

Abstract
Background
Poly(ADP-ribose) polymerase (PARP) signals DNA damage and facilitates DNA repair. PARP inhibitors are being evaluated in cancers with defective DNA repair mechanisms or in combination with cytotoxic therapy or radiation. We evaluated the PARP inhibitor, olaparib, in combination with chemotherapy using in vitro and in vivo pediatric solid tumor models.
Procedure
The IC50 of olaparib alone and in combination with cytotoxic agents was determined in 10 pediatric solid tumor cell lines. Synergy was assessed using the combination index of Chou-Talalay. Olaparib alone and in combination with topotecan/cyclophosphamide was evaluated in xenograft models of Ewing sarcoma (ES) and neuroblastoma (NGP). PAR activity was evaluated in cell lines and tumor lysates.
Results
Olaparib induced growth inhibition, median (range) IC50=3.6 (1–33.8) µM, and inhibited PAR activity in pediatric solid tumor cell lines. The addition of olaparib to DNA damaging agents resulted in additive to synergistic interactions. In ES and NGP xenografts, olaparib inhibited PAR activity by 88% to 100% as a single agent and 100% when administered with cyclophosphamide/topotecan. Although the addition of olaparib did not antagonize the activity of cyclophosphamide/topotecan, clear evidence of synergy could not be demonstrated.
Conclusions
In pediatric solid tumor cell lines, clinically achievable concentrations of single agent olaparib caused growth inhibition. Although the in vitro data demonstrated synergistic efficacy of olaparib when added to the camptothecins and alkylating agents, synergy was not discernible in vivo. Clinical trials of PARP inhibitors in combination DNA damaging agents are necessary to establish the role of PARP inhibitors in childhood cancer.
Keywords: pediatric, PARP inhibitor, olaparib, in vitro, in vivo
INTRODUCTION
Poly(ADP-ribose) polymerase (PARP) is a nuclear enzyme that signals DNA damage and facilitates DNA repair. PARP catalyzes the addition of ADP-ribose to DNA, histone, topoisomerases, and helicases and has a critical function in cellular replication, transcription, differentiation, gene regulation, protein degradation, and spindle maintenance. PARP-1, the primary member of the PARP family, binds to both single and double strand DNA breaks. Inhibition of PARP-1, results in persistent single strand DNA breaks leading to stalled replication forks and double strand DNA breaks. PARP-1 inhibition in cells with innately dysfunctional DNA repair mechanisms, such as BRCA-1 or BRCA-2 deficient cells, results in DNA damage that leads to cell cycle arrest and apoptosis.
PARP inhibitors currently in clinical development include olaparib, niraparib, and veliparib. Early clinical development focused on carcinomas with known defects in DNA repair. This development plan has been based on the hypothesis of synthetic lethality in which the combination of non-lethal events, such as PARP inhibition and tumor-restricted loss of functional DNA repair pathways, could interact synergistically and result in tumor-specific cell death[1,2]. DNA repair mutations, deletions or epigenetic modifications in mesenchymal or neuroectodermal tumors provide the rationale for evaluation of PARP inhibitors in childhood malignancies[3–14].
PARP inhibition may potentiate the activity of DNA damaging agents such as alkylating agents, topoisomerase inhibitors, and radiation therapy[15–21], which are the cornerstones of treatment for childhood cancers. High-grade pediatric central nervous system tumors have enhanced PARP-1 expression[4,6] that may contribute to radiation and chemotherapy resistance [22]. PARP inhibitors enhance the efficacy of temozolomide and topotecan in preclinical models of pediatric leukemia, neuroblastoma, and medulloblastoma[17–19]. In preclinical models of Ewing sarcoma there is an interaction between the genomic fusion products EWS-FLI1 and EWS-ERG and PARP-1[23]. Ewing sarcoma cell lines, primary xenografts, and tumor metastases are sensitive to olaparib, supporting a potential role for PARP inhibition in the treatment[23,24]. A clinical trial of olaparib is currently being conducted in adults with relapsed or refractory Ewing sarcoma (NCT01583543).
Olaparib (AZD2281) is an oral PARP inhibitor that has completed phase 1 and 2 evaluation as a single agent and in combination with cytotoxic chemotherapy [1,2,23,25–29]. In adult phase 1 trials the maximum tolerated dose (MTD) of olaparib, 400mg twice daily, was associated with a maximum plasma concentration (Cmax) of 18µM[28]. Maximum pharmacodynamic activity, PAR inhibition, was demonstrated after administration of 100mg twice daily, and associated with minimum and maximum plasma concentrations of 1µM and 8.5 µM, respectively[28,30]. Phase 2 studies demonstrate clinical benefit at both doses[1,2]. Ongoing trials in adults are designed to evaluate the efficacy and toxicity of olaparib in combination with cytotoxic therapy including carboplatin, cisplatin, topotecan, irinotecan, and doxorubicin. Olaparib in combination with topotecan[29] or olaparib with cisplatin and gemcitabine[23] resulted in dose-limiting myelosuppression requiring significant dose reductions of cytotoxic chemotherapy.
Olaparib, veliparib, and niraparib are all potent inhibitors of PARP catalytic activity (IC50 < 0.1 µM) leading to single-strand DNA breaks, however, they may differ in their ability to trap PARP-DNA complexes[31,32]. One report demonstrated that niraparib followed by olaparib had the greatest potency in trapping PARP, while olaparib was the most potent inhibitor of PARP catalytic activity[33]. Variable toxicity profiles of PARP inhibitors[24,29,34] may be attributed to differences in potency for stabilizing PARP-DNA complexes. PARP-DNA complexes may be more cytotoxic than unrepaired single strand breaks caused by PARP inhibition alone[32] suggesting that the myelosuppression observed olaparib with combination of cytotoxic chemotherapy may be related to the potency of olaparib trapping of PARP-DNA complexes and may not a be class effect.
We evaluated the PARP inhibitor, olaparib, in combination with conventional cytotoxic chemotherapy using both in vitro and in vivo models of pediatric solid tumors. The in vitro cytotoxicity of olaparib alone and in combination with cytotoxic chemotherapy, assessment of the combination index to evaluate drug interactions, and PAR activity were assessed in a panel of pediatric solid tumor cell lines. Based on the established role of PARP-1 inhibition in BRCA deficient cell lines leading to cell cycle arrest and apoptosis, we included a breast carcinoma cell line known to be homozygous deficient for BRCA in our in vitro analysis. Neuroblastoma (NGP) and Ewing sarcoma (ES) xenograft models were used to assess the activity and efficacy and PAR inhibition of olaparib alone and in combination with cyclophosphamide and topotecan. Due to the genomic instability innate to many pediatric solid tumors, we hypothesized that olaparib would demonstrate single agent activity in several tumor types and demonstrate a favorable interaction profile with DNA damaging agents.
MATERIALS AND METHODS
In Vitro Growth Inhibition Assay
Materials
A panel of 11 cell lines representing 5 pediatric solid tumor types and 1 breast cancer cell line was studied (Table I). RD-ES, SK-N-AS, RD, SJCRH-30, MG-63, HOS, HTB-185, HTB-187, and HCC1937 cell lines were purchased from American Type Tissue Collection (Manassa, VA). The following cell lines were kindly provided by the named investigators: NGP[35] (Dr. John Maris, The Children’s Hospital of Philadelphia) and TC-71[36] (Elizabeth Fox, The Children’s Hospital of Philadelphia). The neuroblastoma cell lines were grown in RPMI-1640 containing 10% fetal bovine serum (FBS), 1% glutamine, 1 mM oxaloacetate, 0.45 mM pyruvate, and 0.2 U/ml insulin. All other cell lines were grown in RPMI-1640 containing 10% FBS. All cell lines were negative for mycoplasma contamination using the MycoAlert® detection assay (Cambrex, East Rutherford, NJ).
Table I.
Single Agent IC50 of Olaparib and Cytotoxic Chemotherapy
| Origin | Cell Line | IC50 µM Mean (SD) |
||||||
|---|---|---|---|---|---|---|---|---|
| Olaparib | Irinotecan | SN-38 | Melphalan | Doxorubicin | Carboplatin | Vincristine | ||
| Ewing Sarcoma | TC-71 | 1.5 (0.9) | 0.5 (0.1) | 0.004 (0.001) | 3.2 (0.8) | 0.018 (0.006) | 3.9 (1.3) | 0.006 (0.002) |
| RD-ES | 1.0 (0.3) | 0.008 (0.002) | 0.004 (0.001) | 2.0 (0.6) | 0.020 (0.007) | 5.3 (1.8) | 0.0012 (0.0004) | |
| Medulloblastoma | HTB-185 | 2.1 (0.9) | 0.8 (0.2) | 0.005 (0.001) | 2.2 (0.5) | 0.019 (0.006) | 2.7 (0.9) | 0.004 (0.002) |
| HTB-186 | 2.4 (0.7) | 0.5 (0.1) | 0.002 (0.001) | 1.5 (0.4) | 0.013 (0.004) | 1.6 (0.6) | 0.003 (0.001) | |
| Neuroblastoma | SK-NAS | 33.8 (8.7) | 2.7 (0.5) | 0.019 (0.005) | 4.2 (1.0) | 0.057 (0.008) | 12.5 (3.8) | 0.004 (0.001) |
| NGP | 2.5 (0.8) | 1.0 (0.4) | 0.003 (0.001) | 1.7 (0.3) | 0.010 (0.002) | 9.2 (2.3) | 0.0005 (0.0002) | |
| Rhabdomyosarcoma | SJCRH-30 | 7.7 (2.3) | 1.9 (0.3) | 0.016 (0.005) | 3.8 (0.9) | 0.021 (0.006) | 17.8 (3.0) | 0.003 (0.001) |
| RD | 3.9 (1.3) | 1.4 (0.5) | 0.002 (0.001) | 6.1 (1.3) | 0.019 (0.008) | 12.8 (2.8) | 0.0017 (0.0003) | |
| Osteosarcoma | HOS | 19.6 (2.5) | 1.6 (0.5) | 0.008 (0.002) | 5.2 (1.3) | 0.016 (0.005) | 4.9 (1.6) | 0.006 (0.002) |
| MG-63 | 4.7 (2.0) | 1.5 (0.4) | 0.013 (0.004) | 2.0 (0.7) | 0.035 (0.012) | 9.9 (3.0) | 0.002 (0.0004) | |
| Breast Carcinoma | HCC-1937 | ≈100 | ||||||
Olaparib was provided by AstraZeneca Pharmaceuticals (Wilmington, DE). Melphalan, doxorubicin, SN-38, carboplatin were purchased from Sigma (St. Louis, MO). Irinotecan and vincristine (VCR) were obtained from commercial sources. Irinotecan (20mM), carboplatin (20mM), and vincristine (1mM) were dissolved in sterile water. Doxorubicin was solubilized in phosphate-buffered saline to a final concentration of 3mM. Olaparib and SN38 were solubilized in dimethyl sulfoxide (DMSO) to final concentrations of 10mM and 1mM, respectively. Melphalan was dissolved in ethanol with 2–10% 1N HCl to a final concentration of 80mM or 160mM.
Sulforhodamine B (SRB) Assay
Twenty-four hours after plating in 96-well microtiter plates, cells were exposed to vehicle, olaparib, cytotoxic agent, or combination of olaparib with cytotoxic agent for 120 hours. Drugs, drug combinations, or vehicle control were added to achieve final concentrations: olaparib (0.02–100µM), irinotecan (0.0005–100µM), SN38 (4×10−5 −0.2µM), melphalan (0.06–200µM), doxorubicin (0.0003–0.6µM), carboplatin (0.02–200µM), vincristine (2–10−5 −0.1µM). Growth inhibition was assessed using the SRB assay as previously described[37,38]. The 50% growth inhibitory concentration (IC50) was determined by fitting a 4 parameter logistic equation to the data:
% survival = [(Emax - Emin) / (1 + (dose / EC50)slope] + Emin
where Emax and Emin are the concentrations at which maximum and minimum cytotoxicity are observed, respectively, and EC50 is the concentration at which 50% of the maximum cytotoxicity is attained (Emin + Emax)/2.
Determination of Synergy
The interaction between olaparib and cytotoxic agents was characterized using a constant drug ratio (based on IC50) and analyzed with the combination index (CI) method as previously described[37,39]. The interaction among drugs is characterized by the CI where a CI of < 1 indicates synergism, a CI = 1 indicates additive effects, and a CI of > 1 indicates an antagonistic interaction.
Xenograft Models
Materials
CB17 SCID female mice (Taconic Farms, Germantown, NY) were used to propagate subcutaneously implanted neuroblastoma (NGP) or Ewing sarcoma (RD-ES) tumors as previously described[40]. Olaparib was dissolved in DMSO to final concentration of 50 mg/1mL and then added to 10% 2-hydroxy-propyl-β-cyclodextrin/PBS solution to yield a solution of 2mg/mL (of active compound) and administered at 10µL/g of body weight by i.p. injection. Cyclophosphamide was dissolved in saline to yield a solution of 1mg/mL and administered at 10µL/g of body weight by i.p. injection. Topotecan was dissolved in saline to yield a solution of 0.5mg/mL and administered at 10µL/g of body weight by i.p. injection.
Treatment of Mice
The methods were based on well-established xenograft testing procedures in pediatric cancer models[40]. All experiments were conducted using protocols and conditions approved by the institutional animal care and use committee. Pilot tolerability experiments were performed to determine tolerable doses of topotecan, cyclophosphamide, and olaparib to be used in combination. When tumors reached a size of approximately 200mm3 mice were randomized into 4 groups: (i) olaparib (n=12), (ii) topotecan + cyclophosphamide (n=12), (iii) olaparib + topotecan + cyclophosphamide (n=12), or (iv) vehicle control (10% 2-hydroxy-propyl-β-cyclodextrin/PBS solution) (n=8). All chemotherapeutic agents and vehicle control were administered intraperitoneal injection daily for five days.
Analysis of Tumor Size
Tumor size was measured three times per week using manual spring calipers. Tumor measurements were calculated using the formula: π/6. ((length + width)/2)3. Mice were sacrificed due to tumor growth (≥ 2cm3) or unacceptable toxicity. Mice not meeting these criteria, were sacrificed on day 56.
Pharmacodynamic Assay
Evidence of PARP inhibition was tested in both cell lines and xenograft tumors using the Trevigen® (Gaithersburg, MD) HT PARP In Vivo Pharmacodynamic Assay II. This validated assay measures net PAR level in cellular extracts including tumor lysates from adherent cells in culture and xenografts models using monoclonal PAR capture antibody and quantification of PAR levels using chemiluminescent methods. Twenty-four hours after plating (density of 3×105 cells/mL), RD (rhabdomyosarcoma), RD-ES (Ewing sarcoma), NGP (neuroblastoma), and HCC1937 (BRCA deficient breast cancer) cell lines were exposed to 1µM of olaparib or vehicle control (0.1% DMSO/PBS solution). Following a 48-hour exposure, cells were lysed and evaluated for total protein and PAR activity.
PAR activity was assessed in tumors from both NGP and ES xenograft models following one dose of olaparib or vehicle control. Mice were randomized to receive: (i) olaparib (n=2), (ii) olaparib/topotecan/cyclophosphamide (n=2), or (iii) vehicle control (n=2) at doses and concentrations used in the treatment experiment. Topotecan/cyclophosphamide were administered 30 minutes after olaparib. Forty-five minutes following administration of olaparib or vehicle control mice were sacrificed and tumors were immediately snap frozen and stored in liquid nitrogen.
RESULTS
In vitro activity of olaparib
Growth inhibition in response to olaparib was tested in 10 pediatric solid tumor cell lines and HCC-1937, a breast carcinoma cell line known to be homozygous deficient for BRCA (Table 1, Figure 1). The Ewing sarcoma cell lines were the most sensitive (IC50 ≤ 1.5 µM) to olaparib followed by both medulloblastoma cell lines (IC50 ≤ 2.4 µM). Growth inhibition in the neuroblastoma cell lines and the osteosarcoma cell lines was variable. At clinically achievable olaparib concentrations, no growth inhibition was demonstrated in the BRCA deficient breast carcinoma cell line. In three pediatric solid tumor cell lines (ES, NGP and RD) exposure to olaparib (1µM) for 48hours resulted in 90% (range 86–93%) PARP inhibition and 100% PARP inhibition was achieved in HCC-1937.
Figure 1. Single agent IC50 of Olaparib.
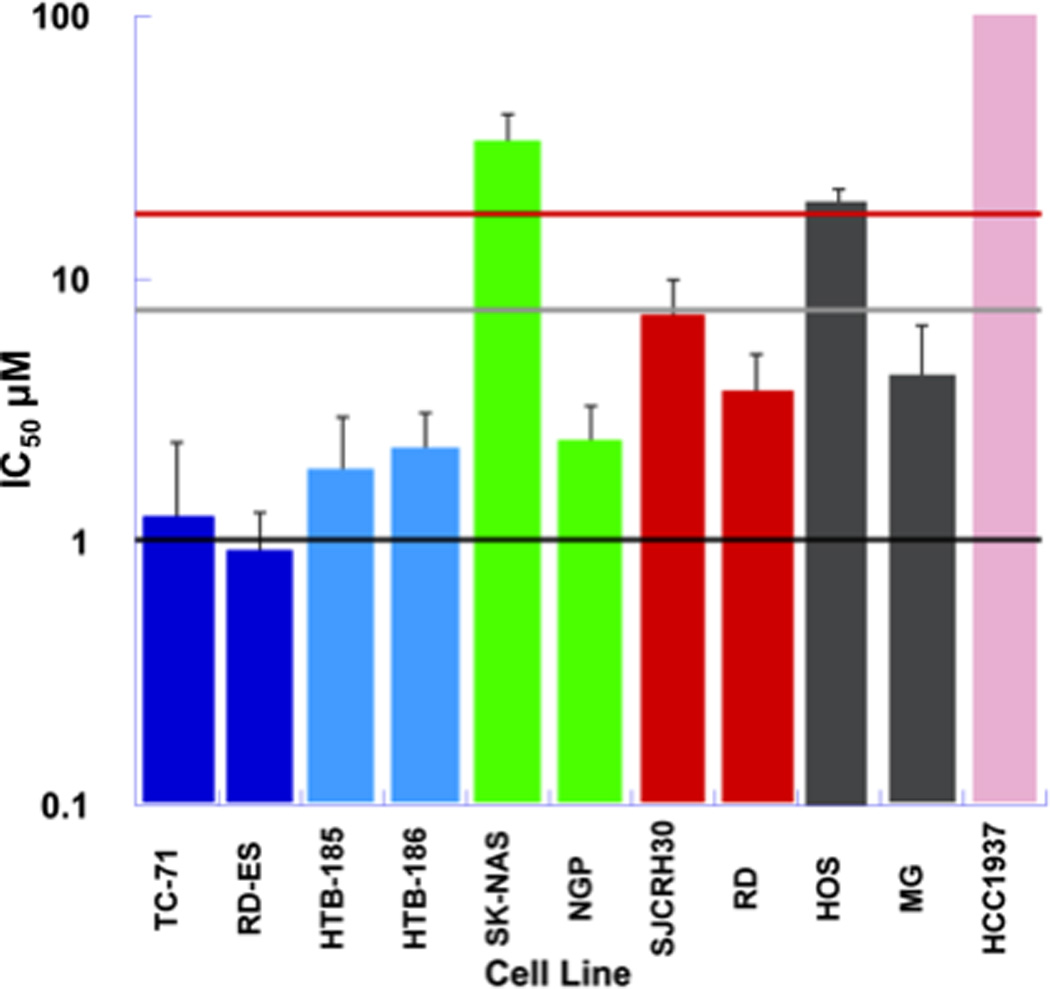
Single agent IC50 of olaparib in a solid tumor cell line panel consisting of Ewing sarcoma (blue), medulloblastoma (light blue), neuroblastoma (green), rhabdomyosarcoma (red), osteosarcoma (grey), and BRCA1 deficient (pink) cell lines. Plasma concentrations achieved in adults receiving olaparib (100 mg BID) are represented by the black line (Cmin=1µM) and light grey line (Cmax = 8.5 µM). The red horizontal line represents the plasma concentration (Cmax =18 µM) associated with maximum tolerated dose (400mg BID) of olaparib when given as a single agent[28].
In vitro activity of cytotoxic agents
In vitro sensitivity to single agent cytotoxic therapy was tested in pediatric solid tumor cell lines (Table I). Sensitivity to melphalan and doxorubicin was comparable across all cell lines. With the exception of the neuroblastoma cell line, NGP, vincristine IC50 ’s (0.002–0.006 µM) were similar; NGP demonstrated increased sensitivity (IC50= 0.0005µM) to vincristine. The medulloblastoma cell lines demonstrated the greatest sensitivity to carboplatin where as the rhabdomyosarcoma cell lines were the least sensitive. The response of cell lines to irinotecan and its active metabolite SN38 was consistent across pediatric solid tumor cell lines. The Ewing sarcoma and medulloblastoma cell lines demonstrated the greatest sensitivity to irinotecan and the sensitivity to SN38 was variable in rhabdomyosarcoma and neuroblastoma cell lines.
In vitro interaction of olaparib with cytotoxic chemotherapy
Evaluation of olaparib in combination with cytotoxic chemotherapy demonstrated a synergistic interaction with the DNA damaging agents. In the combination of olaparib with the camptothecin, irinotecan and its active metabolite SN38, the median (range) CI was 0.4 (0.06–1.0) and 0.2 (0.04–0.7), respectively (Figure 2). The synergistic interaction between olaparib and SN38 was most notable in the medulloblastoma (HTB-185 and HTB-187) and Ewing sarcoma (TC-71) cell lines where growth inhibition of the SN38+olaparib combination was 2 to 3 times that of the sum of the growth inhibition demonstrated when the agents were given alone (Figure 3). The interaction of olaparib with the tubulin inhibitor, vincristine, was disparate across cell lines with favorable interactions found in the neuroblastoma, osteosarcoma, and rhabdomyosarcoma cell lines and antagonistic interactions demonstrated in the Ewing sarcoma and medulloblastoma cell lines. The interaction of olaparib with cytotoxic agents was not evaluated in the BRCA deficient cell line due to the lack of growth inhibition at clinically achievable concentrations or concentrations required for this experimental design.
Figure 2. Interaction of Olaparib and Cytotoxic Agents.

The combination index (symbol) with 95% confidence interval (line) for olaparib in combination with each cytotoxic agent is presented for each cell line. Ewing Sarcoma lines (TC-71 and RD-ES) represented in dark blue, medulloblastoma lines (HTB-185 and HTB-186) represented in light blue, neuroblastoma lines (SK-NAS and NGP) represented in green, rhabdomyosarcoma lines (SJCRH-30 and RD) represented in red, and osteosarcoma lines (HOS and MG-63) represented in grey.
Figure 3. Interaction of Olaparib and SN-38.
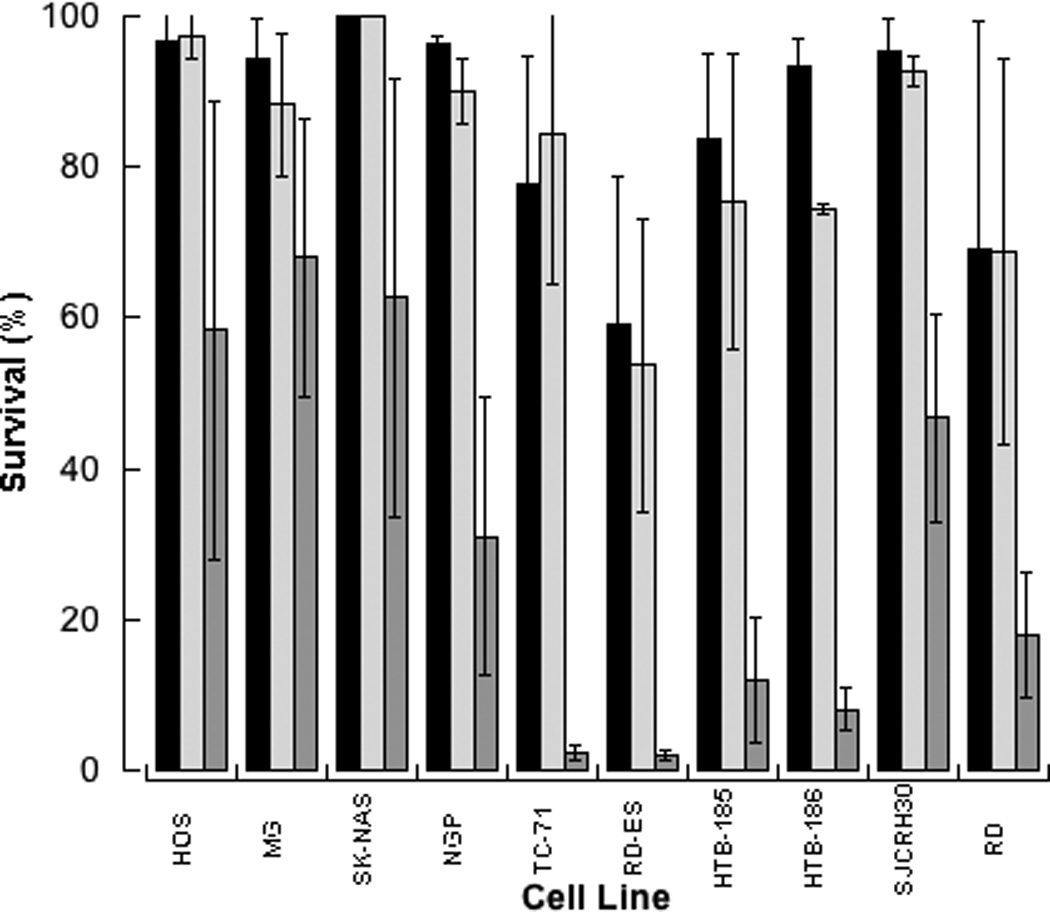
Growth inhibition (mean, SD) following exposure to 1µM olaparib (light grey), cell line specific concentration of SN-38 (black), or concomitant exposure to both agents (dark grey). Concentrations of SN-38, determined by the IC50 ratio of SN38 to olaparib, ranged from 0.00013 to 0.0033 µM and were the same in single agent and combination data presented.
In vivo activity of olaparib
The doses of olaparib, cyclophosphamide, and topotecan were identified in the xenograft models using a limited sample size (n=5) tolerability experiments. The maximum doses tolerated (weight loss/activity) were used in combination studies.
Olaparib administered at the dose required for tolerability in the combination experiments demonstrated negligible single agent activity in the xenograft models (Figures 4A and 4B), and had minimal impact when added to cyclophosphamide and topotecan. No residual tumor was identified at necropsy in the single surviving mouse at the time of sacrifice on day 56. Pharmacodynamic activity of olaparib in the NGP xenograft was demonstrated with a median of 88% (range 35–98%) PARP inhibition in the olaparib group and 100% (range 63–100%) inhibition in the olaparib in combination with topotecan/cyclophosphamide group compared to control. This confirms that PARP inhibition was achieved at doses administered and that the addition of cyclophosphamide/topotecan did not impact the ability of olaparib to inhibit PARP.
Figure 4. Responses of xenografts to topotecan and cyclophosphamide in combination with olaparib. A, Neuroblastoma xenograft (NGP). B, Ewing sarcoma xenograft (ES).
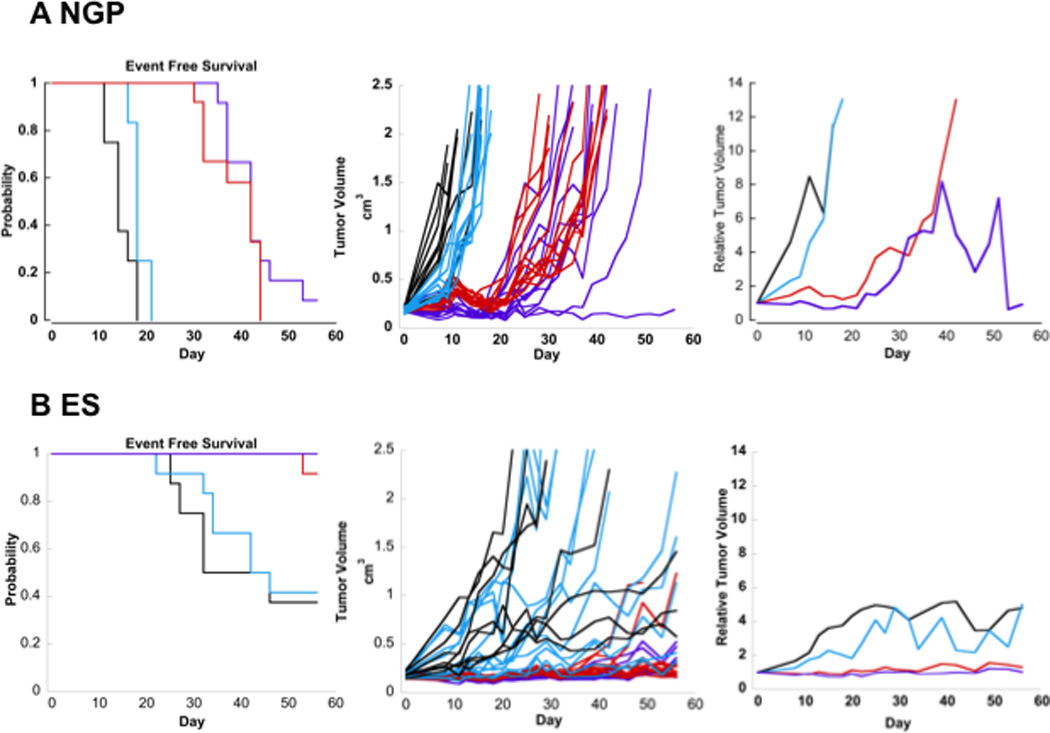
Event-free survival curves (left), individual tumor volume graphs (middle), and median relative tumor volume graphs (right) are shown for tumor xenografts. Tumor bearing mice were treated with a) vehicle control (black), b) olaparib (aqua) (20mg/kg daily ×5 days), c) topotecan (0.5mg/kg daily ×5 days) and cyclophosphamide (10mg/kg daily ×5 days) (red), or d) the combination of topotecan (0.5mg/kg daily ×5 days), cyclophosphamide (10mg/kg daily ×5 days), and olaparib (20mg/kg daily ×5 days) (purple).
Due to the marked efficacy of cyclophosphamide/topotecan regimen in the RD-ES xenograft as demonstrated by 90% survival and no tumor growth at 56 days following a single 5-day course of chemotherapy, improvement in survival for the combination of cyclophosphamide/topotecan and olaparib could not be determined. However, the results demonstrate that the addition of olaparib did not adversely impact the activity of cyclophosphamide/topotecan. Evaluation for PARP inhibition demonstrated 100% (Range 100%) PARP inhibition in both the olaparib as well as olaparib in combination with cyclophosphamide/topotecan groups.
DISCUSSION
This report examined the effect of PARP inhibition on commonly used cytotoxic chemotherapy with differing mechanisms of action in a broad panel of pediatric solid tumor cell lines. Across the pediatric solid tumor histologies evaluated, single agent olaparib caused growth inhibition at clinically achievable concentrations. Of note, growth inhibition was not demonstrated at clinically achievable concentrations in the BRCA deficient breast cancer cell line. Despite significant differences in growth inhibition, ≥ 90% inhibition of PARP was found in the RD-ES, NGP, RD, and HCC1937 cell lines when exposed to 1µM olaparib for 48hours. This observed dissociation between the anti-proliferative activity of olaparib and its inhibition of PARP supports the recent findings by Murai et al. that cytotoxicity of PARP inhibitors may not be solely explained their catalytic inhibition of PARP[33].
Our in vitro analysis demonstrated that olaparib potentiated the activity of DNA damaging agents in this panel of pediatric cell lines. The interactions between olaparib and the camptothecins, irinotecan and SN38 were the most favorable (CI << 1) across all cell lines evaluated. The decision to evaluate both irinotecan as well as SN38 was based on prior publications demonstrating differential sensitivity to the two compounds attributed to decreased intracellular activation of irinotecan by carboxylesterase[41–43]. In addition, olaparib had favorable interaction profile with the DNA damaging agents: melphalan, carboplatin, and doxorubicin. The interaction between olaparib and the microtubule toxin, vincristine whose mechanism of action is independent of DNA damage, was highly variable with synergism found in some lines and antagonism in others.
Based on our in vitro results demonstrating synergy (CI <0.7) between olaparib, a topoisomerase inhibitor or an alkylating agent, we evaluated olaparib in combination with topotecan and cyclophosphamide in xenograft models of Ewing sarcoma and neuroblastoma. In vivo evaluations demonstrated that the addition of olaparib to topotecan/cyclophosphamide did not adversely impact the efficacy of the topotecan/cyclophosphamide regimen. In the neuroblastoma xenograft, NGP, a trend toward favorable interaction was reflected in both tumor size and survival advantage. Due to the efficacy in controlling tumor growth demonstrated by the topotecan/cyclophosphamide regimen, the benefit of olaparib could not be demonstrated. A pharmacokinetic interaction was not directly evaluated, however, based on metabolic pathways of these agents and similar toxicity profiles during the in vivo evaluation, it is unlikely that the benefit of olaparib is related to drug-drug interaction.
Limitations of in vitro and in vivo models in drug development, particularly pediatric drug development, are well known. However, pre-clinical experiments are necessary for new agent prioritization in pediatric cancer and clinical trial optimization, particularly for development of combination regimens[44]. In our experiments, we selected drug exposures based on clinically achievable drug concentrations and screened ten cell lines representing five common pediatric solid tumors. Selection of in vivo models was based on results of in vitro studies. We evaluated the combination of olaparib with cyclophosphamide and topotecan because this is a well-established therapeutic regimen in neuroblastoma and Ewing sarcoma and contains classes of DNA damaging agents that demonstrated in vitro synergy. We did not evaluate topotecan and cyclophosphamide in vitro because their activity requires environmental conditions (pH dependent ring opening and enzyme activation, respectively) that are not feasible in vitro. To optimize the dose of cytotoxic agents in vivo, we performed a pilot study to establish the maximum tolerated doses of topotecan and cyclophosphamide in our xenograft models prior to combination with olaparib. In order to assess the ability of olaparib to improved activity or efficacy of the standard combination chemotherapy regimen, we did not decrease the doses of cyclophosphamide and topotecan in vivo.
Timely validation and implementation of pharmacodynamic endpoints in pre-clinical studies and clinical trials are necessary but remain challenging. In our study we demonstrated that olaparib inhibited PAR activity in vitro and in tumor xenografts. Our results demonstrate that the sensitivity of pediatric solid tumors to PARP inhibition is not limited to Ewing sarcoma. The synergistic interaction between PARP inhibition and DNA damaging agents, most notably the camptothecins, was ubiquitous across the pediatric solid tumor cell lines studied. Evaluation using xenograft models confirmed that the addition of olaparib to cyclophosphamide/topotecan did not adversely impact the efficacy of the combination. These results support a role for olaparib in combination with DNA damaging agents in the treatment of pediatric solid tumors. Further preclinical evaluation should be conducted with the PARP inhibitors, including olaparib, to determine the optimal combination and schedule of PARP inhibition in combination with cytotoxic chemotherapy for early phase clinical evaluation in a spectrum of pediatric solid tumors.
Acknowledgement
This work (REN) was supported by the CTSA K12-KL2RR024134 grants from the National Institute of Health, the Alex’s Lemonade Stand Young Investigator Grant, and The Children’s Hospital of Philadelphia Foerderer Award. This work was supported, in part, by The Children’s Hospital of Philadelphia Pediatric Development Fund (EF).
REFERENCES
- 1.Audeh MW, Carmichael J, Penson RT, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010;376(9737):245–251. doi: 10.1016/S0140-6736(10)60893-8. [DOI] [PubMed] [Google Scholar]
- 2.Tutt A, Robson M, Garber JE, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376(9737):235–244. doi: 10.1016/S0140-6736(10)60892-6. [DOI] [PubMed] [Google Scholar]
- 3.Pollack IF, Hamilton RL, Sobol RW, et al. Mismatch repair deficiency is an uncommon mechanism of alkylator resistance in pediatric malignant gliomas: a report from the Children's Oncology Group. Pediatr Blood Cancer. 2010;55(6):1066–1071. doi: 10.1002/pbc.22634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zarghooni M, Bartels U, Lee E, et al. Whole-Genome Profiling of Pediatric Diffuse Intrinsic Pontine Gliomas Highlights Platelet-Derived Growth Factor Receptor {alpha} and Poly (ADP-ribose) Polymerase As Potential Therapeutic Targets. J Clin Oncol. 2010 doi: 10.1200/JCO.2009.25.5463. [DOI] [PubMed] [Google Scholar]
- 5.Staal FJ, de Ridder D, Szczepanski T, et al. Genome-wide expression analysis of paired diagnosis-relapse samples in ALL indicates involvement of pathways related to DNA replication, cell cycle and DNA repair, independent of immune phenotype. Leukemia. 2010;24(3):491–499. doi: 10.1038/leu.2009.286. [DOI] [PubMed] [Google Scholar]
- 6.Barton VN, Donson AM, Kleinschmidt-DeMasters BK, et al. PARP1 expression in pediatric central nervous system tumors. Pediatr Blood Cancer. 2009;53(7):1227–1230. doi: 10.1002/pbc.22141. [DOI] [PubMed] [Google Scholar]
- 7.Wagner LM, McLendon RE, Yoon KJ, et al. Targeting methylguanine-DNA methyltransferase in the treatment of neuroblastoma. Clin Cancer Res. 2007;13(18 Pt 1):5418–5425. doi: 10.1158/1078-0432.CCR-07-0418. [DOI] [PubMed] [Google Scholar]
- 8.Kennedy RD, D'Andrea AD. DNA repair pathways in clinical practice: lessons from pediatric cancer susceptibility syndromes. J Clin Oncol. 2006;24(23):3799–3808. doi: 10.1200/JCO.2005.05.4171. [DOI] [PubMed] [Google Scholar]
- 9.Thomson B, Tritt R, Davis M, et al. Histology-specific expression of a DNA repair protein in pediatric rhabdomyosarcomas. J Pediatr Hematol Oncol. 2001;23(4):234–239. doi: 10.1097/00043426-200105000-00011. [DOI] [PubMed] [Google Scholar]
- 10.Phillips CL, Gerbing R, Alonzo T, et al. MDM2 polymorphism increases susceptibility to childhood acute myeloid leukemia: a report from the Children's Oncology Group. Pediatr Blood Cancer. 2010;55(2):248–253. doi: 10.1002/pbc.22519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shang X, Vasudevan SA, Yu Y, et al. Dual-specificity phosphatase 26 is a novel p53 phosphatase and inhibits p53 tumor suppressor functions in human neuroblastoma. Oncogene. 2010;29(35):4938–4946. doi: 10.1038/onc.2010.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tabori U, Baskin B, Shago M, et al. Universal poor survival in children with medulloblastoma harboring somatic TP53 mutations. J Clin Oncol. 2010;28(8):1345–1350. doi: 10.1200/JCO.2009.23.5952. [DOI] [PubMed] [Google Scholar]
- 13.Cattelani S, Defferrari R, Marsilio S, et al. Impact of a single nucleotide polymorphism in the MDM2 gene on neuroblastoma development and aggressiveness: results of a pilot study on 239 patients. Clin Cancer Res. 2008;14(11):3248–3253. doi: 10.1158/1078-0432.CCR-07-4725. [DOI] [PubMed] [Google Scholar]
- 14.Strahm B, Malkin D. Hereditary cancer predisposition in children: genetic basis and clinical implications. Int J Cancer. 2006;119(9):2001–2006. doi: 10.1002/ijc.21962. [DOI] [PubMed] [Google Scholar]
- 15.Thomas HD, Calabrese CR, Batey MA, et al. Preclinical selection of a novel poly(ADP-ribose) polymerase inhibitor for clinical trial. Mol Cancer Ther. 2007;6(3):945–956. doi: 10.1158/1535-7163.MCT-06-0552. [DOI] [PubMed] [Google Scholar]
- 16.Calabrese CR, Almassy R, Barton S, et al. Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor AG14361. J Natl Cancer Inst. 2004;96(1):56–67. doi: 10.1093/jnci/djh005. [DOI] [PubMed] [Google Scholar]
- 17.Daniel RA, Rozanska AL, Mulligan EA, et al. Central nervous system penetration and enhancement of temozolomide activity in childhood medulloblastoma models by poly(ADP-ribose) polymerase inhibitor AG-014699. British journal of cancer. 2010;103(10):1588–1596. doi: 10.1038/sj.bjc.6605946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Daniel RA, Rozanska AL, Thomas HD, et al. Inhibition of poly(ADP-ribose) polymerase-1 enhances temozolomide and topotecan activity against childhood neuroblastoma. Clin Cancer Res. 2009;15(4):1241–1249. doi: 10.1158/1078-0432.CCR-08-1095. [DOI] [PubMed] [Google Scholar]
- 19.Horton TM, Jenkins G, Pati D, et al. Poly(ADP-ribose) polymerase inhibitor ABT-888 potentiates the cytotoxic activity of temozolomide in leukemia cells: influence of mismatch repair status and O6-methylguanine-DNA methyltransferase activity. Mol Cancer Ther. 2009;8(8):2232–2242. doi: 10.1158/1535-7163.MCT-09-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng CL, Johnson SP, Keir ST, et al. Poly(ADP-ribose) polymerase-1 inhibition reverses temozolomide resistance in a DNA mismatch repair-deficient malignant glioma xenograft. Mol Cancer Ther. 2005;4(9):1364–1368. doi: 10.1158/1535-7163.MCT-05-0128. [DOI] [PubMed] [Google Scholar]
- 21.Ossovskaya V, Li L, Broude E, et al. Abstract #5552: BSI-201 enhances the activity of multiple classes of cytotoxic agents and irradiation in triple negative breast cancer. AACR Meeting Abstracts. 2009;2009:5552-. (2_Annual_Meeting) [Google Scholar]
- 22.Ratnam K, Low JA. Current development of clinical inhibitors of poly(ADP-ribose) polymerase in oncology. Clin Cancer Res. 2007;13(5):1383–1388. doi: 10.1158/1078-0432.CCR-06-2260. [DOI] [PubMed] [Google Scholar]
- 23.Fenaux P, Chastang C, Chevret S, et al. A randomized comparison of all transretinoic acid (ATRA) followed by chemotherapy and ATRA plus chemotherapy and the role of maintenance therapy in newly diagnosed acute promyelocytic leukemia. The European APL Group. Blood. 1999;94(4):1192–1200. [PubMed] [Google Scholar]
- 24.Ades L, Sanz MA, Chevret S, et al. Treatment of newly diagnosed acute promyelocytic leukemia (APL): a comparison of French-Belgian-Swiss and PETHEMA results. Blood. 2008;111(3):1078–1084. doi: 10.1182/blood-2007-07-099978. [DOI] [PubMed] [Google Scholar]
- 25.Audeh MW, Penson RT, Friedlander M, et al. Phase II trial of the oral PARP inhibitor olaparib (AZD2281) in BRCA-deficient advanced ovarian cancer. ASCO Meeting Abstracts. 2009;27(15S):5500-. [Google Scholar]
- 26.Fong PC, Boss DS, Carden CP, et al. AZD2281 (KU-0059436), a PARP (poly ADP-ribose polymerase) inhibitor with single agent anticancer activity in patients with BRCA deficient ovarian cancer: Results from a phase I study. J Clin Oncol. 2008;26(15_suppl):5510-. (Meeting Abstracts) [Google Scholar]
- 27.Tutt A, Robson M, Garber JE, et al. Phase II trial of the oral PARP inhibitor olaparib in BRCA-deficient advanced breast cancer. ASCO Meeting Abstracts. 2009;27(18S):CRA501-. [Google Scholar]
- 28.Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 29.Sanz MA, Grimwade D, Tallman MS, et al. Management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood. 2009;113(9):1875–1891. doi: 10.1182/blood-2008-04-150250. [DOI] [PubMed] [Google Scholar]
- 30.Fong PC, Yap TA, Boss DS, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 2010;28(15):2512–2519. doi: 10.1200/JCO.2009.26.9589. [DOI] [PubMed] [Google Scholar]
- 31.Chuang HC, Kapuriya N, Kulp SK, et al. Differential anti-proliferative activities of poly(ADP-ribose) polymerase (PARP) inhibitors in triple-negative breast cancer cells. Breast cancer research and treatment. 2012;134(2):649–659. doi: 10.1007/s10549-012-2106-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scribner DR, Jr, Benbrook DM. Retinoids enhance cisplatin-based chemoradiation in cervical cancer cells in vitro. Gynecologic oncology. 2002;85(1):223–225. doi: 10.1006/gyno.2002.6590. [DOI] [PubMed] [Google Scholar]
- 33.Murai J, Huang SY, Das BB, et al. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012;72(21):5588–5599. doi: 10.1158/0008-5472.CAN-12-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sandhu S, Wenham R, Wilding G, et al. First-in-human trial of a poly(ADP-ribose) polymerase (PARP) inhibitor MK-4827 in advanced cancer patients (pts) with antitumor activity in BRCA-deficient and sporadic ovarian cancers. Journal of Clinical Oncology. 2010;28(15s) [Google Scholar]
- 35.Mosse YP, Greshock J, Margolin A, et al. High-resolution detection and mapping of genomic DNA alterations in neuroblastoma. Genes Chromosomes Cancer. 2005;43(4):390–403. doi: 10.1002/gcc.20198. [DOI] [PubMed] [Google Scholar]
- 36.Huang HJ, Angelo LS, Rodon J, et al. R1507, an anti-insulin-like growth factor-1 receptor (IGF-1R) antibody, and EWS/FLI-1 siRNA in Ewing's sarcoma: convergence at the IGF/IGFR/Akt axis. PloS one. 2011;6(10):e26060. doi: 10.1371/journal.pone.0026060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Norris RE, Minturn JE, Brodeur GM, et al. Preclinical evaluation of lestaurtinib (CEP-701) in combination with retinoids for neuroblastoma. Cancer Chemother Pharmacol. 2011;68(6):1469–1475. doi: 10.1007/s00280-011-1623-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Norris RE, Rappaport EF, Adamson PC. Preclinical evaluation of pemetrexed in pediatric solid tumors. Pediatric blood & cancer. 2011;57(7):1233–1235. doi: 10.1002/pbc.23286. [DOI] [PubMed] [Google Scholar]
- 39.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Advances in enzyme regulation. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 40.Kirstein MN, Houghton PJ, Cheshire PJ, et al. Relation between 9-aminocamptothecin systemic exposure and tumor response in human solid tumor xenografts. Clin Cancer Res. 2001;7(2):358–366. [PubMed] [Google Scholar]
- 41.Pisano C, Vesci L, Fodera R, et al. Antitumor activity of the combination of synthetic retinoid ST1926 and cisplatin in ovarian carcinoma models. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO. 2007;18(9):1500–1505. doi: 10.1093/annonc/mdm195. [DOI] [PubMed] [Google Scholar]
- 42.Arrieta O, Gonzalez-De la Rosa CH, Arechaga-Ocampo E, et al. Randomized phase II trial of All-trans-retinoic acid with chemotherapy based on paclitaxel and cisplatin as first-line treatment in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2010;28(21):3463–3471. doi: 10.1200/JCO.2009.26.6452. [DOI] [PubMed] [Google Scholar]
- 43.Goncalves A, Camerlo J, Bun H, et al. Phase II study of a combination of cisplatin, all-trans-retinoic acid and interferon-alpha in squamous cell carcinoma: clinical results and pharmacokinetics. Anticancer research. 2001;21(2B):1431–1437. [PubMed] [Google Scholar]
- 44.Yap TA, Omlin A, de Bono JS. Development of therapeutic combinations targeting major cancer signaling pathways. J Clin Oncol. 2013;31(12):1592–1605. doi: 10.1200/JCO.2011.37.6418. [DOI] [PubMed] [Google Scholar]