

Abstract
Mutations in the SPTLC1 subunit of serine palmitoyltransferase (SPT) cause an adult-onset, hereditary sensory, and autonomic neuropathy type I (HSAN1). We previously reported that mice bearing a transgene-expressing mutant SPTLC1 (tgSPTLC1C133W) show a reduction in SPT activity and hyperpathia at 10 months of age. Now analyzed at a later age, we find these mice develop sensory loss with a distal small fiber neuropathy and peripheral myelinopathy. This phenotype is largely reversed when these mice are crossed with transgenic mice overexpressing wild-type SPTLC1 showing that the mutant SPTLC1 protein is not inherently toxic. Simple loss of SPT activity also cannot account for the HSAN1 phenotype, since heterozygous SPTLC1 knock-out mice have reduced SPT activity but are otherwise normal. Rather, the presence of two newly identified, potentially deleterious deoxysphingoid bases in the tgSPTLC1C133W, but not in the wild-type, double-transgenic tgSPTLC1WT + C133W or SPTLC1+/− mice, suggests that the HSAN1 mutations alter amino acid selectivity of the SPT enzyme such that palmitate is condensed with alanine and glycine, in addition to serine. This observation is consistent with the hypothesis that HSAN1 is the result of a gain-of-function mutation in SPTLC1 that leads to accumulation of a toxic metabolite.
Introduction
Hereditary sensory neuropathies are rare disorders characterized by progressive sensory loss predominantly affecting the lower limbs (Auer-Grumbach et al., 2003; Nicholson, 2006). Sensory loss is often preceded by hyperpathia and spontaneous shooting or lancinating pain. As the disease progresses, sensory loss can give rise to ulcers, mutilation of the fingers and toes, and skin and bone infections. Linkage analysis has identified genetic loci responsible for several autosomal dominant inherited neuropathies (Nicholson, 2006). One of these, hereditary sensory and autonomic neuropathy type I (HSAN1), is caused by missense mutations in the SPTLC1 gene encoding a subunit of the enzyme serine palmitoyltransferase (SPT) (Bejaoui et al., 2001; Nicholson et al., 2001).
To date, three SPTLC1 mutations, C133W, C133Y, and V144D, have been conclusively linked to HSAN1. Each mutation reduces the in vitro activity of the enzyme, as measured by the incorporation of labeled serine with SPT. Despite the impaired in vitro activity of the enzyme, lymphoblasts of HSAN1 patients show no alteration in total sphingolipid levels (Dedov et al., 2004). Similarly, transgenic SPTLC1C133W mice demonstrate a 60% reduction in SPT activity but no significant change in total ceramide levels. By 10 months of age, they develop hyperpathia and motor deficits, peripheral myelin thinning, loss of visceral innervation, and indications of neuronal stress within the dorsal root ganglia (McCampbell et al., 2005).
To determine the basis by which mutations in SPTLC1 result in HSAN1, transgenic mice overexpressing wild-type (WT) and C133W SPTLC1 were generated and examined for SPT activity and neurological phenotypes. In addition, WT and C133W transgenic mice were crossed and F1 double-mutant progeny examined to distinguish between the possibility that the C133W mutant protein is inherently toxic or that reduced SPT activity is responsible for the disease. This latter possibility was also examined by generating heterozygous SPTLC1 knock-out mice.
Materials and Methods
Transgene construction and generation of transgenic mice have previously been described (McCampbell et al., 2005). The final transgene construct consisted of the chicken betaactin promoter with cytomegalovirus immediate early gene-enhancer elements, followed by the SPTLC1 cDNA with hemagglutinin (HA), and the rabbit β-globin poly-adenylation signal. Mice were generated with standard techniques in the BL6/C3H background. Presence of the transgene was detected by PCR amplification of genomic DNA extracted with the DNeasy Tissue Extraction kit (Qiagen). Multiplex PCR was completed with primers for the transgene (F, 5′CGAAAAACCATCCTGCTCTC-3′; R, 5′GGACAGACGG TTC CAG TGTT-3′) and an endogenous locus, the ABCD1 gene (F, 5′-GAGGGAGGTGG AAGGAAAGA-3′; R, 5′GAAGG GTTGTTGCTCTGACC-3′). We performed Western blots for protein levels and measured SPT enzyme activity (brain and liver) in microsomal preparations (McCampbell et al., 2005). Due to the high similarity between hamster SPTLC1 mRNAs and mouse SPTLC1 mRNA (∼89% identical) and the existence of KpnI site in both mRNAs at the same location, cutting with KpnI was unable to distinguish PCR products of the hamster transgenes from endogenous mouse SPTLC1 expression. Therefore, a second digestion of the PCR products with PstI was conducted to distinguish the hamster mRNA from mouse mRNA (PstI exists only in hamster, not in mouse SPTLC1 mRNA). Heterozygous SPT knock-out animals were created with the SPTLC1 knock-out cell line AD0062 obtained from the Sanger Institute Gene trap resource. The cell line was knocked down by insertion of a β geo cassette within intron 2 of the target gene and genotyping of the mice performed by RT-PCR for the gene trap vector.
Behavioral and sensory assays.
Motor function was assessed by rotorod analysis in an accelerating rod paradigm. Mice were acclimated to the rotorod apparatus for 3 d. For use in the trial, a mouse was required to remain on a steadily rotating rod for 1 min for three times. Each mouse was given five trials a day for 3 d to successfully meet the criteria. Eight mice per group successfully completed the trial. Latency to fall was measured as the rod speed was increased from 5 to 40 r.p.m. over 5 min. Each mouse was tested in three trials per day over 3 d. The mean latencies to fall were compared on each day by a one-way ANOVA. Mice were tested at 12 months and 14 months of age. Sensory performance was assessed as previously described (Broom et al., 2004). Mechanical and cold allodynia, as well as mechanical and thermal hyperalgesia, were examined. Mice were tested at 12 months and 14 months of age. Differences were tested by Mann–Whitney (Von Frey and Pin Prick) or ANOVA (acetone and hot plate tests).
Histology and electron microscopy.
Mice at 15 months of age were anesthetized, followed by transcardial perfusion with 0.9% saline and then with Bouin's solution. Whole mouse was postfixed for 2 weeks at 4°C in Bouin's solution. Paraffin embedded sections were cut at 4 mm and stained with hematoxylin and eosin, Luxol fast blue or silver stain. For analysis of sciatic nerve, dorsal root ganglion and spinal cord separate transcardial perfusions with dissections were undertaken. Immunohistochemistry was performed on paraffin-embedded (5 μm), free-floating (35 μm), and frozen (20 μm) sections. We used commercially antibodies (iba-1, CD 68, myelin basic protein, glial fibrillary protein). Some antibodies needed high temperature antigen retrieval, such as steaming in 10 mm citrate buffer, pH 6.0 or 20 mm Tris/0.65 mm EDTA/0.0005% Tween 20, pH 9.0. Negative controls with omission of the primary antibodies were performed.
For the free-floating experiments, the tissue was fixed in formalin, washed in two changes of 0.4 m Sorensen's phosphate buffer for 72 h, protected in 20% glycerol overnight, then frozen and cut using a sliding microtome. The sections were incubated with the primary antibodies for 5 d at 4°C. Double-labeled sections for light microscopy were pretreated as above and incubated overnight at 4°C with two non-cross-reacting primary antibodies (iba-1/CD68, iba-1/MBP). Then first secondary antibodies conjugated with alkaline phosphatase were applied. Fast Red was used as chromogen (Roche Applied Sciences) before sections were incubated with second secondary biotinylated antibodies. Sections were imaged using a Leica DMR (Digital Module R) microscope and an Optronix Magnafire digital camera. Whole slides were scanned using an ArtixScan 4000tf (Microtek) scanner and ScanWizard Pro software.
Mice at 15 months of age were perfused with 3% formaldehyde and 3% glutaraldehyde. The distal sciatic nerves were dissected out and postfixed in 2.5% glutaraldehyde overnight. The nerves were osmificated with 1% osmium tetroxide and embedded in an Araldite resin. For axon distribution, 10 random non-overlapping cross-sectional regions were imaged at 1900× magnification of three to four mice from each condition [wild-type, wild-type SPTLC1 overexpressing, mutant SPTLC1C133W, double-transgenic (dTG), and heterozygote SPTLC1 knock-out mice]. In Photoshop, tracings of axons and myelin sheaths were filled to create a solid black area representing the cross-sectional surface area of myelin and axons. Using the particle analysis function in ImageJ, the surface areas of axon and myelin were computed. Axon diameter, total diameter of myelinated axon, myelin thickness, and g-ratios (axon diameter/total axon and myelin diameter) were calculated from surface areas. Axon diameters grouped by size were plotted to show relative size distribution; the SD from mean was calculated between animal averages. The total number of fibers was counted and fiber density calculated. Statistical significance between the four groups was determined by ANOVA.
Umyelinated fiber bundles were imaged at 25,000× magnification in 25 random non-overlapping cross-sectional regions for two to three mice from each condition (wild-type, wild-type SPTLC1 overexpressing, mutant SPTLC1C133W, double-transgenic, and heterozygote SPTLC1 knock-out mice). The total number of unmyelinated axons was counted and axon diameters calculated as described above. Unmyelinated axon diameters were grouped by size and a relative size distribution for each condition plotted, the SD from mean was calculated between animal averages, and statistical significance between the four groups was determined by ANOVA.
Sperm analysis.
Before transcardial perfusion, testes and epididymis were removed and placed in phosphate buffer saline. The caudal epididymis was separated for sperm analysis. Both testes from each animal were separated and weighted in an electronic balance and fixed with Bouin's solution. The sperm suspension was prepared by mincing the caudal epididymis in 1 ml human tubal fluid with 10% FBS. For analysis, the sperm suspension was in culture for 1 h at 5% CO2 in an air incubator. Sperm count was analyzed in a Makler chamber. Motility was assessed by counting the number of motile sperm with forward progression, motile sperm without forward progression, and nonmotile sperm. Fixed testes were embedded in paraffin and stained with hematoxyline and eosin for histology. Statistical significance was calculated with Student's t test.
Lipid analysis.
Lipid content was analyzed in plasma, brain, spinal cord, sciatic nerve, and testes. The tissue was cut into small pieces and homogenized in ice-cold HET buffer (50 mm HEPES pH 8, 1 mm EDTA, 0.2% TX100) using a Potter–Elvehjem glass homogenizer. The protein concentration of the homogenate was determined using a Bradford reagent (BioRad). The lipids of an homogenate equivalent to 80 μg protein were extracted and subjected to acid and base hydrolysis as described previously (Riley et al., 1999). Extracted lipids were solubilized in 56.7% methanol/33.3% ethanol/10% water and derivatized with ortho-phthaldialdehyd (OPA). The lipids were separated on a C18 column and the OPA derivates analyzed by a serial arrangement of a fluorescence detector (HP1046A; Hewlett Packard) followed by a mass spectrometry detector (LCMS-2010A; Shimadzu). The liquid chromatography (LC) system consisted of an LC-10Ai solvent delivery module (Shimadzu), SIL-10ADvp automatic injector (Shimadzu). Chromatographic conditions were AmmAcetate (5 mm): MetOH (17:83) for 25 min followed by a 5 min wash step with 100% MetOH at a flowrate of 300 μl/min. Atmospheric pressure chemical ionization was used for ionization. Non-natural C17 sphingosine (Avanti Polar Lipids) was used as internal standard.
Results
As previously reported, mice transgenic for either the WT or the mutant (C133W) alleles of SPTLC1 (tgSPTLC1WT and tgSPTLC1C133W) showed high expression of the transgenes in liver, pancreas, and brain (Fig. 1A) that significantly exceed endogenous SPTLC1 levels (Fig. 1B) (McCampbell et al., 2005). More importantly, brain microsomal SPT activity was reduced ∼60% in the tgSPTLC1C133W mice but was unaltered in the tgSPTLC1WT mice, an observation suggesting that SPT activity is limited by SPTLC2 (Fig. 1E). Only heterodimers of SPTLC1/2 are functional, and an excess in SPTLC1 alone is not able to make functional homodimers (Hanada, 2003).
Figure 1.

Overexpression of the wild-type SPT1 subunit rescues the phenotype of HSAN1 mice. Western blots of transgene (A) as well as endogenous SPTLC1 (B) expression in liver (L), brain (B), and pancreatic tissue (P) in WT, wild-type SPTLC1 overexpressing (TGWT), mutant SPTLC1C133W (TGC133W) and dTG mice (arrows point to anticipated molecular weight of SPTLC1). An HA tag was introduced to facilitate identification of the transgene. C, Western blots of liver and brain SPTLC1 in heterozygote knock-out mice (+/−) and wild-type (+/+) animals. D, In the liver, brain, and spinal cord of the double-transgenic mice, the level of WT SPTLC1 transcript is twice as high as those for the mutant SPTLC1 transgene. E, SPT activity in brain tissue of transgenic and knock-out mice: SPT of SPTLC1C133W mice (TGC133W) is inhibited but restored in dTG. In brain, SPT is decreased by 25% in heterozygous knock-out mice (+/−). SPTLC1C133W mice (14-month-old) (TGC133W) were significantly slower to react in the hot plate test at 55°C (p < 0.05; F) and less sensitive to mechanical stimuli (von Frey hair thresholds) (p = 0.3; G). In the dTG animals, statistically significant improvements are seen. H, Rotorod performance is impaired in SPTLC1C133W mice (TGC133W) with a trend toward improvement in the double transgenics. Sperm count in mice at 2 months (I) and 15 months (J). The mutant (TGC133W) animals have significantly reduced sperm count compared with WT. This is restored in the dTG mice.
Aging mutant SPTLC1C133W mice (TGC133W) develop a small fiber neuropathy
Initial characterization of the tgSPTLC1C133W mice showed evidence of hyperpathia at 10 months of age (McCampbell et al., 2005). In the present study, we find that when tested at 12 months of age, these mice are no longer hyperpathic (data not shown), and by 14 months, they become hypopathic, in conjunction with the development of a small fiber sensory neuropathy. The functional sensory deficits at 14 months are evident in both mechanical sensitivity (p < 0.05) and hotplate testing (p < 0.04) relative to wild-type mice (Fig. 1F,G). Moreover, at 14 months, the animals also showed deficits in motor function as gauged by rotorod testing (Fig. 1H). While there was a decreased latency to fall on rotarod testing in the wild-type SPTLC1 overexpressing transgenic mice as well, they showed no abnormalities on sensory testing or axon number or size of myelinated and unmyelinated fibers. As McCampbell et al. (2005) previously reported a slight increase in hindlimb activity at 10 months of age, we speculate whether coordination difficulties may contribute to the poor motor performance of these mice.
When killed at 15 months, electron microscopy of sciatic nerves in the tgSPTLCC133W mice revealed a loss of small unmyelinated axons (<0.2 μm in diameter) (Fig. 2A– D). Myelinated axons showed smaller diameters and abnormal Schwann cells consistent with degeneration of the distal sciatic nerve (Fig. 2B,E). This supports the finding of a mixed motor-sensory, large and small fiber neuropathy. The myelin itself showed the unusual feature of numerous indentures on cross-sectional view, and nearby lipid-laden macrophages were prominent (Fig. 3). Sciatic nerves of TGC133W also showed more TNF-α staining associated with the presence of CD68 positive macrophages compared with the wild-type mice. Reactive microglia, as identified by iba1-positive staining, were also found within the dorsal column of the spinal cord (data not shown).
Figure 2.
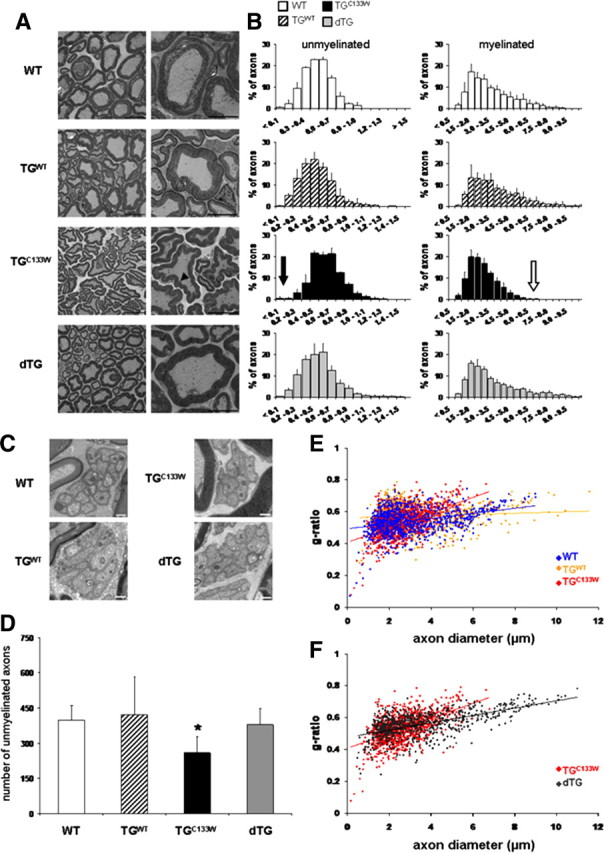
Mutant SPTLC1C133W mice (TGC133W) have smaller axons and myelin abnormalities in the distal sciatic nerve. A, Low- and high-power electron microscopic images show cross-sections of the distal sciatic nerves. Frequent indentures are seen in the myelin sheaths of the mutant SPLTC1 mice (arrowhead) but absent in all other groups; Scale bars: 10 and 5 μm. B, Histogram of the distribution of axon diameter of the distal sciatic nerve in 15-month-old WT, wild-type overexpressing (TGWT), SPTLC1C133W (TGC133W), and dTG mice. Myelinated axons in the SPTLC1C133W mice appear smaller than in wild-type animals (TGC133W mean axon diameter of 2.64 μm vs WT mean of 3.16 μm; p < 0.05; see open arrow). There is a dropout of small unmyelinated axons in the mutant (TGC133W) animals <0.2 μm (see closed arrow in B and EM images in C; scale bar, 50 nm) and a significant reduction in total unmyelinated axons by ∼35% [p < 0.05; D; number of unmyelinated axons in 25 field-of-view (25,000×)]. These findings are reversed in the dTG animals. The g-ratios of mutant (TGC133W) versus WT versus wild-type SPTLC1 (TGWT) overexpressing animals (E) and dTG versus mutant (TGC133W) (F) are plotted relative to the axon diameter with the best fit, linear regression indicated for each of the mice. Myelin thinning occurs in distal sciatic axons of mutant SPTLC1C133W mice and is restored in double-transgenic mice.
Figure 3.
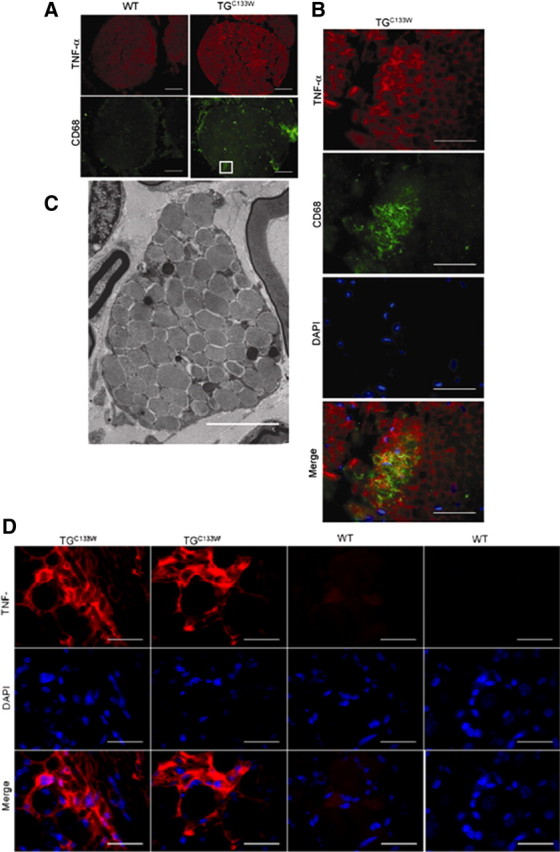
Distal sciatic nerves of mutant SPTLC1C133W mice (TGC133W) reveal a mononuclear phagocytic cell reaction. A, Sciatic nerves of TGC133W show more TNF-α staining compared with the wild-type mice. This is associated with the presence of CD68-positive macrophages; scale bar, 50 μm. B, The boxed area of mutant sciatic nerve is magnified; scale bar, 20 μm. C, Ultrastructural analysis reveals lipid droplets within macrophages in the TGC133W mutant; scale bar, 0.25 μm. D, Dorsal root ganglion neurons at L3 are surrounded by abundant TNF-α in the mutant TGC133W mice.
In addition to the neurologic abnormalities, we noted poor fertility in the mutant tgSPTLC1C133W mice and poor sperm count as early as 2 months of age, which persisted up until 15 months of age (Fig. 1I,J). Sperm motility was reduced by 40% and testes weight by 30% in the mutant compared with the wild-type mice at 2 months of age. Interestingly, at 15 months, the wild-type SPTLC1 overexpressing mice had twice the sperm count compared with the wild type. These findings are consistent with the fact that SPTLC1 is a negative regulator of ABCA1 (Tamehiro et al., 2008) and that ABCA1 overexpressing mice have aspermatogenesis (Selva et al., 2004).
Overexpression of the wild-type SPT1 subunit rescues the phenotype of HSAN1 mice
All the abnormal phenotypes seen in the tgSPTLC1C133W mice were corrected to variable degrees in the dTG mice (Fig. 1). In addition, as seen in Figure 1A, mice overexpressing WT SPTLC1 have higher transgene expression than do the mice overexpressing C133W SPTLC1. We found that in the dTG mice, the level of WT SPTLC1 transcript is twice as high as those for the C133W SPTLC1 transgene (Fig. 1D). This is consistent with our interpretation that preponderance of SPTLC1–SPLTC2 heterodimers are formed with the WT SPTLC1 transgene and not the C133W SPTLC1 transgene, thus contributing to normalization of SPT activity (Fig. 1E). While this result eliminates the possibility that the mutant protein is inherently toxic, it does not prove that simple loss of SPT activity accounts for the HSAN1 pathology.
To directly address whether decreased SPT activity is responsible for the HSAN1 phenotype, the heterozygous knock-out mice were analyzed. Despite the fact that SPTLC1 protein expression (Fig. 1C) and SPT activity (Fig. 1E) were decreased, heterozygous knock-out mice showed no evidence of neuropathy as gauged by testing of temperature sensitivity, mechanical sensitivity, and rotorod at 14 months (data not shown). To ensure that there was no delayed phenotype evolving, we assessed the mice until 22 months of age. Like the dTG mice, detailed behavioral and morphological studies revealed no evidence of a neuropathy (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), and no fertility problems were seen in the heterozygous knock-out mice.
Accumulation of deoxysphingoid bases in C133W mutant transgenic mice
These findings strongly suggest that simple haploinsufficiency cannot explain the HSAN1 phenotype and suggest that the pathogenic mechanism is more complex. In this context, the finding that the deoxysphingoid bases (DSB) are elevated in humans with C133W mutations is of particular interest (Hornemann et al., 2008). We, therefore, investigated whether these novel deoxysphingoid bases were present in the mutant tgSPTLC1C133W mice. Strikingly, these compounds were significantly elevated in plasma, sciatic nerve, and testes of the tgSPTLC1C133W mice as compared with WT mice (Fig. 4). In contrast, unaffected compartments of the nervous system, such as brain and spinal cord, showed little or no accumulation of desoxysphingoid bases. Thus, the presence of desoxysphingoid bases corresponds to the pattern of pathology. Furthermore, the levels of these compounds were also much lower in the dTG mice. This is entirely consistent with the preponderance of WT heterodimers and the lack of a sufficient number of mutant heterodimers to generate toxic levels of these deoxysphingoid bases in the dTG mice. No atypical sphingolipids were detected in heterozygous knock-out mice.
Figure 4.
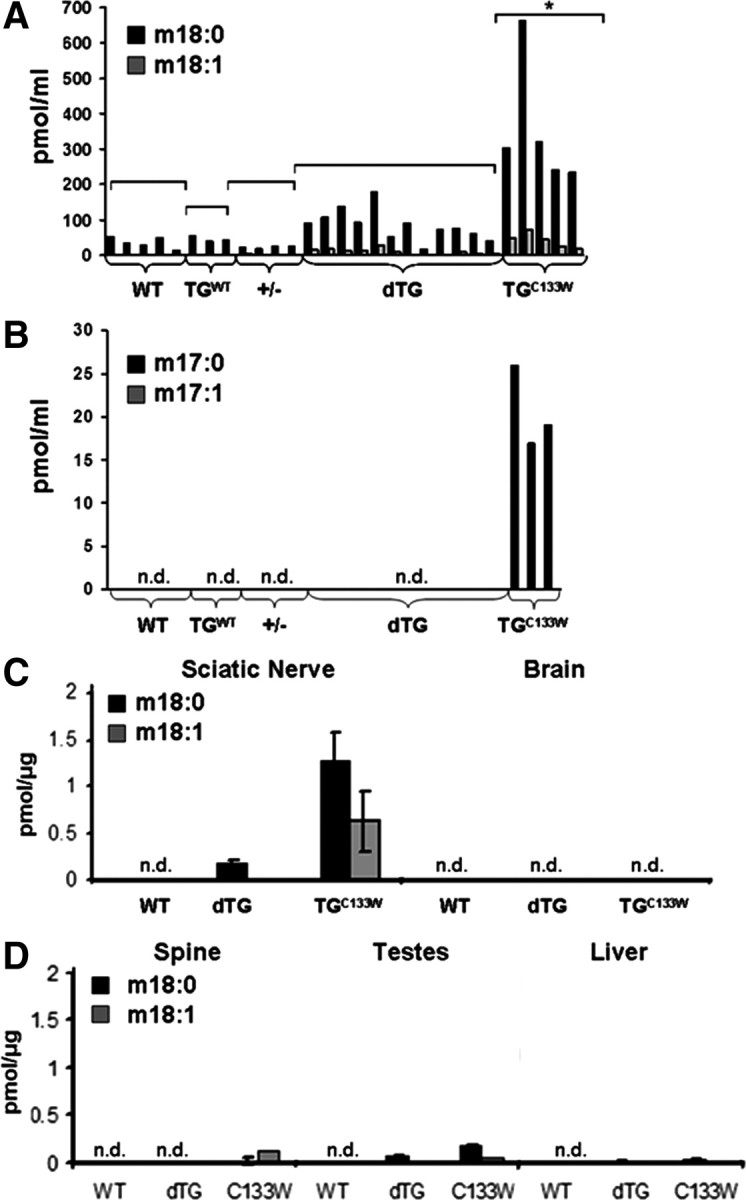
Accumulation of deoxysphingoid bases in C133W mutant transgenic mice. Total lipids from WT, wild-type overexpressing (TGWT), heterozygote knock-out (+/−), SPTLC1C133W mice (TGC133W), and dTG were extracted and subjected to an acid and base hydrolyzes. A, All C133W mutants show plasma levels for m18:0 bases in excess of 200 pmol/ml (p < 0.05, by t test). Some dTG mice have mild accumulation of m18:0 but not beyond this threshold. Levels of m18:0 are <100 pmol/ml in the other conditions. In all mice, levels of 18:0 are dramatically higher than m18:1. B, Similar to humans with the C133W mutation (Hornemann et al., 2008) m17:0 is present in plasma of SPTLC1C133W mutant mice but not other animals. However, levels of m17:1 are not detectable (n.d.). C, D, In SPTLC1C133W mutant mice, high levels of m17:0 are present in the sciatic nerve and lower levels in brain, testes, spinal cord, and liver. Similar to plasma, double-transgenic mice have mild accumulation of m18:0 in sciatic nerve but much reduced compared with the C133W mutant mice.
Discussion
Hereditary sensory and autonomic neuropathy type I (HSAN1) is the most frequent type of hereditary neuropathy that primarily affects sensory neurons (Nicholson, 2006). It is caused by mutations within the SPTLC1 subunit of SPT which catalyzes the pyridoxal 5′-phosphate (PLP)-dependent condensation of l-serine and palmitoyl–coenzyme A, the first and rate-limiting step in the synthesis of sphingolipids (Hanada et al., 1997). We previously reported that transgenic mice expressing the mutant SPTLC1C133W develop hyperpathia and motor deficits, peripheral myelin thinning, and the loss of visceral innervation at the age of 10 months (McCampbell et al., 2005). Here, we report, in follow-up analysis, that these mice are no longer hyperpathic at the age of 12 months and at the age of 14 months, manifest a small fiber sensory neuropathy and deficits in sensory and motor function similar to HSAN1 patients. SPT activity is ∼60% reduced in these animals.
An unexpected finding in our study was the presence of mononuclear phagocytic cells in both distal sciatic nerve and spinal cord of SPTLC1C133W mutant mice. Phagocytosis facilitates Wallerian degeneration of axotomized fibers distal to a nerve lesion and activation of spinal microglia dominates the early glial response in the CNS to peripheral nerve injury (Scholz and Woolf, 2007). The reactive microglial response along sensory nerve pathways, as well as the concurrent proliferation of astrocytes, supports the view that there is selective vulnerability caused by the C133W mutation.
Furthermore, we observe poor fertility, a significantly lower sperm count, impaired sperm mobility, and lower testis weight in the transgenic SPTLC1C133W animals. All these impairments were absent in double-transgenic mice that overexpress both the SPTLC1 wild-type and SPTLC1C133W subunits. These double-transgenic mice have normal SPT activity, no evidence of neuropathy and normal fertility. In addition, heterozygous SPTLC1 knock-out animals, with reduced SPT activity, also show no evidence of neuropathy. Thus, our results exclude the possibility that the C133W mutation creates an inherently toxic protein, as has been reported in other neurodegenerative diseases such as Alzheimer's and Parkinson's disease (Lowe et al., 1988; Mayer et al., 1989). They also eliminate a simple haploinsufficiency as the basis of the disease. Indeed, Hojjati et al. (2005) observed no neurological phenotype with even greater reductions in SPT (M. R. Hojjati, personal communication), and normal individuals have SPT activities that are lower than those in some HSAN1 patients (Dedov et al., 2004). Finally, heterozygous knock-out SPTLC2 mice that also had a 50% reduction in SPT activity were neurologically intact.
The finding that mice and patients expressing the C133W mutant SPTLC1 protein make two unusual DSB provides an attractive alternative hypothesis for the molecular basis of HSAN1. We propose that the HSAN1 mutations in SPTLC1 alter the active site of the heterodimer to allow the mutant SPT to use alanine and glycine as alternative substrates, leading to the generation of 1-deoxy-sphinganine (m18:0) and 1-deoxymethyl-sphinganine (m17:0), respectively. This is consistent with recent homology modeling for the C133W mutant showing a steric clash between W133 and the backbone carbonyl oxygen of the active site lysine residue in SPTLC2 that covalently binds the PLP cofactor (Yard et al., 2007). The PLP cofactor forms the Schiff base with serine, and thus, alterations associated with the HSAN1 mutations could easily alter substrate selectivity of the enzyme. Thus, although it has yet to be proven, it is conceivable that perturbation of geometry of the active site accounts for these changes.
A consequence of the missing C1–OH group in the DSB is the inability of these metabolites to form either complex sphingolipids or to undergo the phosphorylation necessary for degradation. Interestingly, the macrophages identified in peripheral nerve of the mutant mice show prominent lipid droplets (Fig. 3); our analysis reveals that the lipids may at least in part represent sequestered DSB or degenerative myelin products (Fig. 4).
In conclusion, our results provide no evidence that either simple haploinsufficency or an inherent toxicity of the SPTLC1 mutant proteins underlies the pathology of HSAN1. Rather, based on the identification of potentially toxic long-chain bases in mice expressing the mutant protein, we propose that disease-causing mutations result in altered substrate selectivity leading to the accumulation of toxic metabolites. That HSAN1 apparently reflects the acquisition of novel activity of SPT provides an explanation for the very specific mutations that result in this rare disease.
Footnotes
The work was supported by National Institutes of Health Grant K08NS52550 (F.S.E.) and a grant from the Peripheral Neuropathy Society (D.K.). The Day Neuromuscular Research Laboratory receives generous support from the Deater Foundation and the Cecil B. Day Company.
References
- Auer-Grumbach M, De Jonghe P, Verhoeven K, Timmerman V, Wagner K, Hartung HP, Nicholson GA. Autosomal dominant inherited neuropathies with prominent sensory loss and mutilations: a review. Arch Neurol. 2003;60:329–334. doi: 10.1001/archneur.60.3.329. [DOI] [PubMed] [Google Scholar]
- Bejaoui K, Wu C, Scheffler MD, Haan G, Ashby P, Wu L, de Jong P, Brown RH., Jr SPTLC1 is mutated in hereditary sensory neuropathy, type 1. Nat Genet. 2001;27:261–262. doi: 10.1038/85817. [DOI] [PubMed] [Google Scholar]
- Broom DC, Samad TA, Kohno T, Tegeder I, Geisslinger G, Woolf CJ. Cyclooxygenase 2 expression in the spared nerve injury model of neuropathic pain. Neuroscience. 2004;124:891–900. doi: 10.1016/j.neuroscience.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Dedov VN, Dedova IV, Merrill AH, Jr, Nicholson GA. Activity of partially inhibited serine palmitoyltransferase is sufficient for normal sphingolipid metabolism and viability of HSN1 patient cells. Biochim Biophys Acta. 2004;1688:168–175. doi: 10.1016/j.bbadis.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Hanada K. Serine palmitoyltransferase, a key enzyme of sphingolipid metabolism. Biochim Biophys Acta. 2003;1632:16–30. doi: 10.1016/s1388-1981(03)00059-3. [DOI] [PubMed] [Google Scholar]
- Hanada K, Hara T, Nishijima M, Kuge O, Dickson RC, Nagiec MM. A mammalian homolog of the yeast LCB1 encodes a component of serine palmitoyltransferase, the enzyme catalyzing the first step in sphingolipid synthesis. J Biol Chem. 1997;272:32108–32114. doi: 10.1074/jbc.272.51.32108. [DOI] [PubMed] [Google Scholar]
- Hojjati MR, Li Z, Jiang XC. Serine palmitoyl-CoA transferase (SPT) deficiency and sphingolipid levels in mice. Biochim Biophys Acta. 2005;1737:44–51. doi: 10.1016/j.bbalip.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Hornemann T, Penno A, von Eckardstein A. The accumulation of two atypical sphingolipids cause hereditary sensory neuropathy type 1. Chem Phys Lipids. 2008;154S:S62. [Google Scholar]
- Lowe J, Blanchard A, Morrell K, Lennox G, Reynolds L, Billett M, Landon M, Mayer RJ. Ubiquitin is a common factor in intermediate filament inclusion bodies of diverse type in man, including those of Parkinson's disease, Pick's disease, and Alzheimer's disease, as well as Rosenthal fibres in cerebellar astrocytomas, cytoplasmic bodies in muscle, and mallory bodies in alcoholic liver disease. J Pathol. 1988;155:9–15. doi: 10.1002/path.1711550105. [DOI] [PubMed] [Google Scholar]
- Mayer RJ, Lowe J, Lennox G, Doherty F, Landon M. Intermediate filaments and ubiquitin: a new thread in the understanding of chronic neurodegenerative diseases. Prog Clin Biol Res. 1989;317:809–818. [PubMed] [Google Scholar]
- McCampbell A, Truong D, Broom DC, Allchorne A, Gable K, Cutler RG, Mattson MP, Woolf CJ, Frosch MP, Harmon JM, Dunn TM, Brown RH., Jr Mutant SPTLC1 dominantly inhibits serine palmitoyltransferase activity in vivo and confers an age-dependent neuropathy. Hum Mol Genet. 2005;14:3507–3521. doi: 10.1093/hmg/ddi380. [DOI] [PubMed] [Google Scholar]
- Nicholson GA. The dominantly inherited motor and sensory neuropathies: clinical and molecular advances. Muscle Nerve. 2006;33:589–597. doi: 10.1002/mus.20477. [DOI] [PubMed] [Google Scholar]
- Nicholson GA, Dawkins JL, Blair IP, Auer-Grumbach M, Brahmbhatt SB, Hulme DJ. Hereditary sensory neuropathy type I: haplotype analysis shows founders in southern England and Europe. Am J Hum Genet. 2001;69:655–659. doi: 10.1086/323252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley RT, Norred WP, Wang E, Merrill AH. Alteration in sphingolipid metabolism: bioassays for fumonisin- and ISP-I-like activity in tissues, cells and other matrices. Nat Toxins. 1999;7:407–414. doi: 10.1002/1522-7189(199911/12)7:6<407::aid-nt84>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci. 2007;10:1361–1368. doi: 10.1038/nn1992. [DOI] [PubMed] [Google Scholar]
- Selva DM, Hirsch-Reinshagen V, Burgess B, Zhou S, Chan J, McIsaac S, Hayden MR, Hammond GL, Vogl AW, Wellington CL. The ATP-binding cassette transporter 1 mediates lipid efflux from Sertoli cells and influences male fertility. J Lipid Res. 2004;45:1040–1050. doi: 10.1194/jlr.M400007-JLR200. [DOI] [PubMed] [Google Scholar]
- Tamehiro N, Zhou S, Okuhira K, Benita Y, Brown CE, Zhuang DZ, Latz E, Hornemann T, von Eckardstein A, Xavier RJ, Freeman MW, Fitzgerald ML. SPTLC1 binds ABCA1 to negatively regulate trafficking and cholesterol efflux activity of the transporter. Biochemistry. 2008;47:6138–6147. doi: 10.1021/bi800182t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yard BA, Carter LG, Johnson KA, Overton IM, Dorward M, Liu H, McMahon SA, Oke M, Puech D, Barton GJ, Naismith JH, Campopiano DJ. The structure of serine palmitoyltransferase; gateway to sphingolipid biosynthesis. J Mol Biol. 2007;370:870–886. doi: 10.1016/j.jmb.2007.04.086. [DOI] [PubMed] [Google Scholar]