of type I errors due to multiple analyses of the same genetic data. In this report, we have used pointwise significance levels associated with a single analysis. It is likely, however, that complete accounting of multiple comparisons in the context of a genome scan will require prohibitively small significance levels. Further, we believe that the presence of locus heterogeneity is the single most-important impediment to the successful detection and replication of baseline linkage results for complex genetic disorders. Thus, failure to account for locus heterogeneity places a severe limitation on the rate of scientific progress. A balance must therefore be struck between the need for timely gene discovery and the need to be prudent. In general, we recommend careful prior selection of covariates and cautious interpretation of results.
Acknowledgments
This work was supported in part by U.S. Public Health Service grants HG01577, from the National Center for Human Genome Research, and RR03655, from the National Center for Research Resources. Some of the results in this paper were obtained using S.A.G.E., which is supported by National Center for Research Resources grant RR03655. Data were collected in three projects that participated in the NIMH AD Genetic Initiative. From 1991 to 1998, the principal investigators and coinvestigators were: from Massachusetts General Hospital, Boston, Marilyn S. Albert, Ph.D., and Deborah Blacker, M.D., Sc.D. (grant U01 MH46281); from Johns Hopkins University, Baltimore, Susan S. Bassett, Ph.D., Gary A. Chase, Ph.D., and Marshal F. Folstein, M.D. (grant U01 MH46290); and from the University of Alabama, Birmingham, Rodney C. P. Go, Ph.D., and Lindy E. Harrall, M.D. (grant U01 MH46373). Genotypes were generated from biomaterial in NIMH's Alzheimer Disease Genetics Initiative, in the laboratories of Drs. Alison Goate (Department of Psychiatry, Washington University School of Medicine, St. Louis), John Hardy (Mayo Clinic, Jacksonville, FL), and Mike Owen (Department of Psychological Medicine, University of Wales, Cardiff).
Electronic-Database Information
Accession numbers and URLs for data presented herein are as follows:
- Human Genetic Analysis Resource, http://darwin.cwru.edu/ (for S.A.G.E. software)
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for AD [MIM 104300], ApoE [MIM 107741], PSEN1 [MIM 104311], PSEN2 [MIM 633044], CST3 [MIM 119817], and APP [MIM 104760])
- NIMH Center for Genetic Studies, http://zork.wustl.edu/nimh/ (for NIMH Alzheimer Disease Genetics Initiative)
References
- Bertram L, Blacker D, Mullin K, Keeney D, Jones J, Basu S, Yhu S, McInnis MG, Go RCP, Vekrellis K, Selkoe DJ, Saunders AJ, Tanzi RE (2000) Evidence for genetic linkage of Alzheimer’s disease to chromosome 10q. Science 290:2302–2303 [DOI] [PubMed] [Google Scholar]
- Beyer K, Lao JI, Gomez M, Riutort N, Latorre P, Mate JL, Ariza A (2001) Alzheimer’s disease and the cystatin C gene polymorphism: an association study. Neurosci Lett 315:17–20 [DOI] [PubMed] [Google Scholar]
- Blacker D, Haines JL, Rodes L, Terwedow H, Go RC, Harrell LE, Perry RT, Bassett SS,Chase G, Meyers D, Albert MS, Tanzi R (1997) ApoE-4 and age at onset of Alzheimer’s disease: the NIMH genetics initiative. Neurology 48:139–147 [DOI] [PubMed] [Google Scholar]
- Blacker D, Wilcox MA, Laird NM, Rodes L, Horvath SM, Go RCP, Perry R, Watson B Jr, Bassett SS, McInnis MG, Albert MS, Hyman BT, Tanzi RE (1998) Alpha-2 macroglobulin is genetically associated with Alzheimer disease. Nat Genet 19:357–360 [DOI] [PubMed] [Google Scholar]
- Bornebroek M, Haan J, Maat-Schieman ML, van Duinen SG, Roos RA (1996) Hereditarycerebral hemorrhage with amyloidosis-Dutch type (HCHWA-D). I: A review of clinical, radiologic and genetic aspects. Brain Pathol 6:111–114 [DOI] [PubMed] [Google Scholar]
- Collins JS, Perry RT, Watson B, Harrell LE, Acton RT, Blacker D, Albert MS, Tanzi RE, Bassett SS, McInnis MG, Campbell RD, Go RCP (2000) Association of a haplotype for tumor necrosis factor in siblings with late-onset Alzheimer disease: the NIMH Alzheimer Disease Genetics Initiative. Am J Med Genet Neuropsychiatr Genet 96:823–830 [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261:921–923 [DOI] [PubMed] [Google Scholar]
- Crawford FC, Freeman MJ, Schinka JA, Abdullah LI, Gold M, Hartman R, Krivian K,Morris MD, Richards D, Duara R, Anand R, Mullan MJ (2000) A polymorphism in the cystatin C gene is a novel risk factor for late-onset Alzheimer’s disease. Neurology 55:763–768 [DOI] [PubMed] [Google Scholar]
- Daw WE, Payami H, Nemens EJ, Nochlin D, Bird TD, Schellenberg GD, Wijsman EM (2000) The number of trait loci in late-onset Alzheimer disease. Am J Hum Genet 66:196–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertekin-Taner N, Graff-Radford, Younkin LH, Eckman C, Baker M, Adamson J, Ronald J, Blangero J, Hutton M, Younkin SG (2000) Linkage of plasma Aβ42 to a quantitative locus on chromosome 10 in late-onset Alzheimer’s disease pedigrees. Science 290:2303–2304 [DOI] [PubMed] [Google Scholar]
- Finckh U, von der Kammer H, Velden J, Michel T, Andresen B, Deng A, Zhang J, et al (2000) Genetic association of a cystatin C gene polymorphism with late-onset Alzheimer disease. Arch Neurol 57:1579–1583 [DOI] [PubMed] [Google Scholar]
- Goddard KAB, Witte JS, Suarez BK, Catalona WJ, Olson JM (2001) Model-free linkage analysis with covariates confirms linkage of prostate cancer to chromosomes 1 and 4. Am J Hum Genet 68:1197–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehoe P, Wavrant-De Vrieze F, Crook R, Wu WS, Holmans P, Fenton I, Spurlock G, Norton N, Williams H, Williams N, Lovestone S, Perez-Tur J, Hutton M, Chartier-Harlin MC, Shears S, Roehl K, Booth J, Van Voorst W, Ramic D, Williams J, Goate A, Hardy J, Owen MJ (1999) A full genome scan for late onset Alzheimer’s disease. Hum Mol Genet 8:237–245 [DOI] [PubMed] [Google Scholar]
- Lemere CA, Munger JS, Shi GP, Natkin L, Haass C, Chapman HA, Selkoe DJ (1995) The lysosomal cysteine protease, cathepsin S, is increased in Alzheimer’s disease and Down syndrome brain: an immunocytochemical study. Am J Pathol 146:848–860 [PMC free article] [PubMed] [Google Scholar]
- Levy E, Lopez-Otin C, Ghiso J, Geltner D, Frangione B (1989) Stroke in Icelandic patients with hereditary amyloid angiopathy is related to a mutation in the cystatin C gene, an inhibitor of cysteine proteases. J Exp Med 169:1771–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy E, Sastre M, Kumar A, Gallo G, Piccardo P, Ghetti B, Tagliavini F (2001) Codeposition of cystatin C with amyloid-beta protein in the brain of Alzheimer disease patients. J Neuropathol Exp Neurol 60:94–104 [DOI] [PubMed] [Google Scholar]
- Levy-Lehad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu C, Jondro PD, Schmidt SD, Wang K, Crowley AC, Fu Y-H, Guenette SY, Galas D, Nemens E, Wijsman EM, Bird TD, Schellenberg GD, Tanzi RE (1995) Candidate gene for the chromosome 1 familial Alzheimer’s disease disease locus. Science 269:973–977 [DOI] [PubMed] [Google Scholar]
- Maat-Schieman ML, van Diunen SG, Bornebroek M, Haan J, Roos RA (1996) Hereditarycerebral hemorrhage with amyloidosis-Dutch type (HCHWA-D). II: A review of histopathlogical aspects. Brain Pathol 6:115–120 [DOI] [PubMed] [Google Scholar]
- Maruyama H, Izumi Y, Oda M, Torii T, Morino H, Toji H, Sasaki K, Terasawa H, Nakamura S, Kawakami H (2001) Lack of an association between cystatin C gene polymorphisms in Japanese patients with Alzheimer’s disease. Neurology 57:337–339 [DOI] [PubMed] [Google Scholar]
- Munger JS, Haass C, Lemere CA, Shi GP, Wong WS, Teplow DB, Selkow DJ, Chapman HA (1995) Lysosomal processing of amyloid precursor protein to A beta peptides: distinct role for cathepsin S. Biochem J 311:299–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers A, Holmans P, Marshall H, Kwon J, Meyer D, Ramic D, Shears S, Booth J, Wavrant-DeVrieze F, Crook R, Hamshere M, Abraham R, Tunstall N, Rice F, Carty S, Lillystone S, Kehoe P, Rudrasingham V, Jones L, Lovestone S, Perez-Tur J, Williams J, Owen MJ, Hardy J, Goate AJ (2000) Susceptibility locus for Alzheimer’s disease on chromosome 10. Science 290:2304–2305 [DOI] [PubMed] [Google Scholar]
- Olson JM (1999) A general conditional-logistic model for affected-relative-pair linkage studies. Am J Hum Genet 65:1760–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson JM, Goddard KAB, Dudek DM (2001) The amyloid precursor protein locus and very-late-onset Alzheimer disease. Am J Hum Genet 69:895–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parfitt M, Crook R, Roques P, Rossor M, Chartier-Harlin M-C (1993) The cystatin-C gene is not linked to early onset familial Alzheimer’s disease. Neurosci Lett 154:81–83 [DOI] [PubMed] [Google Scholar]
- Pericak-Vance MA, Bass MP, Yamaoka LH, Gaskell PC, Scott WK, Terwedow HA, Menold MM, Conneally PM, Small GW, Vance JM, Saunders AM, Roses AD, Haines JL (1997) Complete genome screen in late-onset familial Alzheimer disease. JAMA 278:1237–1241 [PubMed] [Google Scholar]
- Prelli F, Castano EM, Van Duinen SG, Bots GTAM, Luyendijk W, Frangione B (1988) Different processing of Alzheimer’s β-protein precursor in the vessel wall of patients with hereditary cerebral hemorrhage with amyloidosis-Dutch type. Biochem Biophys Res Commun 151:1150–1155 [DOI] [PubMed] [Google Scholar]
- Risch N (1990) Linkage strategies for genetically complex traits. I. Multilocus models. Am J Hum Genet 46:222–228 [PMC free article] [PubMed] [Google Scholar]
- Saunders AM, Strittmatter WJ, Schmechel D, St George-Hyslop PH, Pericak-Vance MA, Joo SH, Rose BL, Gusella JF, Crapper-MacLachlan DR, Alberts MJ (1993) Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 43:1467–1472 [DOI] [PubMed] [Google Scholar]
- Sherrington R, Rogaev EI, Liang Y, Rogeava EA, Levesque G, Ikeda M, Chi H, et al (1995) Cloning of a novel gene bearing missense mutations in early familial Alzheimer disease. Nature 375:754–760 [DOI] [PubMed] [Google Scholar]
- Steinhoff T, Moritz E, Wollmer MA, Mohajeri MH, Kins S, Nitsch RM (2001) Increased cystatin C in astrocytes of transgenic mice expressing the K670N-M671L mutation of the amyloid precursor protein and deposition in brain amyloid plaques. Neurobiol Dis 8:647–654 [DOI] [PubMed] [Google Scholar]
- Vinters HV, Nishimura GS, Secor DL, Pardridge WM (1990) Immunoreactive A4 and gamma-trace peptide colocalization in amyloidotic arteriolar lesions in brains of patients with Alzheimer’s disease. Am J Pathol 137:233–240 [PMC free article] [PubMed] [Google Scholar]
- Wei L, Berman Y, Castano EM, Cadene M, Beavis RC, Devi L, Levy E (1998) Instability of the amyloidogenic cystatin C variant of hereditary cerebral hemorrhage with amyloidosis, Icelandic type. J Biol Chem 273:11806–11814 [DOI] [PubMed] [Google Scholar]
- Whittemore AS, Tu I-P (1998) Simple, robust linkage tests for affected sibs. Am J Hum Genet 62:1228–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]