

Abstract
Cocaine exposure during pregnancy causes abnormality in fetal brain development, leading to cognitive dysfunction of the offspring, but the underlying cellular mechanism remains mostly unclear. In this study, we examined synaptic functions in the medial prefrontal cortex (mPFC) of postnatal rats that were exposed to cocaine in utero, using whole-cell recording from mPFC layer V pyramidal neurons in acute brain slices. Cocaine exposure in utero resulted in a facilitated activity-induced long-term potentiation (LTP) of excitatory synapses on these pyramidal neurons and an elevated neuronal excitability in postnatal rat pups after postnatal day 15 (P15). This facilitated LTP could be primarily attributed to the reduction of GABAergic inhibition. Biochemical assays of isolated mPFC tissue from postnatal rats further showed that cocaine exposure in utero caused a marked reduction in the surface expression of GABAA receptor subunits α1, β2, and β3, but had no effect on glutamate receptor subunit GluR1. Both facilitated LTP and reduced surface expression of GABAA receptors persisted in rats up to at least P42. Finally, the behavioral consequence of cocaine exposure in utero was reflected by the reduction in the sensitivity of locomotor activity in postnatal rats to cocaine and the dopamine receptor agonist apomorphine. Since the mPFC is an important part of the reward circuit in the rat brain and plays important roles in cognitive functions, these findings offer new insights into the cellular mechanism underlying the adverse effects of cocaine exposure in utero on brain development and cognitive functions.
Introduction
The first National Institute on Drug Abuse National Pregnancy and Health survey conducted in 1992 reported that, each year in the United States, 5.5% of all expectant mothers used an illicit drug at least once during their pregnancy (Mathias, 1995). Among those women surveyed, 1.1% had used cocaine at some time during pregnancy. Cocaine use during pregnancy results in retarded fetal brain growth (Zuckerman et al., 1989), leading to postnatal changes in brain functions (Salisbury et al., 2007; Shankaran et al., 2007). Children who are exposed to high-level cocaine are also likely to show dose-dependent postnatal growth impairment (Mirochnick et al., 1995; Delaney-Black et al., 1996) as well as deficits in postnatal motor function, attention, and language skills (Azuma and Chasnoff, 1993; Nulman et al., 1994; Chiriboga et al., 1995).
Previous animal studies have shown that prenatal cocaine exposure results in changes of cognitive and emotional development of the offspring, including learning and memory (Thompson et al., 2005; Malanga et al., 2007), but cellular and circuit mechanisms underlying these behavioral changes remain unclear. The medial prefrontal cortex (mPFC) is an important part of the reward circuit in the rat brain, with strong reciprocal interactions with the ventral tegmental area (VTA) and nucleus accumbens (NAc)—two regions known to play key functions in the initiation and expression of locomotor sensitization as a result of repeated cocaine exposure (Kalivas et al., 1998; Robinson and Berridge, 2003). Because dopaminergic inputs to mPFC layer V pyramidal cells are involved in attention (Broersen et al., 1996), prenatal cocaine-induced synaptic and neuronal alterations in these neurons may contribute to cognitive impairments and behavioral deficits in postnatal animals.
In the present study, we used a rat model to examine the effects of cocaine exposure in utero on activity-dependent synaptic plasticity in the mPFC. Pregnant rats were given daily intraperitoneal injections with either saline or cocaine for 7 d from embryonic day 15 (E15) to E21. Postnatal rats were examined during postnatal day 8 (P8) to P42 for cocaine-induced alterations in the activity-induced long-term potentiation (LTP) and long-term depression (LTD), the expression of glutamate and GABAA receptors, and the excitability of mPFC layer V pyramidal neurons. We found that cocaine exposure in utero caused a reduction of GABAergic inhibition in layer V pyramidal neurons of rat mPFC, leading to an increased susceptibility of excitatory synapses to LTP induction as well as an elevated spiking activity in response to synaptic excitation or membrane depolarization in these neurons. At the behavioral level, rats exposed to cocaine in utero showed a reduced locomotor sensitivity to cocaine and the dopamine receptor agonist. Together, these results provided new evidence on the cellular mechanisms underlying the effects induced by cocaine exposure in utero.
Materials and Methods
Animals and slice preparation.
Pregnant Sprague Dawley (SD) rats (Charles River) were given intraperitoneal injections of either saline (0.9% NaCl; 1 ml kg−1) or saline containing cocaine (15 mg kg−1 in 1 ml kg−1 of saline) for 7 d from E15 to E21. Offspring from the treated mother were raised by the nontreated mother. No physical difference was shown between the rats exposed to cocaine or saline in utero. Rats were anesthetized with isoflurane and then killed by decapitation. Animal use procedure was approved by the Animal Care and Use Committee at the University of California, Berkeley. Fresh isolated brain slices containing mPFC were prepared from SD rats as previously described (Mansvelder and McGehee, 2000). Coronal slices (250 μm thick) containing mPFC were cut with a Vibratome in a chamber filled with chilled (2–5°C) cutting solution containing the following (in mm): 110 choline-chloride, 25 NaHCO3, 25 d-glucose, 11.6 sodium ascorbate, 7 MgSO4, 3.1 sodium pyruvate, 2.5 KCl, 1.25 NaH2PO4, and 0.5 CaCl2. The slices were then incubated in artificial CSF (ACSF) containing the following (in mm): 119 NaCl, 26.2 NaHCO3, 11 d-glucose, 2.5 KCl, 2.5 CaCl2, 1.5 MgSO4, 1.25 NaH2PO4 at room temperature. The solutions were bubbled with 95% O2 and 5% CO2.
Electrophysiology.
Whole-cell recordings were made from layer V mPFC pyramidal neurons by using a patch-clamp amplifier (MultiClamp 700B; Molecular Devices) under infrared differential interference contrast optics. Microelectrodes were made from borosilicate glass capillaries and had a resistance of 2.5–5 MΩ. Data acquisition and analysis were performed by using a digitizer and pClamp 9 software (DigiData 1322A; Molecular Devices). To stimulate presynaptic fibers, a bipolar tungsten stimulation electrode (WPI) was placed in the layer II/III of the prelimbic region of the mPFC, and pulses of 50 μs duration were applied (at 0.1 Hz). Whole-cell recording was made at 30 ± 1°C in a recording chamber, under the regulation of an automatic temperature controller (Warner Instruments). Series resistance (15–30 MΩ) and input resistance (100–200 MΩ) were monitored throughout the recording. Data were discarded when the change in the series resistance was >20% during the course of the experiment. Mostly, monosynaptic EPSPs were recorded at −70 mV in current-clamp mode. For recording IPSCs, neurons were held at −20 mV in voltage-clamp mode in the presence of 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) (10 μm) and d-2-amino-5-phosphonopentanoic acid (AP5) (25 μm). The intrapipette solution for most whole-cell recordings contained the following (in mm): 140 potassium gluconate, 5 KCl, 10 HEPES, 0.2 EGTA, 2 MgCl2, 4 MgATP, 0.3 Na2GTP, and 10 Na2-phosphocreatine, pH 7.2 (with KOH). The BAPTA-containing intrapipette solution consisted of the following (in mm): 100 potassium gluconate, 5 KCl, 10 HEPES, 20 BAPTA, 2 MgCl2, 4 MgATP, 0.3 Na2GTP, and 10 Na2-phosphocreatine, pH 7.2 (with KOH). For recording biphasic responses (EPSC/IPSC sequences), neurons were voltage clamped at the resting membrane potential (−54 mV). For this experiment, low Cl−-containing intrapipette solution was used (in mm: 145 potassium gluconate, 10 HEPES, 0.2 EGTA, 1 MgCl2, 4 MgATP, 0.3 Na2GTP, and 10 Na2-phosphocreatine, pH 7.2 with KOH) to amplify the IPSCs to obtain biphasic response showed in Figure 5A. For measurements of AMPA/NMDA ratio, the recordings were made in the presence of 1 μm 6-imino-3-(4-methoxyphenyl)-1(6H)-pyridazinebutanoic acid hydrobromide (SR95531) to block GABAergic transmission and 10 μm glycine to coactivate NMDA receptor (NMDAR). First, the neuron was voltage clamped at −70 mV to obtain stable AMPA receptor-mediated currents. Then the neuron was held at +40 mV, and responses were recorded first in the presence of CNQX (10 μm) and then in the presence of both CNQX (10 μm) and AP5 (25 μm). The NMDA receptor-mediated currents were obtained by subtracting the averaged responses obtained in the presence of both CNQX and AP5 from those obtained in the presence of CNQX only. The intrapipette solution contained the following (in mm): 120 CsCH3SO3, 20 HEPES, 0.4 EGTA, 5 TEA-Cl (tetraethylammonium chloride), 2 MgCl2, 2.5 MgATP and 0.3 GTP, 10 Na2-phosphocreatine, 1 QX-314 [N-(2,6-dimethylphenylcarbamoylmethyl)triethylammonium bromide], pH 7.2 (with CsOH). For the treatment of SR95531, data were taken 10 min after the onset of the drug perfusion to allow drug penetration into the slices. Summary data are given as means ± SEM. For assaying EPSP–spike (E–S) coupling, the initial slope of EPSPs was measured for the first 4 ms period of the rising phase (in millivolts per millisecond). In all cases, data from one neuron were collected from each slice. Unless indicated otherwise, statistical tests were performed by using Student's t test or the Kolmogorov–Smirnov test (for cumulative percentage plots).
Figure 5.

Prenatal cocaine exposure reduced GABAA receptor-mediated inhibition to mPFC layer V pyramidal neurons. A, Examples of synaptic currents before and after the application of CNQX (10 μm) and AP5 (25 μm) in response to extracellular stimulation in the mPFC slice from P24 prenatal cocaine-treated rat. Calibration: 50 pA, 10 ms. B, Examples of input–output plot for IPSCs elicited by stimuli of different intensities, from P24 rats prenatally exposed to cocaine and saline, respectively. C, Cumulative percentage for the distribution of the slopes of best fit line for the input–out plots illustrated in B, before and after the application of CNQX (10 μm) and AP5 (25 μm), from P23–P26 rats prenatally exposed to saline (n = 11) and cocaine (n = 10) (*p = 0.017, Kolmogorov–Smirnov test). D, Cumulative percentage for the distribution of the slopes of input–output plots for EPSCs elicited by stimuli of different intensities from the same group of rats as those in C. *p = 0.021, Kolmogorov–Smirnov test. E, The maximal amplitude of IPSCs evoked in the pyramidal neuron by extracellular stimulation was compared between rats (P24–P25) exposed prenatally with either cocaine or saline. *p < 0.05, t test. Number associated with the histogram refers to the total number of cells recorded. Calibration: 200 pA, 50 ms. F, Comparison of paired-pulse ratio (PPR) (IPSC amplitude of the second divided by the first). The IPSCs were evoked by two sequential presynaptic stimuli at an interval of 50 ms, with a sample of IPSC pair (average of 15) shown above. Calibration: 100 pA, 10 ms. Data were obtained from P20–P25 rats prenatally treated with cocaine or saline. p = 0.36, t test. Error bars indicate SEM.
Measurements of transmitter receptor expression.
For biotinylation of membrane proteins, mPFC tissues dissected from freshly isolated brain slices (preincubated for 2 h in ACSF) were incubated in a solution containing sulfo-NHS-S-S-biotin (1 mg/ml; Pierce) for 30 min at 4°C. Unreacted biotinylation reagent was quenched by two successive 20 min washes in ACSF containing 100 mm glycine (or quenching solution), followed by two washes in ice-cold TBS (50 mm Tris, pH 7.5, 150 mm NaCl). The mPFC tissues were lysed in ice-cold homogenate buffer (50 mm Tris-HCl, 100 mm NaCl, 15 mm sodium pyrophosphate, 50 mm sodium fluoride, 5 mm EGTA, 5 mm EDTA, 1 mm phenylmethylsulfonyl fluoride, 0.5% Triton X-100, 2 mm benzamidine, 60 μg/ml aprotinin, and 60 μg/ml leupeptin) and homogenized. To precipitate biotinylated proteins, supernatants of cell lysate (2 mg of total protein) were mixed with immobilized neutravidin beads (Pierce) and kept in a rotator overnight at 4°C. The beads were washed five times with PBS and then eluted with SDS-PAGE sample buffer by boiling for 15 min. Both the total and biotinylated proteins were resolved by SDS-PAGE, transferred to nitrocellulose membranes, and probed with specific antibodies for glutamate receptor subunit GluR1 (Millipore) and for the GABAA receptor subunits α1 and β2 (Alpha Diagnostic International), as well as β1 and β3 (Santa Cruz Biotechnology). The HRP-tagged secondary antibody was used to visualize the specific signals by chemifluorescence substrate. The scanned digital images were quantified with Adobe Photoshop software. For each experiment, tissue lysates from both prenatal cocaine- and saline-treated rats were loaded to the same gel for determining the relative levels of either biotinylated protein or total protein. Immunoblots of endogenous actin (see Fig. 6A) confirmed the effective isolation of the membrane fraction. The ratio of protein levels in the cocaine versus saline samples were determined in three separate experiments, and averaged ratios of biotinylated surface pools and of total protein expression were calculated as shown in Figure 6B.
Figure 6.

Prenatal cocaine exposure reduced the surface expression of GABAA receptor subunits in the mPFC. A, Example immunoblots of the lysates of mPFC tissues obtained from P27 rats that were treated prenatally with saline or cocaine, showing the level of biotinylated membrane-bound fraction (M) and of the total protein (T) for GluR1, GABAA receptor subunits α1, β2 and β3, and actin. B, Densitometric measurements of the band densities of immunoblots for membrane-bound and total levels of various receptor subunits in the mPFC. The data are presented as the ratio of densities measured for cocaine- versus saline-treated rats in the same immunoblot (n = 3). *p < 0.05, **p < 0.01, t test. Error bars indicate SEM.
Locomotion test.
The rat pups exposed to cocaine or saline in utero were born at the same day and transferred to the same nontreated mother, with the left (saline) or right (cocaine) front foot marked. At P20, the rats were placed in the activity chamber immediately after a single injection of saline (0.9% NaCl; 1 ml kg−1) to habituate the chamber for 20 min for 2 d. At the third day, two rats, each from either the saline or cocaine group, were put into the test chamber at the same time for monitoring locomotor activity in 10 (see Fig. 9A) or 4 (see Fig. 9B) 10 min sessions, respectively, with a video tracking system (MED Associates). After the first session, rats were both given an intraperitoneal injection of cocaine (15 mg kg−1 in 1 ml kg−1 of saline) or subcutaneous injection of apomorphine-HCl (2 mg kg−1 body weight; Sigma-Aldrich) dissolved in 0.1% ascorbate/saline in 1 mg ml−1.
Figure 9.

Prenatal cocaine exposure decreased the sensitivity of the locomotor activity to cocaine and apomorphine in postnatal rats. A, Normalized horizontal distances traveled by the rat were measured during ten 10 min sessions at P22. The horizontal distance traveled of each session was normalized to that of the first session. Cocaine (Coc) challenge was given after the first session. (n = 6–16) B, Average horizontal distances traveled by the P24 rat were measured during four 10 min sessions. Apomorphine (Apo) challenge was given after the first session. (n = 11–13). Significant difference between Sal and Coc of the same session is marked by an asterisk: *p < 0.05, t test. Error bars indicate SEM.
Results
Prenatal cocaine exposure facilitates LTP induction
Whole-cell recordings were made from layer V pyramidal cells of the prelimbic region of mPFC to monitor monosynaptic EPSPs in response to extracellular stimulation of axon fibers in layer II/III (Fig. 1A). We first examined the induction of LTP in these pyramidal neurons of fresh brain slices prepared from the offspring of pregnant rats that were intraperitoneally injected daily with either normal saline or cocaine-containing saline for 7 d (E15–E21). We monitored EPSPs at the membrane potential of −70 mV (under current clamp), which is close to the reversal potential of IPSCs determined by the Cl− concentration of the extracellular and intrapipette solution (Liu et al., 2005). The identity of EPSPs was confirmed by their disappearance after bath application of CNQX (10 μm) and AP5 (50 μm), the antagonists for the AMPA and NMDA subtype of glutamate receptors, respectively. We found that repetitive stimulation of presynaptic inputs with a modified theta burst stimulation (mTBS) protocol (Fig. 1B) induced a persistent increase in the amplitude of EPSPs in layer V pyramidal cells in slices from P16–P42 rats that were exposed to cocaine in utero. As shown by the example recording for a P25 rat (Fig. 1C) and the summary of results from 40 rats in the age range of P16–P42 (Fig. 1D), enhanced EPSPs persisted for at least 40 min after application of mTBS. In contrast, the same mTBS only induced a short-term (∼5 min) potentiation in slices obtained from prenatal saline-treated rats of the age range of P16–P42 (Fig. 1E,F). The synaptic potentiation observed for prenatal cocaine-treated rats is similar to LTP found in many brain areas (Dan and Poo, 2004; Malenka and Bear, 2004). It depended on the activation of NMDA receptors and intracellular Ca2+, since no synaptic potentiation was induced in slices from prenatal cocaine-treated rats in the presence of AP5 or BAPTA (supplemental Figs. S1, S2, available at www.jneurosci.org as supplemental material).
Figure 1.

Prenatal cocaine exposure facilitated LTP induction in layer V pyramidal neurons of rat mPFC. A, Images of an acutely isolated coronal slice of P20 rat brain, showing the extracellular stimulating electrode (S) at layer II/III and the whole-cell recording electrode (R) at layer V at the mPFC region (marked by the white box in the inset). Scale bars: box, 1 mm; 200 μm. B, Stimulation protocol for LTP induction (termed mTBS), consisting of presynaptic activation of 10 bursts (each with 5 pulses at 100 Hz) spaced at 200 ms and repeated three times at 10 s intervals and postsynaptic injection of a depolarizing current pulse (1.5 nA, 40 ms) during each burst, with a 5 ms interval between the onset of presynaptic and postsynaptic stimulation. C, An example of LTP induction in a slice prepared from a P25 rat that was exposed to cocaine in utero for 7 d. The data points represent the amplitude of EPSPs recorded before and after application of mTBS (at the time marked by arrow). Sample traces above represent averages of 10 EPSPs at the marked time (arrowhead). Calibration: 6 mV, 50 ms. D, Summary of data from all experiments similar to that in C, showing normalized EPSP amplitudes before and after LTP induction in slices from P16–P42 rats that were prenatally exposed to cocaine (for 7 d from E15 to E21; n = 40). The EPSP amplitudes over 1 min intervals were normalized by the mean amplitude before induction. Error bars indicate SEM. E, F, Same as C and D except that the results were obtained from slices of prenatal saline-treated rats (F) (n = 21).
In contrast to the effect on LTP induction, prenatal cocaine exposure had no effect on the induction of LTD at these excitatory synapses in P20–P25 rats by a spike timing stimulation protocol, involving repetitive presynaptic stimulation after postsynaptic spiking with 8 ms delay (0.2 Hz, 80 pairs) (Fig. 2). In slices obtained from both prenatal cocaine- and saline-exposed rats, we found that the above paired stimulation caused a similar reduction in the EPSP amplitude (p = 0.79, Kolmogorov–Smirnov test). Thus, the cocaine exposure in utero had selectively modified the susceptibility to the induction of LTP in the mPFC, without apparent effect on LTD induction.
Figure 2.

Prenatal cocaine exposure did not affect LTD induction in layer V pyramidal neurons of the mPFC. A, Left, The protocol for LTD induction consisting of 80 pairs of presynaptic and postsynaptic stimulation at 0.2 Hz, with postsynaptic spiking induced at 8 ms before each presynaptic stimulation. A typical postsynaptic response is shown on the right. Calibration: 20 mV, 5 ms. B, C, Top, Examples of LTD induction in the mPFC of P25 rats. Bottom, Summarized results from all 12 experiments on rats treated prenatally with saline (B) or cocaine (C) for 7 d (E15–E21). Data are presented in the same manner as in Figure 1. Horizontal bar, The time of paired stimulation. Calibration: 4 mV, 80 ms. D, Cumulative percentage plot of the distribution of the LTD magnitude, as determined by the mean amplitude of EPSCs (normalized by the mean baseline value of each cell) at 20–30 min after the onset of presynaptic and postsynaptic stimulation for the data set shown in B and C. Sal and Coc, Prenatally exposed to saline and cocaine, respectively. The two sets of data are not significantly different (p = 0.79; Kolmogorov–Smirnov test).
Delayed appearance of LTP facilitation
When LTP induction was performed on slices obtained from younger rats during the age of P8–P15, we found substantial induction of LTP after mTBS in both prenatal cocaine- and saline-treated groups (Fig. 3A–D). To further examine the magnitude of LTP quantitatively, we defined the LTP magnitude as the ratio of the average EPSP amplitude during the 20–30 min period after mTBS to that during the 5 min control period before mTBS. Examination of the changes in the LTP magnitude during the first 6 postnatal weeks showed that LTP became progressively reduced and essentially disappeared after P16 in prenatal saline-treated rats. In contrast, the LTP exhibited progressive increase over the same 6 week period for prenatal cocaine-treated rats (Fig. 3E). This opposite trend resulted in the prominent difference in synaptic plasticity between the two groups after P16, when developmental transition of the GABA action from excitation to inhibition is completed (Owens et al., 1996; Ganguly et al., 2001; Cancedda et al., 2007). As shown by the cumulative percentage plot of the LTP magnitude for prenatal saline- and cocaine-treated groups over two distinct postnatal periods of P8–P15 and P16–P42 (Fig. 3F), the distribution of LTP magnitude showed no significant difference between the two groups of rats during P8–P15 (p = 0.83, Kolmogorov–Smirnov test), but became significantly different during P16–P42 (p < 0.0001, Kolmogorov–Smirnov test). Thus, facilitation of LTP induction by prenatal cocaine exposure occurred only after P16. All following experiments were performed for rats in the age group of P16–P42 unless indicated otherwise.
Figure 3.

The effect of prenatal cocaine exposure on LTP induction appeared during the late postnatal period (after P16). A, B, An example and summarized results (n = 20) on LTP induction in slices prepared from early postnatal rats (P8–P15) that were exposed to cocaine in utero for 7 d (E15–E21). Error bars indicate SEM. C, D, Same as A and B except that the rats were prenatally exposed to saline (n = 20). E, Summary of the magnitude of LTP induced in mPFC pyramidal neurons on different postnatal days (P8–P42), as indicated by the LTP magnitude, which is obtained by dividing the averaged amplitude of EPSPs at 20–30 min after mTBS with that recorded before mTBS. F, Cumulative percentage plot of the distribution of LTP magnitude for the data set shown in E. *p < 0.0001, Kolmogorov–Smirnov test. Sal and Coc, Prenatally exposed to saline and cocaine, respectively.
LTP facilitation is related to reduced GABAergic inhibition
GABAergic activity normally suppresses LTP induction in mature excitatory synapses (Wigström and Gustafsson, 1983; Davies et al., 1991; Paulsen and Moser, 1998; Meredith et al., 2003), after the developmental transition (between P10 and P14 in rat) of the GABA action from excitation to inhibition. Our finding that significant LTP could be induced after P16 for prenatal cocaine-exposed rats (Fig. 3E,F) suggests that reduced GABAergic inhibition may account for the facilitated LTP induction, similar to that found in rat VTA dopaminergic neurons (Liu et al., 2005) and mPFC pyramidal neurons (Huang et al., 2007) after repeated postnatal cocaine treatment. To determine whether prenatal cocaine exposure also causes the reduction of GABAergic inhibition during the postnatal period, we bath-applied the specific inhibitor of GABAA receptor SR95531 during LTP induction. In brain slices from prenatal saline-treated rats on P25, mTBS failed to induce LTP (Figs. 1E, 4A). However, in the presence of SR95531 (0.5 μm), which reduced the amplitude of GABAA receptor-mediated IPSCs by 60% (supplemental Fig. S3, available at www.jneurosci.org as supplemental material), mTBS induced a robust LTP in mPFC layer V pyramidal neurons of prenatal saline-treated rat (Fig. 4B). This result is consistent with the notion that LTP induction is normally suppressed at mature excitatory synapses because of the presence of strong GABAergic inhibition (Wigström and Gustafsson, 1983; Davies et al., 1991; Paulsen and Moser, 1998; Meredith et al., 2003). In contrast, for prenatal cocaine-exposed rats, we found that the presence of SR95531 (0.5 μm) resulted in rightward shifts of the cumulative percentage distribution of LTP magnitude in both saline- and cocaine-treated rats (Fig. 4D), showing that the reduction of inhibition indeed elevated the magnitude of LTP. Importantly, the increase in the magnitude of LTP caused by SR95531 was much larger in prenatal saline-treated rats than that in cocaine-treated rats (Fig. 4C,D), and the SR95531 treatment led to the same extent of LTP elevation in both groups of rats (p = 0.093, Kolmogorov–Smirnov test). Thus, cocaine exposure in utero had resulted in cellular changes similar to the effect induced by SR95531, suggesting that the reduction of GABAergic inhibition underlies the facilitated LTP found in the mPFC of prenatal cocaine-exposed rats.
Figure 4.

Reduction of GABAA receptor-mediated inhibition facilitated LTP induction. A, LTP could not be induced by mTBS in a slice from a P25 rat prenatally treated with saline. B, Robust LTP was induced in another slice from the same brain as that in A, in the presence of a specific GABAA receptor antagonist SR95531 (“SR”) (0.5 μm). Error bars indicate SEM. C, Summary of results on LTP induction from all experiments similar to those shown in A and B for rats prenatally treated with saline (n = 6) or cocaine (n = 8). D, Cumulative percentage for the distribution of the LTP magnitude for the data set shown in C. *p < 0.0001, Kolmogorov–Smirnov test.
Measurements of GABAergic synaptic transmission
To test whether prenatal cocaine exposure reduces the GABA-mediated inhibition postnatally, we applied weak stimuli of different intensities to the layer II/III of the mPFC and measured the amplitude of IPSCs of the disynaptic inhibition (Fig. 5A), by holding the layer V pyramidal neurons at the resting membrane potential (−54 mV). The input–out relationship was determined by plotting the data as stimulus intensity versus IPSC amplitude (Fig. 5B). The slope of the line with best linear fit of the data for prenatal cocaine-treated rats was significantly lower than that for control saline-treated rats, in either the absence (p = 0.017, Kolmogorov–Smirnov test) or the presence (p = 0.017, Kolmogorov–Smirnov test) of CNQX (10 μm) and AP5 (25 μm) (Fig. 5C). In addition, we found that prenatal cocaine exposure increased the slope of the input–output curve for EPSCs (p = 0.021, Kolmogorov–Smirnov test) (Fig. 5D). These results showed that prenatal cocaine exposure weakens the synaptic strength of inhibitory transmission in the pathway in which LTP was induced, supporting the hypothesis that the reduction of GABAergic inhibition underlies the facilitated LTP found in the mPFC of prenatal cocaine-exposed rats.
In addition to the measurements of IPSCs in the disynaptic responses, we also examined whether the effect of prenatal cocaine exposure is reflected by the maximal IPSC amplitude inducible in these pyramidal neurons. We recorded monosynaptic IPSCs (delay of onset <3 ms) at −20 mV in the presence of CNQX (10 μm) and AP5 (25 μm) in response to extracellular stimulation at layer II/III, with stimulus intensity gradually increased to a level that elicited IPSCs with the maximal amplitude. We found that the mean maximal amplitude of IPSCs in slices from rats exposed to cocaine in utero was significantly smaller than that found for rats exposed to saline (“Sal,” 0.96 ± 0.08 nA, n = 20; “Coc,” 0.63 ± 0.04 nA, n = 21; p < 0.05, t test) (Fig. 5E). This finding on the reduction of monosynaptic response agrees with that found for disynaptic inhibition, indicating the reduction of GABAergic inhibition of layer V mPFC pyramidal neurons caused by prenatal cocaine exposure. Furthermore, we found that the paired-pulse depression of IPSCs (at an interpulse interval of 50 ms) was not significantly different between prenatal saline- and cocaine-treated rats (Sal, 0.72 ± 0.02, n = 40; Coc, 0.76 ± 0.03, n = 49; p = 0.36, t test), suggesting postsynaptic rather than presynaptic changes as the cause of reduced inhibition.
Reduced surface expression of GABAA receptors
To directly assess potential postsynaptic changes associated with reduced GABAergic inhibition, we examined the expression of GABAA receptors in the mPFC tissue from postnatal rats at different stages. The amount of surface pool and the total protein level of various GABAA receptor subunits were determined by Western blotting in combination with the biotinylation method (see Materials and Methods). As shown in Figure 6A, we found that, at P27, the surface expression of GABAA receptor α1, β2, and β3 subunits in prenatal cocaine-treated rats was significantly lower than that found in the prenatal saline-treated controls, whereas the total protein levels of these subunits were the same in both groups. Similar reduction was also observed for β1 subunit (data not shown). In contrast to GABAA receptor subunits, the levels of the surface pool and the total protein of the AMPA receptor subunit GluR1 were the same in both groups (Fig. 6A,B). Furthermore, when we examined the change in GABAA receptor subunit α1 expression at five different postnatal ages, we found significant reduction of the surface pool on P17, P27, P33, and P41, but not on P10 (Fig. 6B). Finally, we found that such reduction of surface GABAA receptor subunits was absent in the primary visual cortex (V1) of the same brain tissue used in the above assays for the mPFC at P10, P17, and P27 (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). Thus, cocaine exposure in utero had selectively reduced the surface expression of GABAA receptors in the mPFC during late postnatal period, leading to reduced GABAergic inhibition of layer V pyramidal neurons.
Enhanced excitatory transmission and neuronal excitability
To address whether prenatal cocaine exposure affects the baseline excitatory glutamatergic transmission, we compared the properties of miniature EPSCs (mEPSCs), as well as the paired-pulse ratio and the AMPA/NMDA ratio of EPSCs in layer V mPFC pyramidal neurons between prenatal saline- and cocaine-treated groups. The mean frequency of mEPSCs in the mPFC of prenatal cocaine-treated rats (recorded in the presence of 0.5 μm tetrodotoxin) was higher than that in prenatal saline-treated rats (Sal, 1.8 ± 0.7 Hz, n = 12; Coc, 3.6 ± 0.7, n = 12; p = 0. 016, t test), but the mean amplitude and decay time of mEPSCs did not show any significant difference (Fig. 7A) (amplitude: Sal, 10.7 ± 0.9 pA, n = 12; Coc, 12.7 ± 1.6 pA, n = 12; p = 0. 056, t test; decay time: Sal, 5.3 ± 0.7 ms, n = 12; Coc, 5.3 ± 0.9 ms, n = 12; p = 0.73, t test). Furthermore, for sequentially elicited EPSC pairs at an interval of 50 ms, the paired-pulse ratio of EPSCs from prenatal cocaine-treated rats was smaller than that in prenatal saline-treated rats, suggesting the increase of the probability of presynaptic glutamate release (Fig. 7B) (Sal, 1.2 ± 0.2, n = 30; Coc, 0.9 ± 0.1, n = 29; p = 0. 034, t test). The postsynaptic effect of prenatal cocaine exposure on glutamatergic synapses was also examined by measuring the AMPA/NMDA ratio of EPSCs in these mPFC pyramidal neurons. As shown in Figure 7C, prenatal cocaine exposure did not alter the AMPA/NMDA ratio (Sal, 2.7 ± 0.5, n = 15; Coc, 3.1 ± 0.7, n = 14; p = 0.12, t test).
Figure 7.
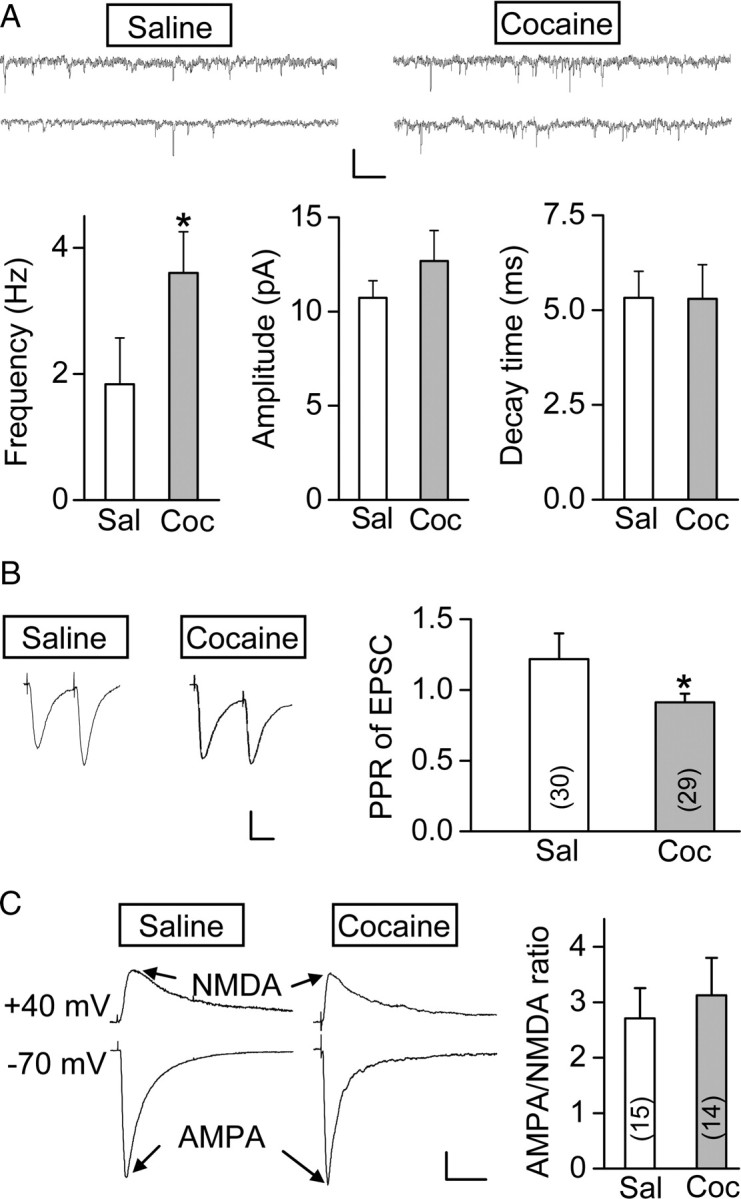
Prenatal cocaine exposure enhanced excitatory glutamatergic transmission of mPFC layer V pyramidal neurons. A, Comparison of the frequency, averaged amplitude, and decay time of mEPSCs from P23–P26 rats prenatally treated with cocaine or saline, with two sample recordings of mEPSCs for each group shown above (n = 12 for each group; *p = 0.016, t test). Calibration: 10 pA, 20 s. B, Left, Sample traces of paired EPSCs (average of 15) in response to two sequential presynaptic stimuli at an interval of 50 ms. Calibration: 100 pA, 25 ms. Right, Summary of PPR (EPSC amplitude of the second divided by the first). Data were obtained from P23–P26 rats prenatally treated with cocaine or saline. Number associated with the histogram refers to the total number of cells recorded. *p = 0.034, t test. C, Left, Sample traces of glutamatergic currents mediated by AMPA and NMDA receptors, recorded at −70 and +40 mV, respectively. Recordings were from P25–P26 rats prenatally treated with saline or cocaine. Calibration: 50 pA, 50 ms. Right, Average AMPA/NMDA ratio obtained by dividing the peak amplitude of AMPA and NMDA receptor-mediated EPSCs. Data were obtained from P24–P26 rats. Number associated with the histogram refers to the total number of cells recorded. Error bars indicate SEM.
The composition of NMDARs in forebrain is known to be developmentally regulated, with the ratio of NR2A to NR2B increased during early postnatal development (for review, see Yashiro and Philpot, 2008). To examine the possibility that prenatal cocaine exposure causes alteration of the postnatal development of NMDARs, we analyzed the decay kinetics of NMDAR-mediated EPSCs, which is known to reflect the composition of the NMDARs—with fast and slow decay for NMDARs containing NR2A and NR2B, respectively. By fitting the decay phase of NMDAR-mediated EPSCs with a double-exponential function, we found no difference in the composition of fast and slow components of the decay phase between prenatal saline- and cocaine-treated rats (the percentages of slow component are 40.3 ± 6.2 and 40.1 ± 5.4% for prenatal saline- and cocaine-treated rats, respectively; n = 14–15; p = 0.98, t test). Therefore, the facilitation of LTP induction by prenatal cocaine exposure is unlikely to involve alteration in the subunit composition of NMDARs. Together, prenatal cocaine exposure appeared to have increased presynaptic release of excitatory synapses in the mPFC without changing postsynaptic properties.
Reduced inhibition and enhanced excitatory transmission of mPFC layer V pyramidal neurons may cause an elevated excitation of these neurons by excitatory synaptic inputs, known as E–S coupling (Lu et al., 2000). Such elevated excitation may in turn facilitate the induction of LTP by mTBS. We have thus examined the E–S coupling by measuring the probability of spike initiation in response to EPSPs of different initial slopes (see Materials and Methods), as elicited by extracellular stimuli of different intensities. As shown by example recordings in Figure 8A, the curve for E–S coupling was left shifted in a P25 rat that was prenatally exposed to cocaine, compared with that of a prenatal saline-exposed P25 rat. The EPSP slope that yields 50% probability of spiking (E50) was determined for each neuron recorded (Fig. 8B). The average value of E50 was significantly different between prenatal cocaine- and saline-treated rats. Moreover, the enhanced E–S coupling caused by cocaine exposure in utero is consistent with a reduced GABAergic inhibition on these pyramidal cells, since blocking GABAergic inhibition with SR95531 (0.5 μm) resulted in an additional leftward shift of the E–S coupling curve to the same level for both groups of rats (Fig. 8A,B). Finally, the elevated excitability of mPFC pyramidal neurons is also reflected by the lower threshold of membrane potential for initiating action potentials (APs) in rats treated with cocaine in utero than those treated with saline (Fig. 8C), and by the increased number of APs initiated by depolarizing current pulses injected into these neurons (Fig. 8D). Thus, prenatal exposure to cocaine had modified the excitability of mPFC layer V pyramidal neurons, an effect that could be mostly attributed to the reduction of GABAergic inhibition (see Discussion).
Figure 8.
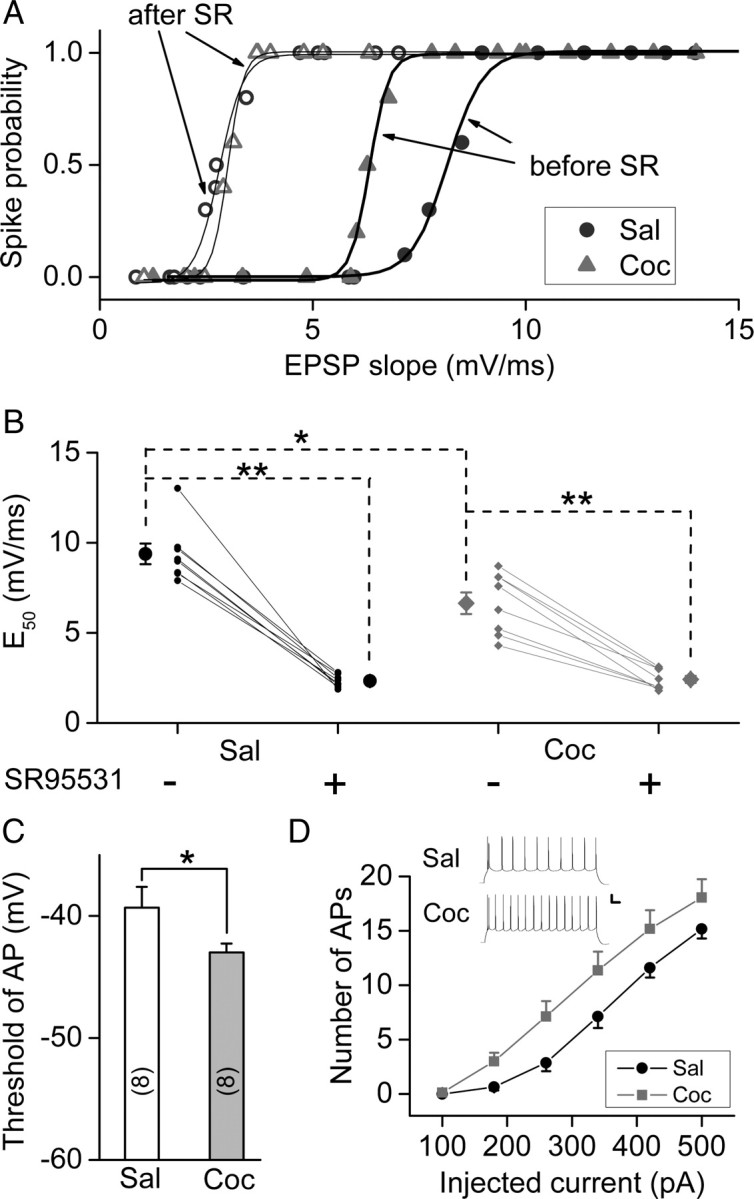
Prenatal cocaine exposure increased the E–S coupling and excitability of mPFC layer V pyramidal neurons. A, E–S coupling studies on rats prenatally exposed to saline (Sal) or cocaine (Coc) in the absence (“before”) and presence (“after”) of SR95531 (SR) (0.5 μm). The curves represent the best fit with the sigmoidal function. B, The values of E50 before and after bath-application of SR95531 (0.5 μm). E50 represents the efficacy of E–S coupling, as defined by the EPSP slope that initiates spiking with a probability of 0.5. Data obtained from the same neuron were connected by the line. Recording were performed in the slices obtained from P24–P27 rats. *p < 0.05, **p < 0.01, t test. C, Summary of the threshold potential for AP initiation. The threshold was determined by the membrane potential at the peak of the maximal subthreshold EPSP. *p < 0.05, t test; n = 9; the same data set as in B. D, Changes of the excitability of mPFC layer V pyramidal neurons resulting from prenatal cocaine exposure, as shown by the number of APs triggered by injection of depolarizing currents of various amplitudes (duration, 800 ms). Data were obtained from the recording on the slices obtained from P19–P27 rats. Inset, Examples of APs elicited by a constant depolarizing current (500 pA, 800 ms) in mPFC neurons from prenatal saline- and cocaine-treated P25 rats (n = 17–24). Calibration: 20 mV, 80 ms. Error bars indicate SEM.
Decreased locomotor sensitivity to cocaine and apomorphine
Layer V mPFC pyramidal neurons provide the main excitatory inputs to VTA dopaminergic neurons (Gabbott et al., 2005), which in turn send dopaminergic projections to the mPFC (for review, see Steketee, 2003, 2005). Thus, higher excitability of these mPFC pyramidal neurons leads to increased excitation of VTA dopaminergic neurons and more dopamine release. This is supported by the finding that the basal dopamine level in mesocorticolimibic area is higher in rat pups exposed to cocaine in utero than prenatal saline-treated control group (Keller and Snyder-Keller, 2000). Given higher basal dopamine level, the prenatal cocaine-exposed rats may become less sensitive to the cocaine challenge when tested postnatally. To examine this idea, we performed locomotion test using P22 rats. The locomotor activity before the intraperitoneal cocaine injection showed no difference between prenatal saline- and cocaine-treated groups during the first 10 min session (supplemental Fig. S5, available at www.jneurosci.org as supplemental material) (p = 0.49, t test). However, as shown in Figure 9A, cocaine-induced increase in locomotor activity was higher in prenatal saline-treated group and lasted for a longer period than that found in the prenatal cocaine-treated group. This result is consistent with that obtained previously by using a mouse model of prenatal cocaine exposure (Crozatier et al., 2003). To further test the hypothesis that the reduced locomotor sensitivity to cocaine in prenatal cocaine-treated rats is related to dopamine system, we examine the effect of apomorphine, a dopamine receptor agonist, on the locomotor activity. Similar to that found for cocaine challenge, prenatal cocaine-treated rats were less sensitive to the apomorphine challenge than prenatal saline-treated rats (Fig. 9B), supporting the hypothesis that the reduced locomotor sensitivity to cocaine is attributable to higher basal dopamine level caused by prenatal cocaine exposure.
Discussion
In this study, we examined the neurophysiological consequence of cocaine exposure of rat in utero and obtained evidence for altered synaptic plasticity in the mPFC of the offspring during the postnatal period. We showed that 7 d repeated prenatal cocaine exposure facilitated activity-induced LTP at excitatory synapses on layer V pyramidal neurons in the mPFC of the rat offspring during the postnatal period P16–P42, whereas no LTP could be induced at these synapses in prenatal saline-treated control rats of the same age unless GABAergic inhibition was reduced. Additional studies showed that this facilitated LTP in prenatal cocaine-exposed rat could be attributed to postnatal reduction of IPSCs and the surface expression of GABAA receptor subunits in the mPFC.
Molecular and cellular changes caused by in utero cocaine exposure
Cocaine exposure in utero results in molecular and cellular changes in several brain areas known to be involved in memory and attention, such as caudate nucleus (Harvey et al., 2001), prefrontal cortex (Morrow et al., 2002, 2007), and hippocampus (Little and Teyler, 1996; Harvey et al., 2001; Morrow et al., 2002, 2007). For example, cocaine exposure in utero results in anatomical changes in the prefrontal cortex of postnatal rats, including a loss of inhibitory projections from parvalbumin-containing GABAergic local circuit neurons to prelimbic pyramidal neurons (Morrow et al., 2005), deficient inhibitory axoaxonic synapses on pyramidal cells (Morrow et al., 2003), and an increased number of spine synapses (Morrow et al., 2007). These structural changes could result in stronger excitation of pyramidal neurons of the prefrontal cortex in response to a given stimulus.
Our study provided strong evidence for another cellular mechanism that could contribute to an elevated excitability of mPFC pyramidal neurons—the reduced GABAergic inhibition. We found a significant reduction in both the amplitude of IPSCs recorded in these pyramidal neurons (Fig. 5) and the surface expression of GABAA receptor subunits in mPFC tissue of prenatal cocaine-treated rats (Fig. 6). This reduced GABAergic inhibition could elevate the excitability of mPFC pyramidal neurons, leading to disruption of physiological cortical rhythms that depend on balanced inhibition and excitation (Shu et al., 2003). In slices from prenatal cocaine-treated rats, an elevated excitation was shown by the higher probability of firing of pyramidal cells in response to membrane depolarizations (Fig. 8), an effect that could mostly be attributed to a reduced GABAergic inhibition and could enhance LTP of excitatory synapses.
We also obtained evidence that prenatal cocaine exposure results in elevated presynaptic release probability at excitatory synapses, as indicated by the increased frequency of mEPSCs and reduced paired-pulse ratio of EPSCs. However, the facilitated LTP appeared to be caused mainly by a reduced GABAergic inhibition rather than enhanced glutamate release. This is based on the following observations: First, blocking GABAergic inhibition with the specific GABAA receptor antagonist SR95531 facilitated LTP induction in slices from prenatal saline-treated control rats (Fig. 4A,B). Second, SR95531 treatments increased the magnitude of LTP of the saline-treated group to the same level as that found for prenatal cocaine-treated rats, and it has no effect on the LTP in the latter group (Fig. 4C,D), indicating that prenatal cocaine exposure had occluded the effect of SR95531 in facilitating LTP. Similarly, SR95531-induced enhancement of EPSP–spike coupling completely occluded the enhanced EPSP–spike coupling found in prenatal cocaine-exposed rats (Fig. 8A). Third, direct measurements of the maximal amplitude of IPSCs in these neurons evoked by extracellular stimulation showed a ∼40% reduction of the total inhibitory input, compared with that found for control rats (Fig. 5E). Fourth, surface biotinylation and Western blotting analysis of mPFC tissues of prenatal saline- or cocaine-treated rats showed that the surface expression of GABAA receptor subunits were significantly reduced in the prenatal cocaine-treated group, with a time course in parallel with that of the LTP facilitation (Fig. 6B). Finally, the effect on LTP was found after P16, when the transition of the GABA action from excitation to inhibition has been completed (Owens et al., 1996; Ganguly et al., 2001; Cancedda et al., 2007) (Fig. 3E). Interestingly, we found that the augmented LTP and the reduced surface expression of GABAA receptor persisted in young rats during P16–P42, suggesting that prenatal cocaine exposure has a long-lasting effect on synaptic plasticity in the mPFC of postnatal rats. This is consistent with previous animal studies showing long-term effects of prenatal cocaine exposure on brain structure and behaviors (for review, see Chae and Covington, 2009). We note that short-term plasticity (within 10 min after LTP induction) was different between prenatal saline- and cocaine-treated rats (Fig. 4C). This may result from the effect of prenatal cocaine exposure on presynaptic glutamate release mechanisms, an aspect not addressed in this study. For assaying the magnitude of LTP, we focused on the mean EPSP amplitude at 20–30 min after LTP induction, after short-term plasticity had subsided. In the postnatal (P30–P40) rabbit hippocampus, prenatal cocaine exposure has been shown to facilitate LTP induction (Little and Teyler, 1996), although whether GABAergic inhibition is altered has not been examined.
Interestingly, the reduction of expression of GABAA receptor α1 subunit caused by prenatal cocaine exposure was observed after P17, but not at P10. There is a progressive increase in the expression of α1 subunit during postnatal development (Fritschy et al., 1994). The change in the expression caused by cocaine exposure in utero may not be detectable at P10 when the normal expression level of α1 subunit is low. Alternatively, the effect of prenatal cocaine exposure takes time to accumulate. The latter idea is supported by the observation that similar reduction in the surface expression of α1 subunit was observed in the mPFC at 1 week (but not on 1 and 3 d) after withdrawal from daily cocaine treatment for 7 d in juvenile rats (our unpublished observation).
Although cocaine exposure in juvenile rats is known to increase mPFC neuronal excitability by modifying the properties of Ca2+ and K+ channels (Dong et al., 2005; Nasif et al., 2005a,b), there is yet no evidence for such changes in rats exposed to cocaine in utero. Based on our results, the elevation of the excitability of the mPFC pyramidal neurons by cocaine exposure in utero may be related to both enhanced baseline excitatory transmission and reduced GABAergic inhibition. However, the latter represents the predominant cause since SR95531 caused a leftward shift of the E–S curve to the same level for both prenatal cocaine- and saline-treated rats (Fig. 8).
Decreased locomotor sensitivity to cocaine
Microdialysis analysis (Keller and Snyder-Keller, 2000) showed that prenatal cocaine treatment caused an elevation of the basal dopamine level in the NAc (of postnatal animals), which receives dopaminergic projections from the VTA. Our finding of the elevated excitability of mPFC layer V pyramidal neurons, which provide the main excitatory inputs to dopaminergic neurons in the VTA (Gabbott et al., 2005), suggests a potential mechanism underlying the higher dopamine release by VTA in these prenatal cocaine-exposed animals. Furthermore, elevated basal dopamine level in these animals may cause desensitization of the reward circuit to postnatal cocaine challenge, since the extent of the dopamine increase in NAc because of postnatal cocaine challenge, when normalized by the basal level, is lower in prenatal cocaine-treated rats than in saline-treated rats (Keller and Snyder-Keller, 2000). As the NAc is the primary brain region responsible for the expression of cocaine sensitization (Robinson and Berridge, 2003; Steketee, 2003), the reduced extent of cocaine-induced dopamine elevation in the NAc of the animals exposed to cocaine in utero may account for their weaker locomotor sensitivity to cocaine (Fig. 9) (Crozatier et al., 2003). Our finding that the dopamine receptor agonist apomorphine exerted similar effect on the locomotor activity as cocaine further supports the involvement of dopamine system.
Cocaine exposure in utero may also alter the sensitivity of the reward system to cocaine through long-term alteration of dopamine receptors. For example, prenatal cocaine exposure results in a sustained impairment in the coupling between D1 dopamine receptors and Gα-protein in cortical neurons (Wang et al., 1995; Friedman et al., 1996; Friedman and Wang, 1998; Jones et al., 2000; Zhen et al., 2001; Stanwood and Levitt, 2007), leading to the reduced D1 receptor functions. Such reduction of D1 receptor functions may also contribute to the reduced locomotor sensitivity to postnatal cocaine challenge.
In conclusion, our results offer new insights into cellular changes caused by repeated cocaine exposure during pregnancy. The finding of the reduced inhibitory input and resultant elevated excitability of mPFC pyramidal neurons may explain the deficits in attention shown by prenatal cocaine-exposed human (Richardson et al., 1996), rats (Garavan et al., 2000), and rabbits (Romano and Harvey, 1996), and support the hypothesis that disruption of excitation/inhibition balance in the prefrontal cortex results in attention deficit hyperactivity disorder (Mattes, 1980; Barkley et al., 1992; Barkley, 1997; Casey et al., 1997). Additional study in identifying the mechanisms underlying these changes may help the development of effective approaches for preventing brain dysfunctions of the fetus caused by drug exposure during pregnancy.
Footnotes
This work was supported by a grant from the National Institutes of Health.
References
- Azuma SD, Chasnoff IJ. Outcome of children prenatally exposed to cocaine and other drugs: a path analysis of three-year data. Pediatrics. 1993;92:396–402. [PubMed] [Google Scholar]
- Barkley RA. Behavioral inhibition, sustained attention, and executive functions: constructing a unifying theory of ADHD. Psychol Bull. 1997;121:65–94. doi: 10.1037/0033-2909.121.1.65. [DOI] [PubMed] [Google Scholar]
- Barkley RA, Grodzinsky G, DuPaul GJ. Frontal lobe functions in attention deficit disorder with and without hyperactivity: a review and research report. J Abnorm Child Psychol. 1992;20:163–188. doi: 10.1007/BF00916547. [DOI] [PubMed] [Google Scholar]
- Broersen LM, Heinsbroek RP, de Bruin JP, Olivier B. Effects of local application of dopaminergic drugs into the medial prefrontal cortex of rats on latent inhibition. Biol Psychiatry. 1996;40:1083–1090. doi: 10.1016/S0006-3223(95)00595-1. [DOI] [PubMed] [Google Scholar]
- Cancedda L, Fiumelli H, Chen K, Poo MM. Excitatory GABA action is essential for morphological maturation of cortical neurons in vivo. J Neurosci. 2007;27:5224–5235. doi: 10.1523/JNEUROSCI.5169-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey BJ, Castellanos FX, Giedd JN, Marsh WL, Hamburger SD, Schubert AB, Vauss YC, Vaituzis AC, Dickstein DP, Sarfatti SE, Rapoport JL. Implication of right frontostriatal circuitry in response inhibition and attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 1997;36:374–383. doi: 10.1097/00004583-199703000-00016. [DOI] [PubMed] [Google Scholar]
- Chae SM, Covington CY. Biobehavioral outcomes in adolescents and young adults prenatally exposed to cocaine: evidence from animal models. Biol Res Nurs. 2009;10:318–330. doi: 10.1177/1099800408330395. [DOI] [PubMed] [Google Scholar]
- Chiriboga CA, Vibbert M, Malouf R, Suarez MS, Abrams EJ, Heagarty MC, Brust JC, Hauser WA. Neurological correlates of fetal cocaine exposure: transient hypertonia of infancy and early childhood. Pediatrics. 1995;96:1070–1077. [PubMed] [Google Scholar]
- Crozatier C, Guerriero RM, Mathieu F, Giros B, Nosten-Bertrand M, Kosofsky BE. Altered cocaine-induced behavioral sensitization in adult mice exposed to cocaine in utero. Brain Res Dev Brain Res. 2003;147:97–105. doi: 10.1016/j.devbrainres.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Dan Y, Poo MM. Spike timing-dependent plasticity of neural circuits. Neuron. 2004;44:23–30. doi: 10.1016/j.neuron.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Davies CH, Starkey SJ, Pozza MF, Collingridge GL. GABA autoreceptors regulate the induction of LTP. Nature. 1991;349:609–611. doi: 10.1038/349609a0. [DOI] [PubMed] [Google Scholar]
- Delaney-Black V, Covington C, Ostrea E, Jr, Romero A, Baker D, Tagle MT, Nordstrom-Klee B, Silvestre MA, Angelilli ML, Hack C, Long J. Prenatal cocaine and neonatal outcome: evaluation of dose-response relationship. Pediatrics. 1996;98:735–740. [PubMed] [Google Scholar]
- Dong Y, Nasif FJ, Tsui JJ, Ju WY, Cooper DC, Hu XT, Malenka RC, White FJ. Cocaine-induced plasticity of intrinsic membrane properties in prefrontal cortex pyramidal neurons: adaptations in potassium currents. J Neurosci. 2005;25:936–940. doi: 10.1523/JNEUROSCI.4715-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman E, Wang HY. Prenatal cocaine exposure alters signal transduction in the brain D1 dopamine receptor system. Ann N Y Acad Sci. 1998;846:238–247. [PubMed] [Google Scholar]
- Friedman E, Yadin E, Wang HY. Effect of prenatal cocaine on dopamine receptor-G protein coupling in mesocortical regions of the rabbit brain. Neuroscience. 1996;70:739–747. doi: 10.1016/s0306-4522(96)83011-9. [DOI] [PubMed] [Google Scholar]
- Fritschy JM, Paysan J, Enna A, Mohler H. Switch in the expression of rat GABAA-receptor subtypes during postnatal development: an immunohistochemical study. J Neurosci. 1994;14:5302–5324. doi: 10.1523/JNEUROSCI.14-09-05302.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbott PL, Warner TA, Jays PR, Salway P, Busby SJ. Prefrontal cortex in the rat: projections to subcortical autonomic, motor, and limbic centers. J Comp Neurol. 2005;492:145–177. doi: 10.1002/cne.20738. [DOI] [PubMed] [Google Scholar]
- Ganguly K, Schinder AF, Wong ST, Poo M. GABA itself promotes the developmental switch of neuronal GABAergic responses from excitation to inhibition. Cell. 2001;105:521–532. doi: 10.1016/s0092-8674(01)00341-5. [DOI] [PubMed] [Google Scholar]
- Garavan H, Morgan RE, Mactutus CF, Levitsky DA, Booze RM, Strupp BJ. Prenatal cocaine exposure impairs selective attention: evidence from serial reversal and extradimensional shift tasks. Behav Neurosci. 2000;114:725–738. [PubMed] [Google Scholar]
- Harvey JA, Romano AG, Gabriel M, Simansky KJ, Du W, Aloyo VJ, Friedman E. Effects of prenatal exposure to cocaine on the developing brain: anatomical, chemical, physiological and behavioral consequences. Neurotox Res. 2001;3:117–143. doi: 10.1007/BF03033234. [DOI] [PubMed] [Google Scholar]
- Huang CC, Lin HJ, Hsu KS. Repeated cocaine administration promotes long-term potentiation induction in rat medial prefrontal cortex. Cereb Cortex. 2007;17:1877–1888. doi: 10.1093/cercor/bhl096. [DOI] [PubMed] [Google Scholar]
- Jones LB, Stanwood GD, Reinoso BS, Washington RA, Wang HY, Friedman E, Levitt P. In utero cocaine-induced dysfunction of dopamine D1 receptor signaling and abnormal differentiation of cerebral cortical neurons. J Neurosci. 2000;20:4606–4614. doi: 10.1523/JNEUROSCI.20-12-04606.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW, Pierce RC, Cornish J, Sorg BA. A role for sensitization in craving and relapse in cocaine addiction. J Psychopharmacol. 1998;12:49–53. doi: 10.1177/026988119801200107. [DOI] [PubMed] [Google Scholar]
- Keller RW, Jr, Snyder-Keller A. Prenatal cocaine exposure. Ann N Y Acad Sci. 2000;909:217–232. doi: 10.1111/j.1749-6632.2000.tb06684.x. [DOI] [PubMed] [Google Scholar]
- Little JZ, Teyler TJ. Prenatal cocaine exposure leads to enhanced long-term potentiation in region CA1 of hippocampus. Brain Res Dev Brain Res. 1996;92:117–119. doi: 10.1016/0165-3806(95)00187-5. [DOI] [PubMed] [Google Scholar]
- Liu QS, Pu L, Poo MM. Repeated cocaine exposure in vivo facilitates LTP induction in midbrain dopamine neurons. Nature. 2005;437:1027–1031. doi: 10.1038/nature04050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YM, Mansuy IM, Kandel ER, Roder J. Calcineurin-mediated LTD of GABAergic inhibition underlies the increased excitability of CA1 neurons associated with LTP. Neuron. 2000;26:197–205. doi: 10.1016/s0896-6273(00)81150-2. [DOI] [PubMed] [Google Scholar]
- Malanga CJ, Pejchal M, Kosofsky BE. Prenatal exposure to cocaine alters the development of conditioned place-preference to cocaine in adult mice. Pharmacol Biochem Behav. 2007;87:462–471. doi: 10.1016/j.pbb.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, McGehee DS. Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron. 2000;27:349–357. doi: 10.1016/s0896-6273(00)00042-8. [DOI] [PubMed] [Google Scholar]
- Mathias R. NIDA survey provides first national data on drug use during pregnancy. NIDA Notes. 1995;10:6–7. [Google Scholar]
- Mattes JA. The role of frontal lobe dysfunction in childhood hyperkinesis. Compr Psychiatry. 1980;21:358–369. doi: 10.1016/0010-440x(80)90017-6. [DOI] [PubMed] [Google Scholar]
- Meredith RM, Floyer-Lea AM, Paulsen O. Maturation of long-term potentiation induction rules in rodent hippocampus: role of GABAergic inhibition. J Neurosci. 2003;23:11142–11146. doi: 10.1523/JNEUROSCI.23-35-11142.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirochnick M, Frank DA, Cabral H, Turner A, Zuckerman B. Relation between meconium concentration of the cocaine metabolite benzoylecgonine and fetal growth. J Pediatr. 1995;126:636–638. doi: 10.1016/s0022-3476(95)70367-5. [DOI] [PubMed] [Google Scholar]
- Morrow BA, Elsworth JD, Roth RH. Male rats exposed to cocaine in utero demonstrate elevated expression of Fos in the prefrontal cortex in response to environment. Neuropsychopharmacology. 2002;26:275–285. doi: 10.1016/S0893-133X(01)00359-1. [DOI] [PubMed] [Google Scholar]
- Morrow BA, Elsworth JD, Roth RH. Axo-axonic structures in the medial prefrontal cortex of the rat: reduction by prenatal exposure to cocaine. J Neurosci. 2003;23:5227–5234. doi: 10.1523/JNEUROSCI.23-12-05227.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow BA, Elsworth JD, Roth RH. Prenatal exposure to cocaine selectively disrupts the development of parvalbumin containing local circuit neurons in the medial prefrontal cortex of the rat. Synapse. 2005;56:1–11. doi: 10.1002/syn.20121. [DOI] [PubMed] [Google Scholar]
- Morrow BA, Hajszan T, Leranth C, Elsworth JD, Roth RH. Prenatal exposure to cocaine is associated with increased number of spine synapses in rat prelimbic cortex. Synapse. 2007;61:862–865. doi: 10.1002/syn.20430. [DOI] [PubMed] [Google Scholar]
- Nasif FJ, Sidiropoulou K, Hu XT, White FJ. Repeated cocaine administration increases membrane excitability of pyramidal neurons in the rat medial prefrontal cortex. J Pharmacol Exp Ther. 2005a;312:1305–1313. doi: 10.1124/jpet.104.075184. [DOI] [PubMed] [Google Scholar]
- Nasif FJ, Hu XT, White FJ. Repeated cocaine administration increases voltage-sensitive calcium currents in response to membrane depolarization in medial prefrontal cortex pyramidal neurons. J Neurosci. 2005b;25:3674–3679. doi: 10.1523/JNEUROSCI.0010-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nulman I, Rovet J, Altmann D, Bradley C, Einarson T, Koren G. Neurodevelopment of adopted children exposed in utero to cocaine. CMAJ. 1994;151:1591–1597. [PMC free article] [PubMed] [Google Scholar]
- Owens DF, Boyce LH, Davis MB, Kriegstein AR. Excitatory GABA responses in embryonic and neonatal cortical slices demonstrated by gramicidin perforated-patch recordings and calcium imaging. J Neurosci. 1996;16:6414–6423. doi: 10.1523/JNEUROSCI.16-20-06414.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen O, Moser EI. A model of hippocampal memory encoding and retrieval: GABAergic control of synaptic plasticity. Trends Neurosci. 1998;21:273–278. doi: 10.1016/s0166-2236(97)01205-8. [DOI] [PubMed] [Google Scholar]
- Richardson GA, Conroy ML, Day NL. Prenatal cocaine exposure: effects on the development of school-age children. Neurotoxicol Teratol. 1996;18:627–634. doi: 10.1016/s0892-0362(96)00121-3. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC. Addiction. Annu Rev Psychol. 2003;54:25–53. doi: 10.1146/annurev.psych.54.101601.145237. [DOI] [PubMed] [Google Scholar]
- Romano AG, Harvey JA. Prenatal exposure to cocaine disrupts discrimination learning in adult rabbits. Pharmacol Biochem Behav. 1996;53:617–621. doi: 10.1016/0091-3057(95)02061-6. [DOI] [PubMed] [Google Scholar]
- Salisbury AL, Lester BM, Seifer R, Lagasse L, Bauer CR, Shankaran S, Bada H, Wright L, Liu J, Poole K. Prenatal cocaine use and maternal depression: effects on infant neurobehavior. Neurotoxicol Teratol. 2007;29:331–340. doi: 10.1016/j.ntt.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankaran S, Lester BM, Das A, Bauer CR, Bada HS, Lagasse L, Higgins R. Impact of maternal substance use during pregnancy on childhood outcome. Semin Fetal Neonatal Med. 2007;12:143–150. doi: 10.1016/j.siny.2007.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y, Hasenstaub A, McCormick DA. Turning on and off recurrent balanced cortical activity. Nature. 2003;423:288–293. doi: 10.1038/nature01616. [DOI] [PubMed] [Google Scholar]
- Stanwood GD, Levitt P. Prenatal exposure to cocaine produces unique developmental and long-term adaptive changes in dopamine D1 receptor activity and subcellular distribution. J Neurosci. 2007;27:152–157. doi: 10.1523/JNEUROSCI.4591-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steketee JD. Neurotransmitter systems of the medial prefrontal cortex: potential role in sensitization to psychostimulants. Brain Res Brain Res Rev. 2003;41:203–228. doi: 10.1016/s0165-0173(02)00233-3. [DOI] [PubMed] [Google Scholar]
- Steketee JD. Cortical mechanisms of cocaine sensitization. Crit Rev Neurobiol. 2005;17:69–86. doi: 10.1615/critrevneurobiol.v17.i2.20. [DOI] [PubMed] [Google Scholar]
- Thompson BL, Levitt P, Stanwood GD. Prenatal cocaine exposure specifically alters spontaneous alternation behavior. Behav Brain Res. 2005;164:107–116. doi: 10.1016/j.bbr.2005.06.010. [DOI] [PubMed] [Google Scholar]
- Wang HY, Runyan S, Yadin E, Friedman E. Prenatal exposure to cocaine selectively reduces D1 dopamine receptor-mediated activation of striatal Gs proteins. J Pharmacol Exp Ther. 1995;273:492–498. [PubMed] [Google Scholar]
- Wigström H, Gustafsson B. Facilitated induction of hippocampal long-lasting potentiation during blockade of inhibition. Nature. 1983;301:603–604. doi: 10.1038/301603a0. [DOI] [PubMed] [Google Scholar]
- Yashiro K, Philpot BD. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology. 2008;55:1081–1094. doi: 10.1016/j.neuropharm.2008.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen X, Torres C, Wang HY, Friedman E. Prenatal exposure to cocaine disrupts D1A dopamine receptor function via selective inhibition of protein phosphatase 1 pathway in rabbit frontal cortex. J Neurosci. 2001;21:9160–9167. doi: 10.1523/JNEUROSCI.21-23-09160.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuckerman B, Frank DA, Hingson R, Amaro H, Levenson SM, Kayne H, Parker S, Vinci R, Aboagye K, Fried LE, Cabral H, Timperi R, Bauchner H. Effects of maternal marijuana and cocaine use on fetal growth. N Engl J Med. 1989;320:762–768. doi: 10.1056/NEJM198903233201203. [DOI] [PubMed] [Google Scholar]