

Abstract
Brain-derived neurotrophic factor (BDNF) is essential for neuronal survival and differentiation during development and for synaptic function and plasticity in the mature brain. BDNF-containing vesicles are widely distributed and bidirectionally transported in neurons, and secreted BDNF can act on both presynaptic and postsynaptic cells. Activity-dependent BDNF secretion from neuronal cultures has been reported, but it remains unknown where the primary site of BDNF secretion is and whether neuronal activity can trigger BDNF secretion from axons and dendrites with equal efficacy. Using BDNF fused with pH-sensitive green fluorescent protein to visualize BDNF secretion, we found that BDNF-containing vesicles exhibited markedly different properties of activity-dependent exocytic fusion at the axon and dendrite of cultured hippocampal neurons. Brief spiking activity triggered a transient fusion pore opening, followed by immediate retrieval of vesicles without dilation of the fusion pore, resulting in very little BDNF secretion at the axon. On the contrary, the same brief spiking activity induced “full-collapse” vesicle fusion and substantial BDNF secretion at the dendrite. However, full vesicular fusion with BDNF secretion could occur at the axon when the neuron was stimulated by prolonged high-frequency activity, a condition neurons may encounter during epileptic discharge. Thus, activity-dependent axonal secretion of BDNF is highly restricted as a result of incomplete fusion of BDNF-containing vesicles, and normal neural activity induces BDNF secretion from dendrites, consistent with the BDNF function as a retrograde factor. Our study also revealed a novel mechanism by which differential exocytosis of BDNF-containing vesicles may regulate BDNF–TrkB signaling between connected neurons.
Introduction
Brain-derived neurotrophic factor (BDNF), a member of neurotrophin family of proteins, is required for neuronal survival and differentiation during development (Leibrock et al., 1989) and for synaptic function and plasticity in the mature brain (McAllister et al., 1999; Poo, 2001). Previous reports have shown that BDNF may act on both presynaptic and postsynaptic cells (Gärtner et al., 2006). Exposure to BDNF increased the frequency but not the amplitude of miniature excitatory synaptic currents (Lohof et al., 1993; Carmignoto et al., 1997). Expression of a dominant-negative TrkB receptor in the presynaptic but not postsynaptic neuron in hippocampal cell cultures blocked BDNF-induced synaptic modifications (Li et al., 1998). Localized application of BDNF induced localized Ca2+ elevation in the presynaptic axon of cultured spinal neurons (Zhang and Poo, 2002) and in dendrites and spines (Kovalchuk et al., 2002; Lang et al., 2007).
Neurotrophins can be transported in the axon in both anterograde and retrograde directions, as demonstrated by radioactive tracer experiments (Hendry et al., 1974; DiStefano et al., 1992; von Bartheld et al., 1996). Bidirectional movements of packets of green fluorescent protein (GFP)-tagged BDNF were also observed along neuritic processes of cultured cortical neurons (Kohara et al., 2001). These findings suggest that secretion and uptake of BDNF occur at both axons and dendrites, either constitutively or in response to neural activity. Regulated secretion of BDNF from the dendrite is of particular functional importance, because a single amino acid substitution (val66met) in the pro-region of BDNF obliterates dendritic trafficking and regulated secretion (Egan et al., 2003) and results in impaired hippocampus-dependent short-term memory (Egan et al., 2003) or anxiety-related behavior (Chen et al., 2006). Activity-induced neurotrophin secretion from cultured neurons has been demonstrated by the presence of increased level of extracellular neurotrophins in response to membrane depolarization (Blöchl and Thoenen, 1995) and high-frequency neuronal spiking (Balkowiec and Katz, 2000; Hartmann et al., 2001; Kojima et al., 2001; Gärtner and Staiger, 2002). Using GFP-tagged BDNF, Kolarow et al. (2007) found that K+-induced depolarization of cultured hippocampal neurons resulted in Ca2+-dependent BDNF secretion from both synaptic and extrasynaptic sites along the dendrite. Conversely, the evidence for presynaptic activity-dependent secretion of neurotrophins is indirect. For example, presynaptic expression of BDNF is selectively required for a late form of long-term potentiation (LTP) in CA3–CA1 hippocampal synapses that could be induced only by high-frequency stimulation (Zakharenko et al., 2003). It is thus of particular interest to determine whether different patterns of presynaptic and postsynaptic activities are required to trigger axonal versus dendritic secretion of neurotrophins, allowing their activity-dependent anterograde and retrograde actions under different physiological conditions. In the present study, we have examined activity-induced BDNF secretion from cultured hippocampal neurons by expressing BDNF tagged with either enhanced GFP (EGFP) or pH-sensitive GFP (pHluorin). Using wide-field epifluorescence and total internal reflection fluorescence (TIRF) microscopy, we found that, under brief spiking activities, full vesicular fusion and BDNF secretion predominantly occur at the dendrite rather than the axon, making the postsynaptic cell the main source of secreted BDNF.
Materials and Methods
Cell culture and transfection.
Low-density cultures of dissociated embryonic hippocampal neurons were prepared as described previously (Shen et al., 2006). Briefly, hippocampi were removed from embryonic rats (embryonic days 18–20) of a Sprague Dawley or Wistar strain and treated with trypsin for 10 min at 37°C, followed by gentle trituration. The dissociated cells were plated at a density of 15,000–25,000/cm2 on poly-l-lysine-coated glass coverslips. The culturing medium was Neurobasal (Invitrogen) supplemented with B-27 (Invitrogen) and glutamine (Invitrogen). We used pyramidal neurons on 14–21 d in vitro (DIV). Cells were transfected on 5 DIV using Lipofectamine2000 (Invitrogen) in Opti-MEM 1 medium (Invitrogen) according to the instructions of the manufacturer. We used a Gal4–UAS bipartite system to drive the expression of the BDNF probes, either alone or together with the axon-targeting synaptic vesicle (SV) protein synaptophysin tagged with the red fluorescent protein mCherry (Syp–Cherry). The ∼3 kb genomic fragment from the zebrafish HuC putative promoter was used to drive the expression of Gal4 activator protein, which in turn binds to the upstream activation sequence (UAS) and induces the expression of the following proteins: mouse BDNF–EGFP, mouse BDNF–pHluorin, rat Syp–Cherry. The cDNA for mouse BDNF encodes precursor pre-proBDNF. The cDNA for BDNF–EGFP was kindly provided by Dr. M. Kojima (National Institute of Advanced Industrial Science and Technology, Osaka, Japan). We replaced EGFP in the BDNF–EGFP construct with superecliptic pHluorin (kindly provided by Dr. G. Miesenboeck, Yale University, New Haven, CT) to generate the UAS–BDNF–pHluorin construct.
Immunoprecipitation and Western blotting.
Cell lysates and conditioned media were prepared from transfected COS-7, HEK293, or 28 DIV hippocampal cultures. Transfection was done with Lipofectamine for COS-7 and HEK293 cells and with Nucleofector (Lonza) for cultured neurons. Immunoprecipitation of pHluorin-tagged proteins in the conditioned media was performed by using custom-made polyclonal anti-GFP antibody. The proteins in the lysates or media were separated by SDS-PAGE, blotted, and probed with polyclonal anti-BDNF (N-20; Santa Cruz Biotechnology) or monoclonal anti-GFP antibody (MBL International). To detect TrkB phosphorylation, we treated cultured cortical neurons at 19 DIV for 5 min with the following factors: the conditioned media collected from COS-7 cells transfected with either EGFP or BDNF–pHluorin, and recombinant human BDNF (Sigma). The cells were lysed and processed for Western blot using a polyclonal antibody for phospho-Trks (Tyr490; Cell Signaling Technology) and anti-pan-Trk antibody (Santa Cruz Biotechnology). Lysates from BDNF knock-out mouse brain (kindly provided by Dr. M. Kojima), wild-type mouse brain, and cultured rat hippocampal neurons were blotted, together with the cell lysates of HEK293 cells expressing BDNF–FLAG–6xHis and recombinant human BDNF as positive controls and probed with the polyclonal anti-BDNF antibody (N-20; Santa Cruz Biotechnology). The blot was reprobed with anti-actin monoclonal antibody (BD Biosciences) to validate the amount of loaded protein. Knockdown of BDNF was performed using a BlockIt miRNA kit (Invitrogen) with the following target sequences: TGAGCGTGTGTGACAGTATTA for miR-BDNF1079, CCAAGTGTAATCCCATGGGTT for miR-BDNF1217, and AGGATAGACACTTCCTGTGTA for miR-BDNF1360. The microRNA against β-galactosidase (lacZ) was used as a negative control.
Immunofluorescence labeling and quantitative analyses.
Cells were fixed with 4% paraformaldehyde and permeabilized with 1% Triton X-100. After being blocked with 10% bovine serum albumin, cells were incubated with primary antibodies for 2 h at room temperature. The primary antibodies used were as follows: polyclonal anti-BDNF antibodies (Santa Cruz Biotechnology or Dr. Katoh-Semba of RIKEN Brain Science Institute, Saitama, Japan) (Katoh-Semba et al., 2001), monoclonal (Millipore Bioscience Research Reagents) or polyclonal (Invitrogen) anti-EGFP (for EGFP and pHluorin), anti-synaptophysin (Millipore Bioscience Research Reagents), anti-PSD-95 (Millipore Bioscience Research Reagents), anti-Smi-312 (Covance), anti-microtubule-associated protein 2 (MAP2) (Novus), and anti-secretogranin II (SecII) (BioDesign). The preparation was washed with PBS and incubated with secondary antibodies conjugated with Alexa 488, 546, or 647 (Invitrogen) at room temperature. The cells were finally washed with PBS and imaged with confocal microscope (Leica) equipped with a 40× [numerical aperture (NA) 1.0] or 63× (NA 1.33) oil-immersion objective.
Because extracellular field stimulation excited all neurons on the coverslip simultaneously, as confirmed by Ca2+ imaging, we were able to determine whether activity-induced changes in BDNF–pHluorin fluorescence of transfected neurons faithfully represent changes in endogenous BDNF in nontransfected neurons on the same coverslip. To compare the immunofluorescence intensities of BDNF puncta between control and stimulated neurons, green fluorescent microspheres (diameter, 0.2 μm; Duke Scientific) were added to the samples after immunostaining and were excited simultaneously with BDNF puncta labeled with Alexa 488 using the argon laser. We normalized the fluorescence intensities of each BDNF punctum by the average fluorescence intensity of adjacent microsphere monitored within the same image (Sugiyama et al., 2005).
Electrical stimulation and time-lapse imaging of BDNF probes.
Coverslips with transfected cells were loaded into a custom-made field stimulation chamber and mounted on the stage of a Nikon E600FN microscope with 40× (NA 0.8) or 60× (NA 1.0) water-immersion objectives. Cells were perfused at 1 ml/min with a normal extracellular solution (in mm: 119 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 30 glucose, and 25 HEPES) warmed at ∼35°C using a temperature controller (TC-344B; Warner Instruments). Cells were illuminated with a 100 W mercury arc lamp with neutral density filter attenuation. The filter set for pHluorin comprised a 470AF40 excitation filter, a 495DRLP dichroic mirror, and a 525AF50 emission filter (Chroma Technology). The filter set consisting of a 560AF40 excitation filter, a 595DRLP dichroic mirror, and a 630AF60 emission filter (Chroma Technology) was used for Syp–Cherry fluorescence. Time-lapse images were acquired (acquisition time, 500 ms) with a 12-bit cooled CCD camera (RetigaEx; Qimaging) at 1 Hz. Extracellular field stimulation was performed via two parallel platinum wires, yielding fields of 10–15 V/cm. Borosilicate glass pipettes (Kimble) filled with an extracellular solution were used for loose-patch stimulation of the soma. Typically, application of 700 mV was enough to elicit action potentials in the patched neurons. Image acquisition and electrical stimulation were synchronized by Master-8 (A.M.P.I.). Theta burst stimulation (TBS) comprised 12 trains at 5 s intervals, each train consisting of five 5 Hz bursts of five 1 ms pulses at 100 Hz. For 3 min TBS, we increased the number of trains to 36. The effectiveness of stimulation was assessed by fluorescence Ca2+ imaging. Hippocampal neurons were incubated in an extracellular solution containing 5 μm Fluo4-AM (Invitrogen) for 30 min and then washed for 30 min. Time-lapse images were acquired with either a confocal or wide-field microscope during stimulation delivered as above.
Total internal reflection fluorescence microscopy.
The method of TIRF microscopy followed those reported previously (Zenisek et al., 2002; Axelrod, 2003). An inverted microscopy (Olympus IX80) equipped with a 100× oil PlanApo TIRF objective (NA 1.45) and a cooled EM-CCD camera (Hamamatsu Photonics) were used to capture total internal reflection fluorescence emitted from BDNF–pHluorin-expressing cultured hippocampal neurons. A 488 nm wavelength beam from a diode laser (Furukawa Electric) was focused off axis onto the back focal plane of the objective. Time-lapse images were acquired with MetaMorph software (Molecular Devices) at 1 Hz (acquisition time, 300–500 ms). Video recording was made at 10 Hz (acquisition time, 100 ms/frame).
Image analysis.
After acquisition, the images were processed for viewing with NIH ImageJ, with subsequent analysis done in MATLAB (MathWorks). We set a region of interest (ROI) with a circle of a fixed size (diameter, 540 nm) over all the BDNF–EGFP or BDNF–pHluorin puncta observed at the axon or dendrite within the entire field of view of the image. To cancel out possible variation of expression levels of BDNF–pHluorin and image acquisition conditions among preparation, we presented data as normalized fluorescence changes (ΔF/F0), in which fluorescence changes (ΔF) at a given time were divided by the baseline fluorescence before stimulation (F0). Puncta with the amplitude of fluorescence reduction ≥2 SDs from the baseline fluorescence were categorized as undergoing full fusion, and those showing a fluorescence increase ≥2 SDs from the baseline fluorescence were categorized as undergoing a transient event. The rest of the events were defined as no fusion.
For automatic identification of puncta (BDNF probes, Syp–Cherry, SecII), single frames or stacks of images were thresholded and segmented by the maximum entropy algorithm to define ROIs. This was done by using Nucleus Counter or Object Counter 3D plug-ins for NIH ImageJ. For the colocalization analyses of BDNF–pHluorin puncta and SecII puncta, we performed object-based colocalization analysis as described previously (Bolte and Cordelières, 2006). Briefly, pixel noises in confocal z-stack images of BDNF–pHluorin and SecII immunofluorescence were removed by a Gaussian filter. BDNF–pHluorin puncta were identified as centroids based on their fluorescence intensity, and SecII puncta were identified as particles. The degree of colocalization was calculated as the percentage of the BDNF–pHluorin centroids colocalized with SecII particles.
Solutions and pharmacological reagents.
An acidic solution with a final pH of 5.2 was prepared by replacing HEPES with 2-[N-morpholino]ethane sulfonic (MES) (Sigma) acid (pKa of 6.1). Ammonium chloride solution was prepared by substituting 50 mm NaCl in the normal extracellular solution with NH4Cl (Sigma) with a final pH of 7.4. All other components in the saline remained unchanged. 6-Cyano-7-nitroquinoxaline-2,3-dione (CNQX) and (2R)-amino-5-phosphonopentanoate (APV) were purchased from Tocris Bioscience. K252a (9S, 10R, 12R)-2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-9, 12-epoxy-1H-diindolo[1,2,3-fg:3`,2`,1`-kl]pyrrolo[3,4-i][1,6] benzodiazocine-10-carboxylic acid methyl ester, tetanus toxin (TeNT), nimodipine, and CdCl2 were from Sigma. Tetanus toxin (whole protein) was reconstituted in 1 m KH2PO4 and added to perfusion solution containing 0.1% BSA. Bafilomycin A1 was purchased from Calbiochem. Recombinant human TrkB-Fc chimera (R & D Systems) was added to the extracellular solution containing 0.1% BSA. Dynasore was kindly provided by Dr. T. Kirchhausen (The CBR Institute for Biomedical Research, Boston, MA) or purchased from Sigma.
Statistical analyses.
To decide the statistical test for the comparison between two datasets, we first examined whether the data in each set are normally distributed (Jarque-Bera test). If both datasets showed normal distribution, we used the parametric test (t test); otherwise, we used the nonparametric test (Kolmogorov–Smirnov test). For comparison involving multiple datasets, we performed nonparametric ANOVA (Friedman's test) to examine whether at least one dataset is different from the rest and then further examined the difference between two individual datasets using two-tailed unpaired Student's t test or Kolmogorov–Smirnov test, depending on whether the data are normally distributed.
Results
Expression of endogenous and fluorescently tagged BDNF
In cultured hippocampal neurons, immunostaining studies showed that endogenous BDNF is distributed predominantly in the form of clusters (or puncta) in both dendrites and axons, which were identified as thick processes stained positive for MAP2 and thin processes devoid of MAP2, respectively (Fig. 1A). For studying activity-induced BDNF secretion, we transfected cultures on 6 DIV with a construct expressing BDNF tagged with either EGFP or its pH-sensitive variant pHluorin and examined the cells 9–16 d after transfection. The distribution of BDNF–EGFP or BDNF–pHluorin, as shown by coimmunostaining for EGFP and MAP2, was found to be similar to that of endogenous BDNF stained with a polyclonal antibody to BDNF (N-20) (supplemental Note 1 and Fig. 1, available at www.jneurosci.org as supplemental material), showing punctate patterns in both axons and dendrites (Fig. 1B,C). Neuronal processes that were not stained with MAP2 antibody were further confirmed as axons by costaining with an antibody against the axon-specific microtubule-associated protein Smi-312 (Fig. 1D). Reconstructed images through the axial plane of the axon and dendrite further supported the granular nature of the fluorescent punctate staining (Fig. 1D).
Figure 1.
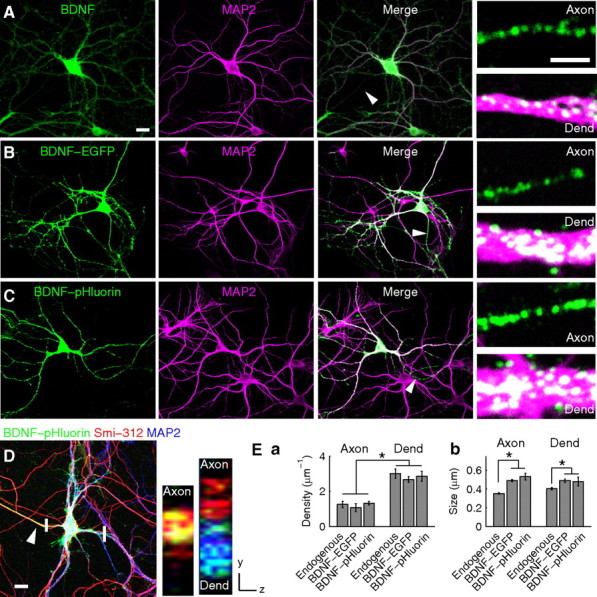
Expression and localization of endogenous BDNF and BDNF–EGFP/pHluorin in cultured hippocampal neurons. A, Coimmunostaining of endogenous BDNF (green) and MAP2 (magenta) in cultured hippocampal neurons. Arrowhead, Axon. Small panels, Higher-magnification images of an axon and a dendrite (Dend). Scale bars: left, 20 μm; small panels, 2 μm. B, C, Neurons expressing either BDNF–EGFP (B) or BDNF–pHluorin (C) were immunostained for EGFP (green) and MAP2 (magenta). Arrowheads, Axons. Small panels, Higher magnification of axons and dendrites. Scale bars are the same as in A. D, A neuron triple immunostained with antibodies to EGFP (green), axon (arrowhead)-specific Smi-312 (red), and MAP2 (blue). Small panels, Reconstructed images along the axial plane (at regions marked by white bars) of a single BDNF–pHluorin-expressing axon (left) and of an axon not expressing BDNF–pHluorin (right) together with a BDNF–pHluorin-expressing dendrite (right). Scale bar, 20 μm. E, Bar graphs showing the density (Ea) and size (Eb) of BDNF-containing fluorescence puncta. *p < 0.01, significant difference between axon and dendrite by two-way ANOVA and t test.
The density of endogenous BDNF puncta in axons and dendrites of control (untransfected) neurons was not significantly different from that found in neurons expressing either BDNF–EGFP or BDNF–pHluorin (p = 0.32) (Fig. 1Ea). In all cases, the density of BDNF puncta in the dendrite was significantly higher than that in the axon. The apparent size of the puncta in neurons expressing BDNF probes was slightly larger than that found in control neurons (Fig. 1Eb), presumably reflecting the elevated amount of BDNF in the puncta attributable to overexpression. The average fold increases in BDNF immunoreactivity attributable to BDNF–pHluorin overexpression were 2.0 ± 0.42 and 1.9 ± 0.38 at the axon and dendrite, respectively (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Above all, overexpression of BDNF–pHluorin increased the amount of BDNF to a similar degree in axons and dendrites. Each BDNF–pHluorin punctum is likely to consist of many BDNF-containing vesicles, because the average size of BDNF–pHluorin puncta (∼490 nm) is significantly larger than that of large dense core vesicles (LDCVs) in neurons (∼160 nm) measured by electron microscopy (Morris et al., 1978).
Coimmunostaining of BDNF–pHluorin-expressing neurons with antibodies against EGFP and the synaptic vesicle protein synaptophysin revealed a subpopulation (28 ± 0.1%; n = 432 puncta, 4 neurons) of BDNF–pHluorin puncta in the dendrite that were juxtaposed to synaptophysin-positive presynaptic boutons (supplemental Fig. 3Aa, available at www.jneurosci.org as supplemental material). Similarly, immunostaining with antibodies to PSD-95 demonstrated that a subpopulation (35 ± 0.1%; n = 422 puncta, 6 neurons) of BDNF–pHluorin puncta in the axon was juxtaposed to postsynaptic PSD-95 puncta (supplemental Fig. 3Ab, available at www.jneurosci.org as supplemental material). Thus, only a minor population of BDNF-containing vesicles is located at synaptic sites in both axon and dendrite, and a large amount of BDNF resides in vesicles at extrasynaptic sites.
After its synthesis in the Golgi apparatus, BDNF is targeted to and stored in LDCVs (Thomas and Davies, 2005). To further investigate the subcellular localization of expressed BDNF–pHluorin, we double-labeled transfected neurons using an antibody against SecII (Huttner et al., 1991; Ozawa and Takata, 1995), a marker for large dense core vesicles in the secretory pathway. We found a high degree of colocalization of BDNF–pHluorin puncta with SecII-positive puncta in axons (88 ± 0.4%; n = 318 puncta, 3 axons) and dendrites (90 ± 1.4%; n = 197 puncta, 3 dendrites), and there was no difference in the degree of colocalization between axon and dendrite (p = 0.38, t test) (supplemental Fig. 3B, available at www.jneurosci.org as supplemental material). Thus, BDNF-containing vesicles represent a population of secretory granules in these cultured hippocampal neurons.
To examine whether BDNF–pHluorin was properly processed and remained biologically active after secretion, we performed Western blot analyses using an anti-BDNF (N-20) antibody on cultured hippocampal neurons. We found that the expressed BDNF probe was efficiently processed into the mature form in the cytoplasm (for details, see supplemental Note 2 and Fig. 4, available at www.jneurosci.org as supplemental material), consistent with previous reports (Pang et al., 2004; Matsumoto et al., 2008). Furthermore, secreted BDNF–pHluorin in the conditioned medium was capable of inducing tyrosine phosphorylation of Trk receptors in cultured cortical cells (supplemental Fig. 4C, available at www.jneurosci.org as supplemental material).
Figure 4.

Partial versus full fusion of BDNF–pHluorin vesicles at axon and dendrite. A, C, Wide-field fluorescence images of an axon expressing BDNF–pHluorin and fluorescent debris from a broken cell nearby (dashed circle, A) or in the same field of view (box, C). Trace below, BDNF–pHluorin puncta fluorescence changes with time (normalized by the basal fluorescence) at a single axon (black) and at a piece of cell debris (blue), before and after field stimulation (10 Hz, 300 pulses) or in response to extracellular perfusion with an MES-buffered acidic solution (pH 5.2; open bars). Numbers refers to the time the images above were collected. B, D, The average trace of the changes in pHluorin fluorescence after field stimulation at time marked as “10 Hz”, showing the sensitivity of the fluorescence decay at the axon (B) or of the tail of fluorescence reduction at the dendrite when the cultures were treated with bafilomycin-A1 (Baf), dynasore (Dyn), and K252a. Dynasore and K252a applied 20 min before and bafilomycin applied immediately before stimulation. Error bars indicate SEM.
BDNF–pHluorin as a probe for BDNF vesicle fusion
To study BDNF secretion from cultured neurons, we used BDNF–pHluorin as a pH-sensitive probe for vesicular exocytosis (Miesenböck et al., 1998). The pHluorin fluorescence was quenched to a considerable degree as a result of the acidity of the vesicle lumen but rapidly increased during the formation of the fusion pore at the plasma membrane as a result of deacidification of the vesicular lumen (Miesenböck et al., 1998; Sankaranarayanan and Ryan, 2001; Granseth et al., 2006). When expressed in cultured hippocampal neurons, BDNF–pHluorin puncta were sparsely distributed in both axon and dendrite (Figs. 2, 3). We found that bath application of NH4Cl solution (50 mm, pH 7.4) (Sankaranarayanan et al., 2000), which deacidifies the vesicular lumen and thereby unquenches BDNF–pHluorin fluorescence, resulted in a marked increase in the fluorescence intensity at each punctum and unveiled a large number of BDNF–pHluorin puncta in both axon and dendrite (Fig. 3). This observation suggests that BDNF–pHluorin was indeed quenched in a majority of vesicles, although vesicular pH was apparently higher in a minority of vesicles. Notably, the amplitude of the change in BDNF–pHluorin fluorescence during exposure to NH4Cl solution (pH 7.4) was larger than that for BDNF–EGFP puncta by sevenfold to ninefold (Fig. 3B), consistent with the lower pKa of EGFP than pHluorin (Sankaranarayanan et al., 2000, their Fig. 2). Thus, BDNF–pHluorin is a more sensitive probe for luminal pH changes than BDNF–EGFP and was used in the present study to monitor the exocytosis of BDNF-containing vesicles.
Figure 2.
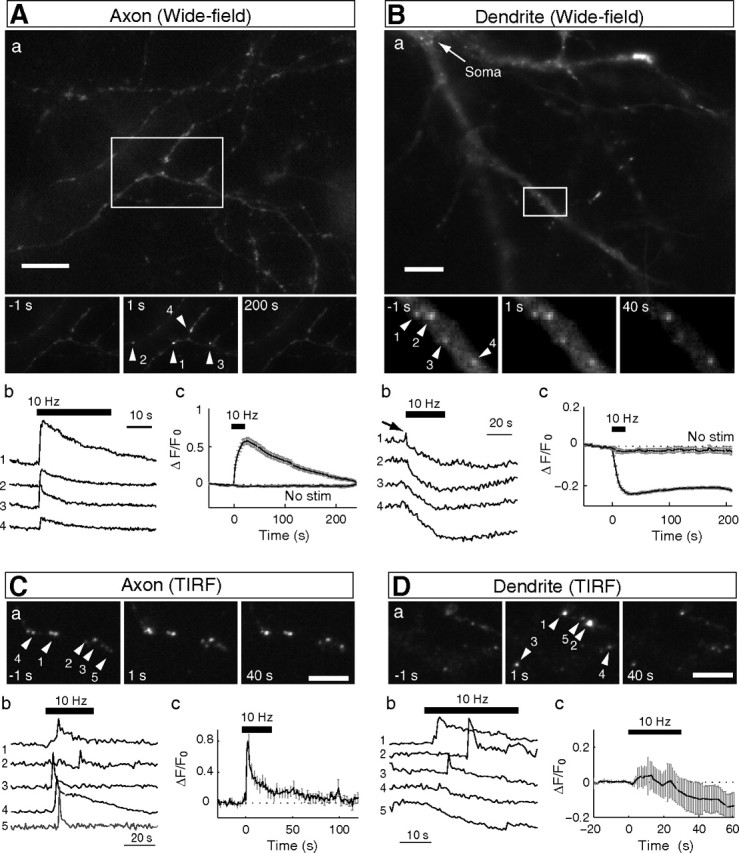
Activity-induced fusion of BDNF-containing vesicles in axon and dendrite. A, B, a, Wide-field fluorescence microscopic images of BDNF puncta at the axon (A) and dendrite (B) of cultured hippocampal neurons expressing BDNF–pHluorin. Insets, Magnified views of BDNF–pHluorin puncta in boxed areas at 1 s before and 1 and 200 s after field stimulation (at 10 Hz for 300 pulses). Scale bar, 20 μm. b, Sample traces depict the fluorescence change (ΔF) with time at individual puncta at the axon (A) and dendrite (B). Numbers refer to the corresponding numbered puncta shown in the images above. c, Fluorescence changes normalized by the basal fluorescence (F0) and the average below traces represent the mean ± SEM pHluorin fluorescence change at the axon (A; n = 594 puncta, 16 cells) and dendrite (B; n = 300 puncta, 7 cells). No Stim, No field stimulation applied, showing insignificant bleaching over the 4 min recording period. C, D, TIRF microscopic images and pHluorin fluorescence changes at axon and dendrite. a, Images of BDNF–pHluorin puncta at 1 s before and 1 and 40 s after field stimulation. b, c, Same as in A and B, except that the fluorescence changes (ΔF) were monitored with TIRF microscopy.
Figure 3.

NH4Cl and acid perfusion of neurons expressing BDNF–EGFP/pHluorin. A, Representative images of the axon and dendrite (Dend) of neurons expressing either BDNF–EGFP or BDNF–pHluorin, after the perfusion of various solutions. Scale bar, 10 μm. B, Time course of the fluorescence changes of BDNF–EGFP or BDNF–pHluorin, averaged over 50 puncta in single neurons, induced by the perfusion with NH4Cl (pH 7.4) or MES-buffered acidic solution (pH 5.2). C, a, Schematic diagram depicting the measured quantities (ΔFNH4Cl and ΔFMES) in perfusion-induced changes in the pHluorin fluorescence. Because pHluorin is completely quenched at pH 5.2 (Sankaranarayanan et al., 2000), ΔFNH4Cl represents the total amount of BDNF–pHluorin in the vesicular compartment that was unquenched by perfusion with NH4Cl. ΔFMES represents the quenched surface fraction (fsur) of BDNF–pHluorin by perfusion with MES-buffered acidic solution (pH 5.2). The surface fraction thus can be calculated as fsur = ΔFMES/(ΔFNH4Cl + ΔFMES). b, Bar graph showing the calculated fraction (mean ± SEM) for BDNF–EGFP and BDNF–pHluorin on the cell surface, from measurements of ΔFNH4Cl and ΔFMES.
Large surface fraction of pHluorin-based probes would artificially lower the amplitude of fluorescence changes associated with fusion events when the changes were normalized by the baseline fluorescence intensity (ΔF/F0) (Sankaranarayanan et al., 2000, their Fig. 5B). We have estimated the fraction of BDNF–pHluorin on the cell surface by a combination of perfusion with the acidic (pH 5.2) and NH4Cl (pH 7.4) solutions (for details, see legend for Fig. 3). The surface fraction of BDNF–pHluorin was found to be small and similar in axon (4.7 ± 1.7%; n = 4) and dendrite (4.8 ± 1.7%; n = 3) (Fig. 3C). Thus, we have used the fractional change in fluorescence (ΔF/F0) to normalize data obtained from different neurons. The computed surface fractions could be underestimated, because some of the surface-bound BDNF–pHluorin might have been washed away by the acid perfusion, which reduces the binding affinity of BDNF (Du et al., 2000).
Figure 5.
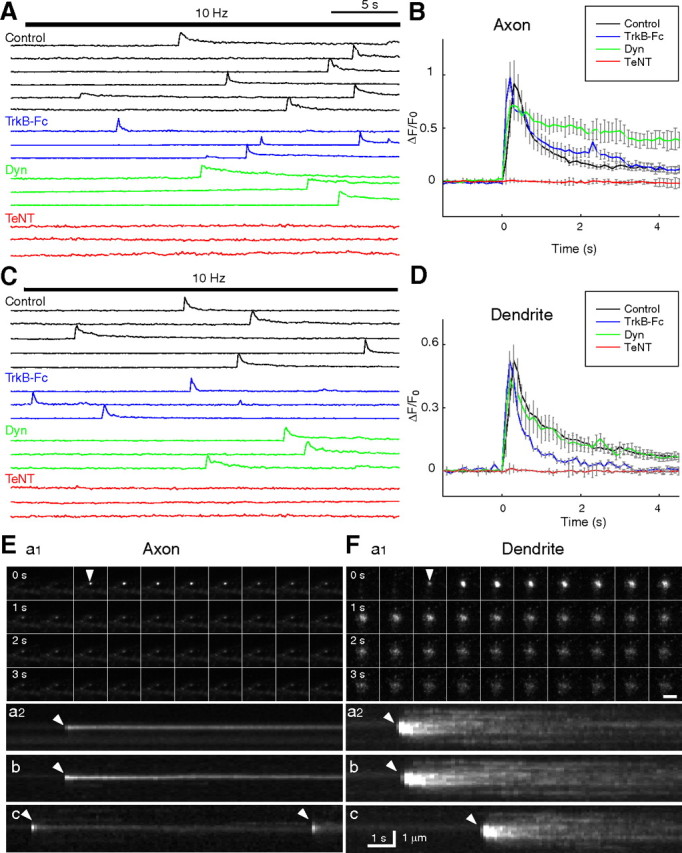
TIRF video recording of the fusion of single BDNF–pHluorin vesicles. A, C, Sample traces of BDNF–pHluorin fluorescence changes (ΔF) measured at the center of each vesicle at the axon (A) and dendrite (C) of control neurons, neurons perfused with TrkB-Fc-containing solution (4 ng/ml), or neurons pretreated with dynasore (Dyn; 80 mm) or TeNT (10 nm). Each line represents a single vesicle. Bar indicates field stimulation at 10 Hz for 300 pulses. B, D, Average traces of BDNF–pHluorin fluorescence transients at axon (B) and dendrite (D) as those shown in A and C, normalized by the basal fluorescence of each puncta (F0) and with the onset time of the transients aligned. E, F, Representative images of single vesicles at the axon (E) and dendrite (F) under 10 Hz (300 pulses) stimulation. a1, Consecutive images of a fusion event at the axon taken at 4 s interval. Scale bar, 2 μm. a2, Pseudo-line scan images for 12 s along the center of the spot for the vesicle shown in a1. b, c, Two other examples of vesicles undergoing fusion events. Repeated opening of the fusion pore was found for the vesicle shown in c. Arrowhead, Opening of fusion pore.
Partial versus full fusion of BDNF–pHluorin-containing vesicles
Activity-induced exocytosis of BDNF–pHluorin-containing vesicles in cultured hippocampal neurons was first examined by monitoring changes in pHluorin puncta fluorescence, using wide-field epifluorescence microscopy at a sampling rate of 1 Hz (Fig. 2A,B). The axon/dendrite identity was determined by the neurite morphology and further confirmed in some cases by post hoc immunostaining with somatodendritic marker MAP2 or by coexpressing the axon-targeting synaptic vesicle protein synaptophysin tagged with the red fluorescent protein mCherry (Syp–Cherry) (supplemental Fig. 5C, available at www.jneurosci.org as supplemental material). During repetitive extracellular field stimulation of the culture (10 Hz, 300 pulses, 1 ms pulse duration), with each pulse eliciting an action potential in the neuron (see Materials and Methods), the fluorescence intensity of BDNF–pHluorin puncta in the axon showed an immediate increase, followed by a slow decay toward the original level (Fig. 2Ab,Ac). The activity-induced transient increase in the puncta fluorescence suggests the formation of fusion pores by BDNF-containing vesicles in the axon, analogous to that observed during synaptic vesicle exocytosis at presynaptic nerve terminals (Gandhi and Stevens, 2003; Harata et al., 2006a). The subsequent decay of fluorescence to a level close to that observed before stimulation suggests no net loss of BDNF–pHluorin within the puncta.
In contrast to that found in the axon, the same field stimulation triggered a gradual decrease in the pHluorin puncta fluorescence in the dendrite (Fig. 2Bb). Two types of fluorescence changes were observed: a monotonic decay followed by a gradual recovery to an intermediate level in most cases, and a monotonic decay to a stable low level in a minority of cases (Fig. 2Bb). The overall reduction of fluorescence (Fig. 2Bc) indicates a substantial release of BDNF–pHluorin from the puncta. Because each punctum consists of many BDNF–pHluorin-containing vesicles (see below), the remaining puncta fluorescence may reflect vesicles that failed to undergo full fusion, whereas the slow fluorescence recovery suggests partial endocytic reuptake of secreted BDNF–pHluorin (see below). Unlike that found in the axon, we observed transient fluorescence elevation before the decay only in a small fraction of puncta. This may be attributable to asynchronous opening of fusion pores superimposed by the reduction of fluorescence associated with BDNF–pHluorin release.
Consistent with a previous report (Adachi et al., 2005), some of the visible puncta exhibited movement (44% in axons, n = 328; 40% in dendrites, n = 272, 4 neurons) and others were immobile during the period of observation (∼5 min). During field stimulation (300 pulses at 10 Hz), most immobile BDNF–pHluorin puncta in the axon showed transient fluorescence increases, whereas most moving puncta did not change their fluorescence (ΔF/F0, 0.61 ± 0.24 vs −0.049 ± 0.084; p < 0.01) (supplemental Fig. 5A,B, available at www.jneurosci.org as supplemental material). Mobile BDNF–pHluorin puncta may represent clusters of BDNF-containing vesicles that were transported along the axon. We found that 95% of identified Syp–Cherry puncta were immobile (n = 329, 3 neurons), suggesting that they represent presynaptic sites. Among all immobile BDNF–pHluorin puncta, puncta that colocalized with Syp–Cherry showed much higher average amplitude of stimulation-induced fluorescence increase than those not colocalized with Syp–Cherry (ΔF/F0, 0.65 ± 0.06 vs 0.063 ± 0.007; p < 0.01) (supplemental Fig. 5D, available at www.jneurosci.org as supplemental material), consistent with the idea that stimulation-induced fusion of BDNF-containing vesicles occurs mostly at presynaptic sites.
To better examine the exocytosis of BDNF–pHluorin vesicles that are docked at the plasma membrane, we used TIRF microscopy, which provides a means to selectively excite fluorophores in the evanescent field above the cover glass (≤100 nm) (Axelrod, 2003), leading to a reduced background fluorescence. That TIRF captures individual vesicle fusion events is supported by the observation that the fluorescence increase at the pHluorin puncta triggered by perfusion with NH4Cl solution (pH 7.4) was similar in amplitude to that induced by the field stimulation (supplemental Fig. 6B, available at www.jneurosci.org as supplemental material). In contrast, under wide-field microscopy, the fluorescence increase at each BDNF–pHluorin punctum caused by NH4Cl perfusion was ∼10-fold of that induced by the same field stimulation (for detailed computation, supplemental Fig. 6A, available at www.jneurosci.org as supplemental material), indicating that each punctum visible by the wide-field microscopy represents a cluster of many BDNF-containing vesicles.
Using TIRF microscopy, we found that the field stimulation (300 pulses at 10 Hz) triggered a transient increase of the BDNF–pHluorin puncta fluorescence in the axon, with a much more rapid rise and an apparent two-phase decay (Fig. 2C). Notably, the delay of onset of fluorescence transients after the stimulus onset was variable, suggesting asynchronous fusion of vesicles triggered by the stimulation. The fluorescence transient averaged over a large number of puncta is shown in Figure 2Cc. Individual fusion events were also observed at the dendrite with TIRF microscopy as fluorescence transients with a viable delay of onset after the stimulus (Fig. 2Db). Unlike that observed by the wide-field microscopy, TIRF microscopy now revealed many events with initial fluorescence increase, followed by a two-phase decay. The average trace showed that the field stimulation triggered an overall reduction of BDNF–pHluorin fluorescence, consistent with substantial BDNF secretion.
Transient partial fusion of BDNF-containing vesicles at the axon
Results described above suggest that stimulation-induced BDNF–pHluorin fluorescence increase at the puncta reflects the deacidification of the vesicular lumen during fusion pore formation. This idea was further tested by rapid perfusion of the cultured neuron with an acidic solution (pH 5.2). Before electrical stimulation, acid perfusion resulted in a slight fluorescence decrease (Fig. 4A), presumably attributable to quenching of surface BDNF–pHluorin fluorescence (Fig. 3). As a control, the same perfusion reversibly quenched the fluorescence of pHluorin-positive cell debris in a nearby region of the imaged field (Fig. 4A, dashed circle). Acid quenching after electrical stimulation, however, resulted in a large fluorescence reduction with a time course similar to that found for the cell debris, consistent with the presence of fusion pores that allowed the extracellular access of the vesicular lumen. The slow decay of BDNF–pHluorin fluorescence after the initial rapid increase depended on the reacidification of vesicles that had closed their fusion pores, because bath application of bafilomycin A1 (1 μm), an inhibitor of the vesicular proton ATPase, before electrical stimulation markedly suppressed the decay of pHluorin fluorescence (Fig. 4B). Consistent with this idea, preventing endocytic closure of the fusion pore by inhibiting dynamin with dynasore (80 μm) (Macia et al., 2006) also abolished the decay (Fig. 4B). Moreover, this endocytic process does not involve the high-affinity BDNF receptor TrkB, because pretreatment of neurons with K252a (200 nm), which inhibits TrkB-mediated endocytosis (Santi et al., 2006), did not affect the fluorescence decay (Fig. 4B). This result argues against the idea that there was full vesicular fusion/collapse at the axon, but subsequent endocytosis allowed full recovery of BDNF–pHluorin attributable to the tight binding of secreted BDNF with TrkB at the cell surface, because K252a would have significantly reduced the amount of endocytosed BDNF–pHluorin.
Full fusion of BDNF-containing vesicles at the dendrite
Field stimulation (10 Hz, 300 pulses) of the cultured hippocampal neurons expressing BDNF–pHluorin resulted in a gradual reduction of pHluorin puncta fluorescence at the dendrite (Fig. 4C,D), reflecting full fusion of BDNF-containing vesicles and BDNF–pHluorin secretion. The plateau fluorescence of the puncta induced by stimulation represents the remaining unfused vesicles within the puncta that were inaccessible to the extracellular space. This was further shown by the perfusion with the acidic solution (pH 5.2), which caused only a small change in the plateau pHluorin fluorescence (Fig. 4C) compared with that for the cell debris (Fig. 4C, box). Therefore, unlike those at the axon, the fusion pore opening led to full fusion of vesicle and substantial BDNF secretion at the dendrite (Fig. 4C). The absence of endocytic closure of fusion pores at the dendrite was further supported by the finding that bafilomycin A1 did not affect the stimulation-induced reduction of pHluorin puncta fluorescence (Fig. 4D).
We observed that a majority of dendritic BDNF–pHluorin puncta exhibited partial fluorescence recovery after the initial stimulation-induced decrement, reaching a plateau level within 5 min (Fig. 2Bb). This recovery of fluorescence suggests the recruitment of additional BDNF–pHluorin vesicles into the puncta after stimulated BDNF–pHluorin release. Because a substantial amount of the secreted neurotrophin is known to be bound to the cell surface (Blöchl and Thoenen, 1995), immediate receptor-mediated endocytic salvage of a portion of secreted BDNF may contribute to the recovery of the depleted pool of BDNF-containing vesicles within the puncta. This idea was further supported by the finding that this partial fluorescence recovery was abolished by bath application of either dynasore or K252a (Fig. 4D).
TIRF recording of single fusion events
Fusion pore opening at BDNF–pHluorin puncta was further examined with TIRF video microscopy at a higher time resolution of 10 Hz. As shown by sample traces of fluorescence intensity changes at individual puncta (Fig. 5A,C) and onset-aligned average traces over a large number of puncta (Fig. 5B,D), the fluorescence transients associated with the fusion events exhibited similar rise time and decay time at axon versus dendrite during the first 4 s after the onset of the fusion. These events were completely absent at both axon and dendrite when the neurons were pretreated for 20 min with TeNT (10 nm), which is known to cleave the vesicle-associated protein synaptobrevin and prevents vesicle fusion (Schiavo et al., 1992). This TeNT sensitivity precluded the possibility that the fluorescence transient is attributable to vesicle movement into and out of the evanescent field. The fluorescence transients exhibited two distinct phases in both axon and dendrite. The decay phase of fluorescence transients could be described as the sum of two exponentials, with 84% of the fluorescence lost with a characteristic time constant of τfast = 0.45 s and 16% lost with a time constant of τslow = 5.9 s at the axon, and corresponding τfast = 0.36 s (74%) and τslow = 3.4 s (26%) at the dendrite. However, both phases of the decay at the axon was increased by the presence of dynasore (37% with τfast = 0.70 s; 63% with τslow = 31 s), which inhibited dynamin and apparently impeded the endocytic closure of fusion pores and effective reacidification of resealed vesicles (Fig. 5B). In contrast, the decay of fluorescence at the dendrite was unaffected by dynasore (Fig. 5D), consistent with the full vesicle fusion and the idea that the fluorescence decay at the dendrite was attributable to dispersion of secreted BDNF–pHluorin. This interpretation was further supported by the finding that TrkB-Fc, a soluble scavenger of BDNF (Cabelli et al., 1997), enhanced the fluorescence decay at the dendrite, presumably by facilitating dissociation of BDNF–pHluorin from the cell surface but had no effect at the axon because of its failure in passing through the fusion pore (Fig. 5B,D) (see Discussion).
Additional time-lapse TIRF observations of individual fusion events revealed distinct spatial profiles of fluorescence changes at the axon versus dendrite. Field stimulation triggered fluorescence transients at the axon that began with the appearance of a fluorescent spot of distinct contour that remained throughout the entire transient (Fig. 5E) (for fluorescence changes and respective plots, see supplemental Movie 1, available at www.jneurosci.org as supplemental material). In contrast, fluorescence transients at the dendrite began with a spot of a similar size as that at the axon, but the spot rapidly dilated to a larger size, accompanied by a gradual dilution of fluorescence (Fig. 5F). The spatiotemporal profile of fusion events before and after the onset of the fluorescence transient was further plotted as pseudo-line scan through the center of the spot (Fig. 5E,F). The persistent fluorescence spot at the axon after the onset of the fusion event suggests the presence of a fusion pore without changing the size of BDNF–pHluorin-containing vesicle, whereas the rapid dilation of fluorescence at the dendrite is consistent with full vesicle fusion and diffusion of BDNF–pHluorin from the fusion site (supplemental Movie 2, available at www.jneurosci.org as supplemental material). Importantly, the area over which BDNF–pHluorin fluorescence spread was larger than the diameter of the dendrite. Thus, the spreading fluorescence is likely to represent diffusion of secreted BDNF–pHluorin in the extracellular space in addition to the lateral diffusion of surface-bound BDNF–pHluorin. Repetitive transients (Fig. 5Ec) were observed occasionally at the axon during video recording of the same punctum but were not seen at the dendrite. Finally, we note that full fusion events were occasionally observed at the axon with a very low frequency (14 of 353 vesicles) during the 10 Hz field stimulation, suggesting that BDNF–pHluorin secretion is possible at the axon (see below).
Notably, the mean and peak amplitudes of fluorescence increases in the region of vesicles were larger at the axon than that at the dendrite (Fig. 5B,D) (supplemental Fig. 7A–C, available at www.jneurosci.org as supplemental material). However, the sum of fluorescence changes at dendritic vesicles were larger than that at the axon (supplemental Fig. 7D, available at www.jneurosci.org as supplemental material), suggesting that a higher amount of total BDNF–pHluorin was present in the evanescent field, presumably as a consequence of the vesicle and BDNF–pHluorin release. Because the expression levels of BDNF–pHluorin appeared similar in axons and dendrites (Fig. 1E) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), the higher amplitude of fluorescence transients at the axon reflected the spatial constraint of BDNF–pHluorin within the vesicle during the fusion pore opening rather than a higher accumulation of BDNF–pHluorin in axonal vesicles.
Exocytic fusion of BDNF vesicles is Ca2+ dependent
To examine the Ca2+ dependence of stimulation-induced fusion of BDNF-containing vesicles, we varied the extracellular Ca2+ concentration ([Ca2+]o) and measured the change in pHluorin punctum fluorescence induced by field stimulation (10 Hz, 40 pulses, 1 ms pulse width). Under wide-field epifluorescence microcopy, we found that increasing [Ca2+]o from 1.0 to 3.5 mm led to a significant increase in the peak amplitude of puncta fluorescence changes at both axon (Fig. 6A) and dendrite (Fig. 6B). Bath application of Cd2+, a potent competitor of Ca2+ for permeation through calcium channels, reversibly inhibited stimulation-induced fluorescence changes in a dose-dependent manner (Fig. 6C). Thus, Ca2+ entry in response to neuronal activity is required for the fusion of BDNF-containing vesicles at both axon and dendrite.
Figure 6.

Calcium dependence in activity-induced fusion of BDNF–pHluorin vesicles. A, B, Average changes of BDNF–pHluorin puncta fluorescence changes induced by field stimulation (40 pulses, 10 Hz) at different [Ca2+]o at axons (±SEM; n = 215 puncta in 9 axons) and dendrites (n = 231 puncta in 8 dendrites). Insets, Average peak fluorescence change, normalized to that observed at 2 mm [Ca2+]o. C, Average changes of BDNF–pHluorin puncta fluorescence recorded from a single axon and dendrite, in the presence of various concentrations of Cd2+ in the culture medium. Bar graphs, Average peak fluorescence change, normalized to that at 0 mm [Cd2+]o. D, E, Average changes of peak BDNF–pHluorin puncta fluorescence at axons and dendrites induced by either field stimulation of the culture or loose-patch stimulation of individual soma, in the presence of various blockers of glutamate receptors and L-type Ca2+ channels, and normalized by those observed in the absence of drug treatment (n = 3–7 neurons for each group). Nim, Nimodipine.
We also examined the contribution of glutamate receptors and voltage-gated calcium channels (VGCCs) to the fusion of BDNF-containing vesicles induced by field stimulation, which activates both presynaptic and postsynaptic neurons simultaneously. We found that blocking AMPA and/or NMDA subtypes of glutamate receptors with CNQX and APV, respectively, had no effect on the amplitude of stimulation-evoked BDNF–pHluorin fluorescence transients at the axon (Fig. 6D) but greatly diminished amplitude of fluorescence reduction at the dendrite (Fig. 6E). Furthermore, blocking L-type VGCCs with nimodipine also suppressed the dendritic fluorescence reduction without affecting axonal fluorescence transients. Thus, Ca2+ influxes mediated by both synaptic NMDA receptors and L-type VGCCs contribute to the full fusion of BDNF vesicles at the dendrite but not partial fusion of BDNF vesicles at the axon. The latter is presumably mediated by other Cd2+-sensitive Ca2+ channels, e.g., N- and P/Q-type VGCCs.
In addition to field stimulation, we also examined the effect of loose-patch stimulation of the soma (at 10 Hz for 300 pulses) of these cultured neurons, a procedure that reduces synaptic activation of the neuron. Loose-patch stimulation induced BDNF–pHluorin fluorescence changes with an efficacy similar to that found for field stimulation (supplemental Fig. 8, available at www.jneurosci.org as supplemental material). Importantly, we found that the soma stimulation-induced pHluorin fluorescence reduction at the dendrite was not affected by glutamate receptor blockers, consistent with the absence of presynaptic activation by the soma stimulation (Fig. 6E). Conversely, blocking L-type VGCCs diminished soma stimulation-induced fluorescence reduction to a similar extent as that found with field stimulation (Fig. 6E), indicating that Ca2+ influx through L-type VGCCs indeed plays a major role in triggering full fusion of BDNF vesicles at the dendrite.
Differential activity dependence for vesicle fusion at axon versus dendrite
We next examine the relationship between the pattern of neuronal activity and BDNF secretion at the axon versus dendrite. We first tested the effect of tonic stimulation of different frequencies (2–50 Hz) but a constant total number of stimuli (300), as well as TBS (total of 300 pulses) (see Materials and Methods), which simulates physiological activities in many brain regions. For tonic stimulation, we found that 2 Hz stimulation triggered only small changes in BDNF–pHluorin fluorescence at both axon and dendrite (Fig. 7A–C). The amplitude and the rate of fluorescence rise at the axon increased with stimulation frequency (from 2 to 50 Hz), corresponding to more transient fusion events within the puncta (Fig. 7A,D). At the dendrite, increasing stimulation frequency from 2 to 10 or 50 Hz resulted in larger reduction of pHluorin puncta fluorescence, reflecting more full fusion events within the puncta. Application of TBS did not cause any significant change in axonal BDNF–pHluorin puncta but was highly effective in triggering the reduction of pHluorin puncta fluorescence at the dendrite (Fig. 7A–C). Blocking NMDA receptors during TBS reduced the amplitude of BDNF–pHluorin fluorescence changes at the dendrite but not at the axon (Fig. 7C), consistent with the contribution of NMDA receptor activation to Ca2+ elevation in the dendrite.
Figure 7.

Activity pattern-dependent partial and full fusion of BDNF–containing vesicles. A, B, Average changes of BDNF–pHluorin puncta fluorescence evoked by various conditions of stimulation at axon (A) and dendrite (B). C, Peak amplitude of BDNF–pHluorin fluorescence changes plotted against the frequency of tonic field stimulation and after TBS (1 min). +APV, TBS applied in the presence of APV (50 μm). D, The fraction of BDNF–pHluorin puncta exhibiting partial fusion, full fusion, and no fusion at axon and dendrite, under different stimulation conditions. Error bars indicate SEM. E, Average changes of BDNF–pHluorin puncta fluorescence at the axon, in response to tonic stimulation at 50 Hz (a) and TBS (b) for three different durations (n = 3–5 neurons each).
Interestingly, significant reduction of BDNF–pHluorin fluorescence could be induced at the axon by prolonged stimulation. After tonic stimulation at 50 Hz or TBS lasting for 3 min, BDNF–pHluorin punctum fluorescence at the axon showed a delayed decrease to a level comparable with that found at the dendrite (Fig. 7E), suggesting full fusion and exocytic BDNF secretion at the axon. There appeared to be a threshold for the duration of stimulation required to induce such “delayed secretion” of BDNF–pHluorin at the axon. For tonic stimulation at 50 Hz, delayed secretion was rarely observed when the stimulation lasted for 1 min or less but was found in five of seven cases when stimulation lasted for 3 min (Fig. 7Ea). Similarly, TBS of 36 burst trains (900 pulses over 3 min) successfully triggered the delayed secretion in three of five axons, but TBS for 2 min or shorter duration was ineffective (Fig. 7Eb). Thus, the delayed secretion was not simply attributable to the latency of the response but reflected the requirement of longer duration of sustained stimulation. This requirement was further supported by the fact that we failed to observe delayed secretion from the axon in response to three episodes of TBS spaced at 3 min intervals with each containing 12 trains of stimuli (900 pulses over 9 min). This delayed secretion could be induced repeatedly in the same neuron, suggesting that it was not attributable to a deleterious condition of the cell. Furthermore, partial or full fusion events at the axon could be triggered by stimulation of different patterns in random sequence, indicating that neurons may alternate between different modes of exocytic secretion, depending on the pattern of activity.
Activity-induced secretion of endogenous BDNF
To verify that the reduction of BDNF–pHluorin fluorescence reports faithfully the secretion of endogenous BDNF, we examined the change in the level of endogenous BDNF triggered by electrical stimulation. The intensity of immunostained BDNF puncta in control and TBS-exposed cultures was measured for those neurons that were not expressing BDNF–pHluorin in the culture and normalized by the average fluorescence intensity of adjacent fluorescent microspheres that were added into the specimen after immunostaining (see Materials and Methods). As shown in the example traces in Figure 8, A and B, 1 min of TBS resulted in a reduction of fluorescence for most BDNF–pHluorin puncta at the dendrite but not at the axon, whereas 3 min of TBS was effective in causing pHluorin reduction in both compartments. Subsequent immunostaining of these cultures with the N-20 antibody showed that the puncta intensity of endogenous BDNF in the axon was lower than that found in control unstimulated cultures after 3 min but not 1 min TBS (Fig. 8C), whereas the puncta intensity at the dendrite was lower than that of the control neurons after both 1 and 3 min TBS (Fig. 8D). These results indicate differential secretion of endogenous BDNF from axon versus dendrite after 1 min TBS (Fig. 8E) and a good correlation between the reduction of endogenous BDNF immunoreactivity and the amplitude of stimulation-induced reduction in BDNF–pHluorin fluorescence. Importantly, these results were reproduced by immunocytochemical experiments using another custom-made antibody to BDNF (supplemental Fig. 9 and Note 1, available at www.jneurosci.org as supplemental material). Thus, activity-induced changes in BDNF–pHluorin fluorescence indeed reflected the behavior of endogenous BDNF.
Figure 8.
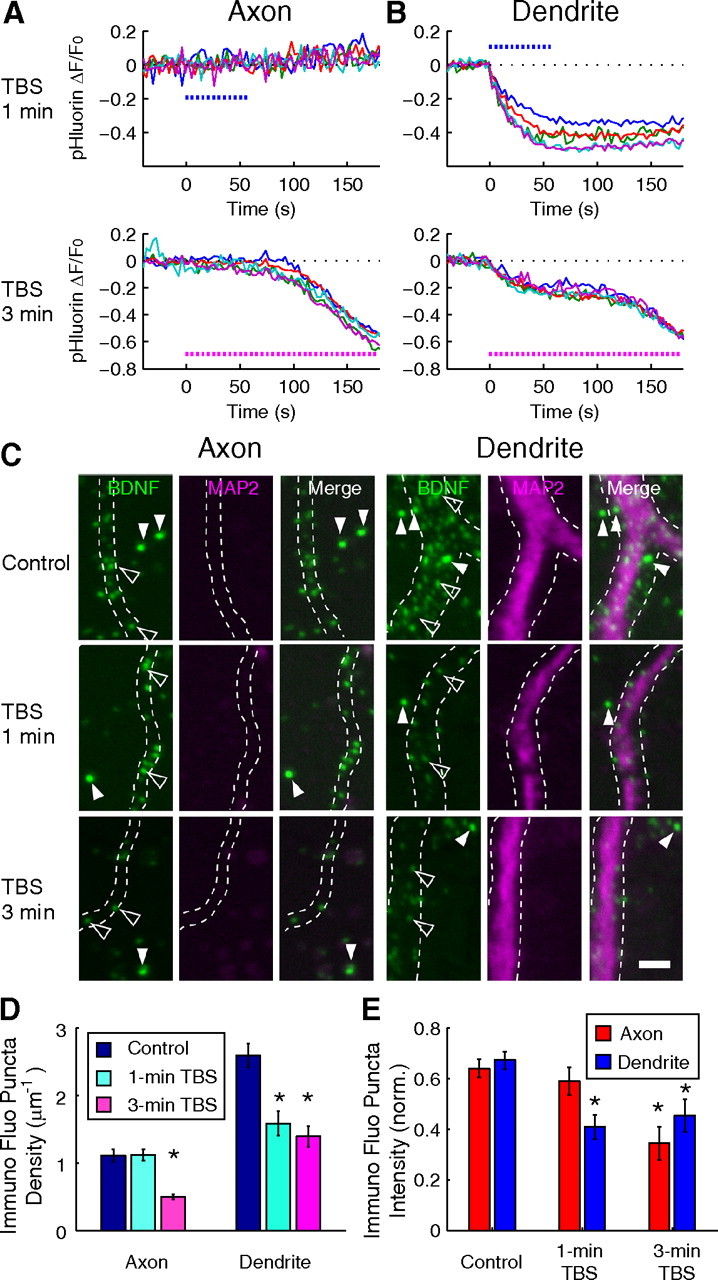
Endogenous BDNF exhibits similar activity-induced secretion as BDNF–pHluorin. A, B, Sample traces of fluorescence changes at individual BDNF–pHluorin puncta induced by 1 or 3 min TBS at axon (A) and dendrite (B), before immunostaining for endogenous BDNF. C, Neurons in the same culture as those in A and B were immunostained for endogenous BDNF immediately after 1 or 3 min TBS. Open arrowheads, Puncta of immunostained endogenous BDNF. Filled arrowheads, Green fluorescent beads added to the culture after immunostaining for normalization. Scale bar, 2 μm. D, E, Bar graphs showing the density of endogenous immunostained BDNF puncta (D) and the intensity of endogenous BDNF puncta fluorescence, normalized by that of the microspheres (E), with or without exposure to 1 or 3 min TBS, for axons (n = 5 neurons) and dendrites (n = 5 neurons). Error bars indicate SEM.
Discussion
We have examined how electrical activity triggers BDNF secretion from the axon and dendrite of cultured hippocampal neurons. We found that BDNF–pHluorin is an excellent reporter for exocytic fusion of BDNF-containing vesicles in these neurons. Using this optical probe, we showed that BDNF-containing vesicles exhibited distinct modes of activity-dependent fusion at the axon versus dendrite. Under most stimulation conditions, we observed incomplete fusion without BDNF secretion at the axon but full fusion with substantial BDNF secretion at the dendrite. However, sustained high-frequency stimulation resulted in full fusion of BDNF-containing vesicles at the axon. These results support the notion that dendritic BDNF release represents the predominant form of activity-induced BDNF secretion from these cultured hippocampal neurons.
Differential fusion events at axon versus dendrite
The wide-field and TIRF microscopy provided information on stimulation-induced fusion behaviors of vesicles en masse and as individuals, respectively. At a single vesicle level, dynamin-dependent endocytosis immediately follows the exocytic fusion induced by electrical stimulation at the axon but not at the dendrite (Fig. 5B,D). Thus, endocytosis is immediately linked to exocytosis in situ at the axon, a hallmark of partial (incomplete) fusion. Imaging of a population of vesicles revealed that a dynamin-dependent process was evident in the recovery phase of the pHluorin fluorescence after the termination of stimulation, and secreted BDNF was internalized by TrkB receptor at the dendrite (Fig. 4D). Such receptor-mediated endocytosis may help local replenishment and recycling of BDNF at the dendrite (Santi et al., 2006). Finally, we note that TIRF microscopy captured only vesicles that were docked at the plasma membrane in direct contact with the glass substratum, suggesting that the observed fluorescent vesicles are extrasynaptic. Nevertheless, the distinct activity-induced partial versus full fusion at the axon versus dendrite observed by TIRF are consistent with those found for synaptic markers-associated BDNF–pHluorin puncta observed under wide-field microscopy.
It is well established that two modes of exocytosis exist: one accompanying full collapse of vesicles that results in the complete extrusion of the vesicle content and retrieval of membrane components by clathrin-mediated endocytosis, and the other involving rapid recapture of the vesicle after the opening of the fusion pore that preserves the vesicle identity (Harata et al., 2006a). The latter is called “kiss-and-run” exocytosis in the terminology for SVs (Aravanis et al., 2003; Gandhi and Stevens, 2003; Harata et al., 2006b) or “cavicapture” for endocrine granules (Henkel and Almers, 1996). Glutamate (147 Da) in SVs may penetrate even partial fusion pores, and synaptic efficacy may be regulated by altering neurotransmitter release kinetics through changes in the size and opening dynamics of the fusion pore (Choi et al., 2000; Renger et al., 2001; An and Zenisek, 2004). Because dense core vesicles release substance with larger molecular weights (such as hormones and peptides), the size and kinetics of fusion pore opening have more impact on the release of vesicular content. Indeed, PC-12 cells and chromaffin cells exhibit both complete and incomplete release of peptides in the same cells, depending on the content of the vesicles (Holroyd et al., 2002; Taraska et al., 2003; Perrais et al., 2004); neuropeptide Y (7–15 kDa) was released within a second, but tissue plasminogen activator (70 kDa) was retained (Perrais et al., 2004). The partial fusion resembles the fusion event of BDNF–pHluorin-containing vesicles at the axon in that it is dynamin dependent (Holroyd et al., 2002). Our TIRF images further indicate that the initial fusion pore formed at the axon failed to undergo full fusion. The size of the mature BDNF homodimer (27 kDa) has a dimension of 6 × 2.5 × 1.5 nm (McDonald et al., 1991; Lou et al., 2005), which is larger than the size of the initial fusion pore (∼1.4 nm) of insulin-containing vesicles estimated by aqueous phase tracers (Takahashi et al., 2002). This accounts for the failure of both endogenous BDNF and BDNF–pHluorin secretion at the axon. However, the initial fusion pore of insulin-containing vesicles has a lifetime of ∼1.8 s and subsequently dilated to 12 nm as the vesicle collapsed to release insulin (Takahashi et al., 2002). This sequence of events resembles those of BDNF–pHluorin vesicles at the dendrite observed by TIRF (Fig. 5F). Bath-applied TrkB-Fc (240–260 kDa) failed to scavenge BDNF at the axon, presumably attributable to its failure to pass through the small fusion pore at the axon. However, under normal stimulation conditions, other smaller components costored in BDNF-containing vesicles in the axon may still be released via partial fusion events in the absence of full vesicle collapse (Obermüller et al., 2005).
Differential activity dependence at axon versus dendrite
Electrical stimulation enables us to examine exocytic release of BDNF in relation to the frequency and number of spikes, which was not achieved by high K+ stimulation used in previous studies (Kolarow et al., 2007; Xia et al., 2009). The frequency of transient fusion events increased with stimulation frequency from 2 to 50 Hz at the axon, with the total number of stimuli (300 pulses) kept constant. In contrast, the frequency of full fusion at the dendrite reached an apparent maximum with 10 Hz stimulation. Most notably, a brief period of TBS (1 min, 300 pulses) was highly effective for inducing full fusion and BDNF secretion at the dendrite but was ineffective at the axon. Previous studies on activity-induced BDNF secretion from cultured neurons have not addressed the difference in BDNF secretion between axon and dendrite under the same stimulation condition. High-frequency burst stimulation (16 bursts of 1 s duration at 50 Hz with 2.5 s interval) triggered dendritic BDNF secretion from cultured hippocampal neurons expressing BDNF–EGFP (Hartmann et al., 2001). Measurement of the BDNF level in the culture medium with ELISA, in which secretion from axon versus dendrite could not be distinguished, revealed that LTP-inducing stimulation (tetanic stimulation at 50 or 100 Hz and TBS) led to substantial BDNF release, whereas low-frequency stimulation at 1 Hz was ineffective (Gärtner and Staiger, 2002). For primary cultures of sensory neurons, ELISA in situ measurements showed that prolonged stimulation (60 min) of increasing frequencies (from 5 to 50 Hz) led to increasing amounts of BDNF secretion (Balkowiec and Katz, 2000). Based on our results, we suggest that BDNF secretion observed in these previous studies was mainly attributable to dendritic secretion, except for the case using prolonged stimulation, which may have triggered full fusion of BDNF-containing vesicles at the axon.
Mechanisms and physiological implications of differential BDNF secretion
Our findings indicate that exocytic fusion of BDNF-containing vesicles at both axon and dendrite depends on the Ca2+ influx and elevation of [Ca2+]i. Consistent with a previous report (Kuczewski et al., 2008), loose-patch somatic stimulation was sufficient to induce dendritic release of BDNF, presumably by triggering backpropagating action potential and Ca2+ influx via VGCCs. However, under field stimulation that activates both presynaptic and postsynaptic sites, synaptic activation of postsynaptic NMDA receptor channels also contributes to a large extent to the Ca2+ influx at the dendrite. Interestingly, changes in [Ca2+]o in the range of 1.0 to 3.5 mm did not alter the mode of exocytic events. Differences in the local spatiotemporal dynamics of [Ca2+]i or downstream effectors of Ca2+ signals at the axon versus dendrite may result in differential fusion efficacy. For example, postsynaptic BDNF secretion depends on the activation of α-calcium/calmodulin-dependent protein kinase II (Kolarow et al., 2007), which is known to be concentrated in postsynaptic dendritic spines. Finally, dendrite- versus axon-targeting BDNF-containing vesicles may represent two distinct populations of secretory granules endowed with different sets of associated proteins, e.g., synaptotagmins (Lynch et al., 2008; Dean et al., 2009), hence differential fusion properties. Prolonged high-frequency spiking activity may build up global “residual” Ca2+ in the cytoplasm, a condition favorable for neuropeptide secretion from the presynaptic nerve terminal (Peng and Zucker, 1993; Muschol and Salzberg, 2000) and thereby overcome the less favorable condition inherent in the axon, resulting in BDNF secretion from the axon. The relative contribution of these mechanisms to the binary choice of partial versus full fusion remains to be elucidated.
Hippocampal neurons in vivo are normally exposed to tonic activities ranging from 1 to 20 Hz and brief high-frequency (>50 Hz) bursting activities (Bland, 1986). Under these conditions, BDNF is secreted predominantly from the dendrite and acts on presynaptic terminals as a retrograde signal or on the postsynaptic cell as an autocrine factor. The preference for the fusion of BDNF-containing vesicle at axon versus dendrite was a common feature under other experimental conditions: in cultured cortical neurons (data not shown) or by depolarization with high K+ (supplemental Note 3 and Fig. 10, available at www.jneurosci.org as supplemental material). However, other neural circuits may exhibit different preferences for BDNF secretion, particularly in a more intact preparation in vivo. Full fusion of BDNF-containing vesicles at the axon required sustained high-frequency tonic or bursting activities for a few minutes, a condition found during pathological conditions, i.e., status epilepticus. Finally, our studies have not addressed the possibility that full fusion and BDNF secretion from the axon could occur constitutively or could be regulated by factors other than electrical activity and serve for normal physiological functions as well.
Footnotes
This work is supported in part by a grant from National Institutes of Health (EY014949). We thank Pei-lin Cheng for preparation of hippocampal cultures, Masami Kojima for technical advice, the BDNF–EGFP construct, and brain lysate from BDNF knock-out mice, Shingo Suzuki for technical advice, Gero Miesenboeck for the pHluorin plasmid, Roger Tsien for the mCherry plasmid, Tomas Kirchhausen for dynasore, Junichi Nabekura for an electrical stimulator, and Naoki Takahashi for technical assistance.
References
- Adachi N, Kohara K, Tsumoto T. Difference in trafficking of brain-derived neurotrophic factor between axons and dendrites of cortical neurons, revealed by live-cell imaging. BMC Neurosci. 2005;6:42. doi: 10.1186/1471-2202-6-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An S, Zenisek D. Regulation of exocytosis in neurons and neuroendocrine cells. Curr Opin Neurobiol. 2004;14:522–530. doi: 10.1016/j.conb.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Aravanis AM, Pyle JL, Tsien RW. Single synaptic vesicles fusing transiently and successively without loss of identity. Nature. 2003;423:643–647. doi: 10.1038/nature01686. [DOI] [PubMed] [Google Scholar]
- Axelrod D. Total internal reflection fluorescence microscopy in cell biology. Methods Enzymol. 2003;361:1–33. doi: 10.1016/s0076-6879(03)61003-7. [DOI] [PubMed] [Google Scholar]
- Balkowiec A, Katz DM. Activity-dependent release of endogenous brain-derived neurotrophic factor from primary sensory neurons detected by ELISA in situ. J Neurosci. 2000;20:7417–7423. doi: 10.1523/JNEUROSCI.20-19-07417.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bland BH. The physiology and pharmacology of hippocampal formation theta rhythms. Prog Neurobiol. 1986;26:1–54. doi: 10.1016/0301-0082(86)90019-5. [DOI] [PubMed] [Google Scholar]
- Blöchl A, Thoenen H. Characterization of nerve growth factor (NGF) release from hippocampal neurons: evidence for a constitutive and an unconventional sodium-dependent regulated pathway. Eur J Neurosci. 1995;7:1220–1228. doi: 10.1111/j.1460-9568.1995.tb01112.x. [DOI] [PubMed] [Google Scholar]
- Bolte S, Cordelières FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- Cabelli RJ, Shelton DL, Segal RA, Shatz CJ. Blockade of endogenous ligands of trkB inhibits formation of ocular dominance columns. Neuron. 1997;19:63–76. doi: 10.1016/s0896-6273(00)80348-7. [DOI] [PubMed] [Google Scholar]
- Carmignoto G, Pizzorusso T, Tia S, Vicini S. Brain-derived neurotrophic factor and nerve growth factor potentiate excitatory synaptic transmission in the rat visual cortex. J Physiol. 1997;498:153–164. doi: 10.1113/jphysiol.1997.sp021848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZY, Jing D, Bath KG, Ieraci A, Khan T, Siao CJ, Herrera DG, Toth M, Yang C, McEwen BS, Hempstead BL, Lee FS. Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science. 2006;314:140–143. doi: 10.1126/science.1129663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S, Klingauf J, Tsien RW. Postfusional regulation of cleft glutamate concentration during LTP at “silent synapses.”. Nat Neurosci. 2000;3:330–336. doi: 10.1038/73895. [DOI] [PubMed] [Google Scholar]
- Dean C, Liu H, Dunning FM, Chang PY, Jackson MB, Chapman ER. Synaptotagmin-IV modulates synaptic function and long-term potentiation by regulating BDNF release. Nat Neurosci. 2009;12:767–776. doi: 10.1038/nn.2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiStefano PS, Friedman B, Radziejewski C, Alexander C, Boland P, Schick CM, Lindsay RM, Wiegand SJ. The neurotrophins BDNF, NT-3, and NGF display distinct patterns of retrograde axonal transport in peripheral and central neurons. Neuron. 1992;8:983–993. doi: 10.1016/0896-6273(92)90213-w. [DOI] [PubMed] [Google Scholar]
- Du J, Feng L, Yang F, Lu B. Activity- and Ca2+-dependent modulation of surface expression of brain-derived neurotrophic factor receptors in hippocampal neurons. J Cell Biol. 2000;150:1423–1434. doi: 10.1083/jcb.150.6.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, Zaitsev E, Gold B, Goldman D, Dean M, Lu B, Weinberger DR. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell. 2003;112:257–269. doi: 10.1016/s0092-8674(03)00035-7. [DOI] [PubMed] [Google Scholar]
- Gandhi SP, Stevens CF. Three modes of synaptic vesicular recycling revealed by single-vesicle imaging. Nature. 2003;423:607–613. doi: 10.1038/nature01677. [DOI] [PubMed] [Google Scholar]
- Gärtner A, Staiger V. Neurotrophin secretion from hippocampal neurons evoked by long-term-potentiation-inducing electrical stimulation patterns. Proc Natl Acad Sci U S A. 2002;99:6386–6391. doi: 10.1073/pnas.092129699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gärtner A, Polnau DG, Staiger V, Sciarretta C, Minichiello L, Thoenen H, Bonhoeffer T, Korte M. Hippocampal long-term potentiation is supported by presynaptic and postsynaptic tyrosine receptor kinase B-mediated phospholipase Cγ signaling. J Neurosci. 2006;26:3496–3504. doi: 10.1523/JNEUROSCI.3792-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granseth B, Odermatt B, Royle SJ, Lagnado L. Clathrin-mediated endocytosis is the dominant mechanism of vesicle retrieval at hippocampal synapses. Neuron. 2006;51:773–786. doi: 10.1016/j.neuron.2006.08.029. [DOI] [PubMed] [Google Scholar]
- Harata NC, Aravanis AM, Tsien RW. Kiss-and-run and full-collapse fusion as modes of exo-endocytosis in neurosecretion. J Neurochem. 2006a;97:1546–1570. doi: 10.1111/j.1471-4159.2006.03987.x. [DOI] [PubMed] [Google Scholar]
- Harata NC, Choi S, Pyle JL, Aravanis AM, Tsien RW. Frequency-dependent kinetics and prevalence of kiss-and-run and reuse at hippocampal synapses studied with novel quenching methods. Neuron. 2006b;49:243–256. doi: 10.1016/j.neuron.2005.12.018. [DOI] [PubMed] [Google Scholar]
- Hartmann M, Heumann R, Lessmann V. Synaptic secretion of BDNF after high-frequency stimulation of glutamatergic synapses. EMBO J. 2001;20:5887–5897. doi: 10.1093/emboj/20.21.5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendry IA, Stöckel K, Thoenen H, Iversen LL. The retrograde axonal transport of nerve growth factor. Brain Res. 1974;68:103–121. doi: 10.1016/0006-8993(74)90536-8. [DOI] [PubMed] [Google Scholar]
- Henkel AW, Almers W. Fast steps in exocytosis and endocytosis studied by capacitance measurements in endocrine cells. Curr Opin Neurobiol. 1996;6:350–357. doi: 10.1016/s0959-4388(96)80119-x. [DOI] [PubMed] [Google Scholar]
- Holroyd P, Lang T, Wenzel D, De Camilli P, Jahn R. Imaging direct, dynamin-dependent recapture of fusing secretory granules on plasma membrane lawns from PC12 cells. Proc Natl Acad Sci U S A. 2002;99:16806–16811. doi: 10.1073/pnas.222677399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttner WB, Gerdes HH, Rosa P. The granin-(chromogranin/secretogranin) family. Trends Biochem Sci. 1991;16:27–30. doi: 10.1016/0968-0004(91)90012-k. [DOI] [PubMed] [Google Scholar]
- Katoh-Semba R, Takeuchi IK, Inaguma Y, Ichisaka S, Hata Y, Tsumoto T, Iwai M, Mikoshiba K, Kato K. Induction of brain-derived neurotrophic factor by convulsant drugs in the rat brain: involvement of region-specific voltage-dependent calcium channels. J Neurochem. 2001;77:71–83. doi: 10.1046/j.1471-4159.2001.t01-1-00138.x. [DOI] [PubMed] [Google Scholar]
- Kohara K, Kitamura A, Morishima M, Tsumoto T. Activity-dependent transfer of brain-derived neurotrophic factor to postsynaptic neurons. Science. 2001;291:2419–2423. doi: 10.1126/science.1057415. [DOI] [PubMed] [Google Scholar]
- Kojima M, Takei N, Numakawa T, Ishikawa Y, Suzuki S, Matsumoto T, Katoh-Semba R, Nawa H, Hatanaka H. Biological characterization and optical imaging of brain-derived neurotrophic factor-green fluorescent protein suggest an activity-dependent local release of brain-derived neurotrophic factor in neurites of cultured hippocampal neurons. J Neurosci Res. 2001;64:1–10. doi: 10.1002/jnr.1080. [DOI] [PubMed] [Google Scholar]
- Kolarow R, Brigadski T, Lessmann V. Postsynaptic secretion of BDNF and NT-3 from hippocampal neurons depends on calcium calmodulin kinase II signaling and proceeds via delayed fusion pore opening. J Neurosci. 2007;27:10350–10364. doi: 10.1523/JNEUROSCI.0692-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalchuk Y, Hanse E, Kafitz KW, Konnerth A. Postsynaptic induction of BDNF-mediated long-term potentiation. Science. 2002;295:1729–1734. doi: 10.1126/science.1067766. [DOI] [PubMed] [Google Scholar]
- Kuczewski N, Porcher C, Ferrand N, Fiorentino H, Pellegrino C, Kolarow R, Lessmann V, Medina I, Gaiarsa JL. Backpropagating action potentials trigger dendritic release of BDNF during spontaneous network activity. J Neurosci. 2008;28:7013–7023. doi: 10.1523/JNEUROSCI.1673-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang SB, Stein V, Bonhoeffer T, Lohmann C. Endogenous brain-derived neurotrophic factor triggers fast calcium transients at synapses in developing dendrites. J Neurosci. 2007;27:1097–1105. doi: 10.1523/JNEUROSCI.3590-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibrock J, Lottspeich F, Hohn A, Hofer M, Hengerer B, Masiakowski P, Thoenen H, Barde YA. Molecular cloning and expression of brain-derived neurotrophic factor. Nature. 1989;341:149–152. doi: 10.1038/341149a0. [DOI] [PubMed] [Google Scholar]
- Li YX, Xu Y, Ju D, Lester HA, Davidson N, Schuman EM. Expression of a dominant negative TrkB receptor, T1, reveals a requirement for presynaptic signaling in BDNF-induced synaptic potentiation in cultured hippocampal neurons. Proc Natl Acad Sci U S A. 1998;95:10884–10889. doi: 10.1073/pnas.95.18.10884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohof AM, Ip NY, Poo MM. Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature. 1993;363:350–353. doi: 10.1038/363350a0. [DOI] [PubMed] [Google Scholar]
- Lou H, Kim SK, Zaitsev E, Snell CR, Lu B, Loh YP. Sorting and activity-dependent secretion of BDNF require interaction of a specific motif with the sorting receptor carboxypeptidase E. Neuron. 2005;45:245–255. doi: 10.1016/j.neuron.2004.12.037. [DOI] [PubMed] [Google Scholar]
- Lynch KL, Gerona RR, Kielar DM, Martens S, McMahon HT, Martin TFJ. Synaptotagmin-1 utilizes membrane bending and SNARE binding to drive fusion pore expansion. Mol Biol Cell. 2008;19:5093–5103. doi: 10.1091/mbc.E08-03-0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell. 2006;10:839–850. doi: 10.1016/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Matsumoto T, Rauskolb S, Polack M, Klose J, Kolbeck R, Korte M, Barde YA. Biosynthesis and processing of endogenous BDNF: CNS neurons store and secrete BDNF, not pro-BDNF. Nat Neurosci. 2008;11:131–133. doi: 10.1038/nn2038. [DOI] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity. Annu Rev Neurosci. 1999;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- McDonald NQ, Lapatto R, Murray-Rust J, Gunning J, Wlodawer A, Blundell TL. New protein fold revealed by a 2.3-A resolution crystal structure of nerve growth factor. Nature. 1991;354:411–414. doi: 10.1038/354411a0. [DOI] [PubMed] [Google Scholar]
- Miesenböck G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- Morris JF, Nordmann JJ, Dyball RE. Structure-function correlation in mammalian neurosecretion. Int Rev Exp Pathol. 1978;18:1–95. [PubMed] [Google Scholar]
- Muschol M, Salzberg BM. Dependence of transient and residual calcium dynamics on action-potential patterning during neuropeptide secretion. J Neurosci. 2000;20:6773–6780. doi: 10.1523/JNEUROSCI.20-18-06773.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermüller S, Lindqvist A, Karanauskaite J, Galvanovskis J, Rorsman P, Barg S. Selective nucleotide-release from dense-core granules in insulin-secreting cells. J Cell Sci. 2005;118:4271–4282. doi: 10.1242/jcs.02549. [DOI] [PubMed] [Google Scholar]
- Ozawa H, Takata K. The granin family: its role in sorting and secretory granule formation. Cell Struct Funct. 1995;20:415–420. doi: 10.1247/csf.20.415. [DOI] [PubMed] [Google Scholar]
- Pang PT, Teng HK, Zaitsev E, Woo NT, Sakata K, Zhen S, Teng KK, Yung WH, Hempstead BL, Lu B. Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science. 2004;306:487–491. doi: 10.1126/science.1100135. [DOI] [PubMed] [Google Scholar]
- Peng YY, Zucker RS. Release of LHRH is linearly related to the time integral of presynaptic Ca2+ elevation above a threshold level in bullfrog sympathetic ganglia. Neuron. 1993;10:465–473. doi: 10.1016/0896-6273(93)90334-n. [DOI] [PubMed] [Google Scholar]
- Perrais D, Kleppe IC, Taraska JW, Almers W. Recapture after exocytosis causes differential retention of protein in granules of bovine chromaffin cells. J Physiol. 2004;560:413–428. doi: 10.1113/jphysiol.2004.064410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- Renger JJ, Egles C, Liu G. A developmental switch in neurotransmitter flux enhances synaptic efficacy by affecting AMPA receptor activation. Neuron. 2001;29:469–484. doi: 10.1016/s0896-6273(01)00219-7. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S, Ryan TA. Calcium accelerates endocytosis of vSNAREs at hippocampal synapses. Nat Neurosci. 2001;4:129–136. doi: 10.1038/83949. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S, De Angelis D, Rothman JE, Ryan TA. The use of pHluorins for optical measurements of presynaptic activity. Biophys J. 2000;79:2199–2208. doi: 10.1016/S0006-3495(00)76468-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santi S, Cappello S, Riccio M, Bergami M, Aicardi G, Schenk U, Matteoli M, Canossa M. Hippocampal neurons recycle BDNF for activity-dependent secretion and LTP maintenance. EMBO J. 2006;25:4372–4380. doi: 10.1038/sj.emboj.7601303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavo G, Benfenati F, Poulain B, Rossetto O, Polverino de Laureto P, DasGupta BR, Montecucco C. Tetanus and botulinum-B neurotoxins block neurotransmitter release by proteolytic cleavage of synaptobrevin. Nature. 1992;359:832–835. doi: 10.1038/359832a0. [DOI] [PubMed] [Google Scholar]
- Shen W, Wu B, Zhang Z, Dou Y, Rao ZR, Chen YR, Duan S. Activity-induced rapid synaptic maturation mediated by presynaptic Cdc42 signaling. Neuron. 2006;50:401–414. doi: 10.1016/j.neuron.2006.03.017. [DOI] [PubMed] [Google Scholar]
- Sugiyama Y, Kawabata I, Sobue K, Okabe S. Determination of absolute protein numbers in single synapses by a GFP-based calibration technique. Nat Methods. 2005;2:677–684. doi: 10.1038/nmeth783. [DOI] [PubMed] [Google Scholar]
- Takahashi N, Kishimoto T, Nemoto T, Kadowaki T, Kasai H. Fusion pore dynamics and insulin granule exocytosis in the pancreatic islet. Science. 2002;297:1349–1352. doi: 10.1126/science.1073806. [DOI] [PubMed] [Google Scholar]
- Taraska JW, Perrais D, Ohara-Imaizumi M, Nagamatsu S, Almers W. Secretory granules are recaptured largely intact after stimulated exocytosis in cultured endocrine cells. Proc Natl Acad Sci U S A. 2003;100:2070–2075. doi: 10.1073/pnas.0337526100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas K, Davies A. Neurotrophins: a ticket to ride for BDNF. Curr Biol. 2005;15:R262–R264. doi: 10.1016/j.cub.2005.03.023. [DOI] [PubMed] [Google Scholar]
- von Bartheld CS, Byers MR, Williams R, Bothwell M. Anterograde transport of neurotrophins and axodendritic transfer in the developing visual system. Nature. 1996;379:830–833. doi: 10.1038/379830a0. [DOI] [PubMed] [Google Scholar]
- Xia X, Lessmann V, Martin TFJ. Imaging of evoked dense-core-vesicle exocytosis in hippocampal neurons reveals long latencies and kiss-and-run fusion events. J Cell Sci. 2009;122:75–82. doi: 10.1242/jcs.034603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakharenko SS, Patterson SL, Dragatsis I, Zeitlin SO, Siegelbaum SA, Kandel ER, Morozov A. Presynaptic BDNF required for a presynaptic but not postsynaptic component of LTP at hippocampal CA1-CA3 synapses. Neuron. 2003;39:975–990. doi: 10.1016/s0896-6273(03)00543-9. [DOI] [PubMed] [Google Scholar]
- Zenisek D, Steyer JA, Feldman ME, Almers W. A membrane marker leaves synaptic vesicles in milliseconds after exocytosis in retinal bipolar cells. Neuron. 2002;35:1085–1097. doi: 10.1016/s0896-6273(02)00896-6. [DOI] [PubMed] [Google Scholar]
- Zhang X, Poo MM. Localized synaptic potentiation by BDNF requires local protein synthesis in the developing axon. Neuron. 2002;36:675–688. doi: 10.1016/s0896-6273(02)01023-1. [DOI] [PubMed] [Google Scholar]