

Abstract
Increasing evidence implicates the orexin 1 (OX1) receptor in reward processes, suggesting OX1 antagonism could be therapeutic in drug addiction. In a program to develop an OX1 selective antagonist, we designed and synthesized a series of substituted tetrahydroisoquinolines and determined their potency in OX1 and OX2 calcium mobilization assays. Structure-activity relationship (SAR) studies revealed limited steric tolerance and preference for electron deficiency at the 7-position. Pyridylmethyl groups were shown to be optimal for activity at the acetamide position. Computational studies resulted in a pharmacophore model and confirmed the SAR results. Compound 72 significantly attenuated the development of place preference for cocaine in rats.
Keywords: Orexin, antagonist, selective, tetrahydroisoquinoline
Introduction
Orexin A and B, also known as hypocretin 1 and 2, are hypothalamic neuropeptides independently discovered by two groups in 1998.1, 2 They are the endogenous ligands for two G protein-coupled receptors (GPCRs), orexin 1 (OX1) and orexin 2 (OX2).3, 4 Orexin-expressing neurons are located predominantly in a small area of the hypothalamus.2, 5-7 However, the nerve fibers of orexin neurons project throughout the central nervous system (CNS).1, 7-9 Interestingly, orexin receptors have different patterns of expression: Some brain regions express predominantly OX1, others predominantly OX2,2, 10, 11 suggesting that these receptors might modulate many unrelated functions as also suggested by antagonist and knock-out mouse studies.3, 4, 12 The orexin system has been shown to play a role in a variety of important biological processes, including sleep/wake cycle,13, 14 feeding,2 and energy homeostasis.2
Recently, the orexin system, particularly the OX1 receptor, was implicated in drug reward, reinstatement of drug seeking and psychomotor sensitization.15-18 This is consistent with the findings that orexin neurons have a prominent input to basal ganglia and forebrain structures (central amygdala, ventral bed nucleus of the stria terminalis, nucleus accumbens shell, ventral pallidum and ventral tegmental area) that underlie motivation, reward, stress and addiction-related behaviors.19 Orexinergic signaling seems to be involved in the rewarding effects of natural rewards and some drugs. Mice lacking orexins not only have reduced appetite but also show much less addiction-like sequelae associated with exposure to morphine and amphetamines.20, 21 Chemical activation of lateral hypothalamus (LH) neurons reinstates extinguished morphine seeking in rats, and the effect is blocked by the selective OX1 antagonist SB334867 (1).16 Blockade of orexin A transmission also decreases alcohol self-administration and cue-induced reinstatement of extinguished alcohol and cocaine seeking,22, 23 and attenuates stress-induced reinstatement of extinguished cocaine and alcohol seeking.19, 24 Moreover, antagonism of orexin A transmission at the OX1 receptor decreases nicotine self-administration in rats and the motivation to obtain the drug.25
Early efforts to modulate the orexin system have been focused on the blockade of both receptors, or OX2 selectively, because of their well-recognized implications in the pathophysiology of sleep disorders. As a result, several structural classes of dual OX1/OX2 antagonists and OX2 selective antagonists were developed.12, 26-29 The dual antagonists almorexant (2), SB-649868 (3) and suvorexant (4) entered clinical trials for insomnia.27, 28 However, relatively few OX1 selective antagonists have been reported to date. Compound 1, developed by GlaxoSmithKline, was the first selective OX1 antagonist described.30, 31 It has a potency of ~40 nM at OX1 and is at least 50-fold selective for OX1 over OX2. Structure-activity relationship (SAR) studies have resulted in analogs with improved OX1 selectivity,32 but 1 remains an important tool to probe the pharmacology and function of OX1. Despite its wide application, 1 has limitations including a less than desirable pharmacokinetic profile and the recently discovered hydrolytic instability where the 2-methylbenzoxazole undergoes ring opening under acidic conditions.31, 33 Other antagonists with OX1 selectivity or preference are the pyrrolidine-based SB674042 and disubstituted piperidines based on the structure of 3.34, 35 Two compounds of the latter series showed OX1 selectivity or preference, though significant OX2 activity remains for most.35 A similar approach by Rottapharm resulted in a number of spiro-piperidines and spiro-pyrrolidines with reasonable OX1 potency and selectivity.36 During the preparation of this manuscript, a phenylglycine-amide substituted tetrahydroisoquinoline derivative (ACT-335827) was reported by Actelion that was potent and OX1 selective; however some OX2 activity remained (IC50 OX1 = 6 nM, OX2 = 417 nM).37
Here we report our efforts in developing OX1 selective antagonists based on a tetrahydroisoquinoline scaffold as present in both 2 and TCS-OX2-29 (5), a selective OX2 antagonist.38 Compound 6, identified in a high throughput screening campaign by Actelion which led to the discovery of 2, showed reasonable OX1 activity and selectivity (IC50 OX1 = 119 nM, OX2 = 8100 nM).39 Initial SAR studies by the same group identified several positions that could be modified to improve OX1 activity. For example, replacement of the methoxy group at the 7-position with larger groups such as ethoxy or propoxy further increased OX1 potency while little OX2 activity was observed. The present work further examines SARs within the tetrahydroisoquinoline series, with the aim to improve potency and selectivity over OX2 and identify a viable tool compound for pharmacological studies. Our investigation looked specifically at the structural requirement at the 7-position of the tetrahydroisoquinoline, as well as the substitution pattern at the acetamide.
Results and Discussion
Chemistry
7-Alkoxytetrahydroisoquinolines were synthesized according to Scheme 1. Thus, amide coupling between amine 7 and acid 8 using HBTU yielded 9. Bischler-Napieralski reaction of 9 with phosphorus oxychloride in toluene at 90 °C gave the intermediate dihydroisoquinoline,40 which was readily reduced to tetrahydroisoquinoline 10 using sodium borohydride. N-Alkylation with α-bromo-benzylacetoamide gave the desired tetrahydroisoquinoline derivative 11.41, 42 The 7-position could then be alkylated as desired using potassium carbonate as base and the addition of tetrabutylammonium iodide as appropriate. For the trifluoroethyl analog 24, better results were obtained using cesium carbonate at 50 °C. Sulfonates (22-23, 25-26) were prepared using the appropriate sulfonyl chloride in dichloromethane with triethylamine as base.
Scheme 1.

Synthesis of 6-Methoxy-7-Alkoxy-N-benzylacetamidotetrahydroisoquinolinesa
To prepare compounds varying in the N-acetamide (Scheme 2), the base tetramethoxy-tetrahydroisoquinoline (31) was made according to literature procedures and then N-alkylated by ethyl bromoacetate to give ester 32.39 The ester could then be hydrolyzed and the acid converted to the amide using BOP and the corresponding amine. Further elaboration of the 4-nitrophenyl derivative gave a series of acylated anilines 57-59. The benzyl ester 40 was also made by alkylation on the acid with benzyl bromide.
Scheme 2.

Synthesis of N-acetamidotetrahydroisoquinolinesa
To prepare the 7-alkoxy derivatives, with greater variety in the acetamide, a series of compounds were synthesized via Scheme 3. Phenol 10 was alkylated with ethyl bromoacetate to give the tetrahydroisoquinoline acetate 65 in good yield. The phenol was then alkylated with the appropriate alkyl halide, with either potassium or cesium carbonate as base. Hydrolysis of the ester 66-68 with aqueous sodium hydroxide in ethanol gave the acid, which could then be coupled with the desired amine using BOP as coupling agent.
Scheme 3.

Synthesis of tetrahydroisoquinoline derivatives varied at both acetamide and 7 positionsa
Activity of the target compounds at the OX1 and OX2 receptors was evaluated in a calcium mobilization based functional assay. The apparent dissociation constant Ke was calculated from compound-mediated inhibition of orexin A activity as previously described.32 Briefly, EC50 curves of the agonist, orexin A, were obtained alone and together with the test compound, respectively, and the right-shift of the agonist curve was measured. Ke values were then calculated using the equation Ke = [L]/ ((EC50−/EC50+)-1), where [L] is the test compound concentration, EC50– is the EC50 of orexin A alone, and EC50+ is the EC50 of orexin A in the presence of test compound. In these assays, The EC50 for orexin A at OX1 and OX2 is 0.13 ± 0.02 nM and 4.2 ± 0.2 nM, respectively. All the compounds that had OX1 Ke values < 1 μM were also tested alone at 10 μM as agonists at the OX1 receptor. None of them showed any agonist activity. Due to the lack of commercially available radioligands, binding studies were not performed on these compounds.
Our studies on this series focused on two primary areas of the molecule, the 7-position of the tetrahydroisoquinoline moiety itself and the acetamide side chain. Table 1 shows changes at the 7-position. The unsubstituted 7-hydroxy analog 11 showed micromolar potency at both OX1 and OX2. The OX1 potency increased as the size of the alkoxy group increased (6, 12, 13), in agreement with the literature report.39 However, the potency started to drop as the chain length further increased (14, 15), and the isopentyl (16) was equipotent with hexyl (15). Finally, compound 17 with the space demanding cyclohexylmethyl group had no OX1 activity up to 10 μM, suggesting a limited bulk tolerance at this position. In an attempt to probe possible hydrogen bonding effects, heteroatoms as H-acceptors were introduced on the alkyl chain, and this resulted in significant decrease of potency (18, 19, 20). Aromatic substituents were then examined. While the benzyl (21) was similar in potency to the hexyl (15), the two pyridylmethyl analogs (22, 23) showed a modest increase in potency over the benzyl 21. Interestingly, when a phenoxybutyl group was introduced, 24 displayed equipotency to the butyl analog. This may suggest additional aromatic stacking interaction in this region, probably further away from the tetrahydroisoquinoline core.
Table 1.
Effect of the 7-position substitution on OX antagonism
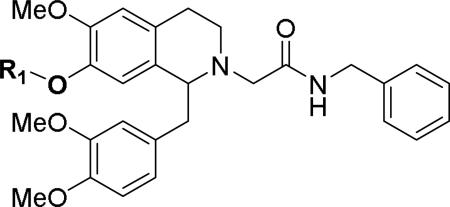
| ||||
|---|---|---|---|---|
| No. | R1 | Ke (OX1, nM)b | Ke (OX2, nM)c | OX2/OX1 |
| 11 | H | 1530±530 | 5740±96 | 3.8 |
| 6 | Methyl | 199±47 | >10000 | >50.3 |
| 12 | Ethyl | 30.0±9.0 | >10000 | >333 |
| 13 | n-Propyl | 17.3±3.1 | >10000 | >578 |
| 14 | n-Butyl | 54.7±23 | 1890±480 | 34.6 |
| 15 | n-Hexyl | 315±81 | >10000 | >31.7 |
| 16 | Isopentyl | 326±46 | 3920±1200 | 12.0 |
| 17 | CyclohexylCH2 | >10000 | a | |
| 18 | Me2N-CH2CH2 | 4740±440 | a | |
| 19 | Me2N-(CH2)4 | 5740±3600 | a | |
| 20 | N-Piperidinyl-CH2CH2 | 8040±5400 | a | |
| 21 | Benzyl | 402±58 | a | |
| 22 | 2-Pyridine-CH2 | 101±56 | a | |
| 23 | 3-Pyridine-CH2 | 277±75 | a | |
| 24 | PhO-(CH2)4 | 43.5±9.2 | >10000 | >230 |
| 25 | CH3CH2CH2CO | 78.5±24 | >10000 | >127 |
| 26 | CH3SO2 | 7.3±2.7 | 2170±790 | 297 |
| 27 | CF3SO2 | 10.0±2.8 | >10000 | >1000 |
| 28 | CF3CH2 | 9.4±1.7 | >10000 | >1060 |
| 29 | PhSO2 | 30.5±9.1 | 2900±191 | 95.1 |
| 30 | 4-MePhSO2 | 118±54 | a | |
< 30% inhibition at 10 μM
Values are the mean of at least three independent experiments in duplicate
Values are the mean of at least two independent experiments in duplicate; for compounds with Ke < 100 nM at OX1 at least three independent experiments in duplicate were performed.
Electronic properties at the 7-position were also investigated. The butyryl analog 25 showed similar OX1 activity as the butyl compound 14, suggesting electron withdrawing groups are tolerated. Interestingly, two highly electron withdrawing sulfonate analogs (26, 27) showed excellent potency, further confirming the preference of electronegativity at this position. Trifluoroethyl (28), a group that can be considered a bioisostere for acetyl or mesylate because of its electron deficient character and similar size, gave low nanomolar potency and over 1000-fold selectivity over the OX2 receptor. This also suggests that the observed high potency is a result of the electronic properties, other than potential hydrogen bonding interactions with the presence of the additional oxygen atoms. Finally, the phenyl (29) and tolyl (30) sulfonates both had higher potency than the benzyl analog.
At the acetamide position (Table 2), extending the benzyl to a phenethyl (33) or phenylpropyl (34) lowered potency at OX1. Conformational restriction of the aromatic group led to analogs such as the tetrahydroisoquinoline 35, the piperidines 36 and 37, and the piperazine 38, but none of these compounds showed any appreciable OX1 activity. It was posited that the NH group might be required for potency, and, indeed, its importance was highlighted by the N-methyl 39 and the benzyl ester 40, which both had low potency at OX1. None of these compounds showed any activity at OX2.
Table 2.
Effect of N-alkylation on OX antagonism
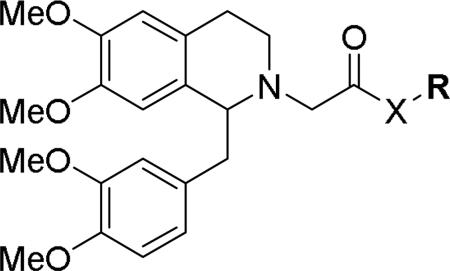
| ||||
|---|---|---|---|---|
| No. | X-R | Ke (OX1, nM)b | Ke (OX2, nM)c | OX2/OX1 |
| 6 |
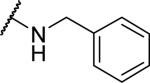
|
199±47 | >10000 | >50.3 |
| 33 |
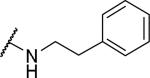
|
3250±230 | a | |
| 34 |
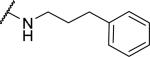
|
2370±240 | a | |
| 35 |
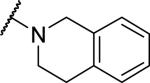
|
3030±170 | 9000±760 | 3.0 |
| 36 |
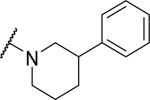
|
3530±650 | 9030±3800 | 2.6 |
| 37 |
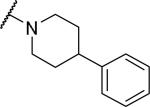
|
>10000 | a | |
| 38 |
|
>10000 | a | |
| 39 |
|
2090±330 | >10000 | >4.8 |
| 40 |
|
>10000 | a | |
| 41 |
|
732±250 | a | |
| 42 |
|
4220±1100 | a | |
| 43 |
|
4510±430 | a | |
| 44 |
|
944±160 | 1760±370 | 1.9 |
| 45 |
|
8980±2200 | a | |
| 46 |
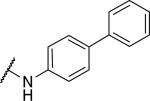
|
2240±110 | a | |
| 47 |
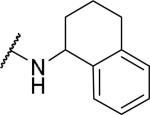
|
41.1±6.0 | 655±190 | 15.9 |
| 48 |
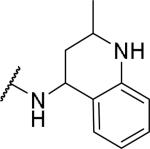
|
7170±390 | a | |
| 49 |
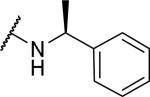
|
>l0000 | a | |
| 50 |
|
80.0±12 | >l0000 | >l25 |
| 51 |
|
162±16 | 2270±110 | 14.0 |
| 52 |
|
279±81 | 3460±1000 | 12.4 |
| 53 |
|
261±120 | >10000 | >38.3 |
| 54 |
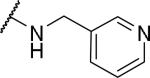
|
152±41 | >10000 | >65.8 |
| 55 |
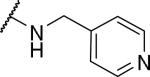
|
1010±160 | a | |
| 56 |
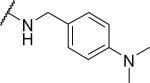
|
3230±1300 | >10000 | >3.1 |
| 57 |
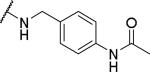
|
>10000 | a | |
| 58 |
|
>10000 | 3790±670 | <0.4 |
| 59 |
|
3680±1800 | 2440±670 | 0.7 |
| 60 |
|
589±230 | 2310±540 | 3.9 |
| 61 |
|
>10000 | a | |
| 62 |
|
6630±350 | a | |
| 63 |
|
7640±1100 | a | |
| 64 |
|
>10000 | a | |
< 30% inhibition at 10 μM
Values are the mean of at least three independent experiments in duplicate
Values are the mean of at least two independent experiments in duplicate; for compounds with Ke < 100 nM at OX1 at least three independent experiments in duplicate were performed.
Several fused aromatic or biphenyl systems were examined where the second aromatic ring occupies a similar position as that in the benzyl group but in a planar confirmation. The naphthalene 41 had a Ke of 732 nM, around 3.6-fold less potent than the benzyl compound. Heteroatom containing aromatic systems such as the quinolines 42 and 43 gave a further reduction in potency, suggesting a preference for a non-planar conformation for the aromatic ring. We next examined a series of biphenyl analogs 44-46, with each acting as a phenyl spacer to the second aromatic ring. However, only 2-biphenyl 44 showed sub-micromolar OX1 potency, with some activity at OX2. These results suggest that a non-aromatic spacer is preferred for OX1 potency. Indeed, the tetrahydronaphthalene (47) gave a Ke of 41 nM, significantly more potent than the naphthalene 41. Interestingly, the tetrahydroquinoline 48 was inactive, which shows that hydrophobic interactions might be preferred in this region of the molecule.
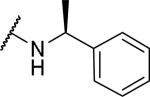
With the chiral center at the 1-position of the tetrahydroisoquinoline, substitution with a methyl at the α-position of the benzyl group (S-α–methylbenzyl) gave two diastereomers, which were separated by HPLC into compounds 49 and 50. While 50 showed good OX1 potency and selectivity, 49 was inactive at 10 μM at both receptors. This is consistent with previous findings that the S confirmation at the 1-position of the tetrahydroisoquinoline is required in 2 and its analogs.28, 43
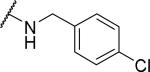
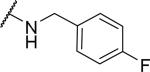
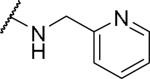
Substituents on the phenyl ring were generally well tolerated, with chloride 51 and fluoride 52 about equipotent with the benzyl (6), suggesting that electron withdrawing groups are tolerated. Among the pyridylmethyl analogs, the 3-pyridyl (54) showed similar potency as the 2-analog (53), both of which were more potent than the 4-analog (55). Interestingly, the electron rich dimethylamino (56) showed limited OX1 potency in the μM range. To determine whether this activity loss is due to electronic or steric reasons, substituents including amides and ureas were introduced onto the nitrogen. Surprisingly, the acetyl group (57) resulted in a total loss of OX1 activity. Large amide (58) and urea (59) showed no or little potency, suggesting limited or no tolerance for size at the 4-position of the benzyl group.
Finally, the requirement for aromaticity was investigated at the acetamide position. The straight chain heptyl analog 60 showed modest potency at 500 nM though was poorly selective over OX2. Attempts to introduce heteroatoms or polar groups (61-64) all resulted in dramatic loss of OX1 potency. Taken together, these findings suggest an aromatic group is generally needed for OX1 potency and selectivity.
Although none of the acetamide substituents gave a significant improvement in potency from the parent benzyl compound 6, the pyridylmethyl analogs 54 and 55 showed most promise given their improved physicochemical properties due to the possibility of salt formation. Thus, several pyridylmethyl analogs were studied in conjunction with the more potent 7-position substituents (Table 3). Such analogs include 2- and 3-pyridines combined with the 7-ethoxy, propoxy and trifluoroethoxy derivatives (69-74). All compounds showed good to excellent OX1 potency (8-87 nM) and high selectivity over OX2, with none of the compounds showing any activity at 10 μM.
Table 3.
Effect of multiple changes on OX antagonism
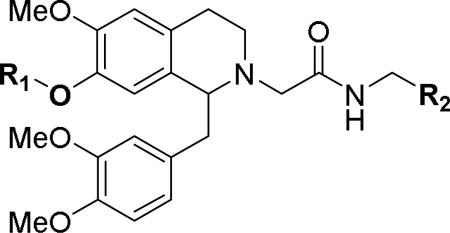
| |||||
|---|---|---|---|---|---|
| No | R1 | R2 | Ke (OX1, nM)a | Ke (OX2, nM)b | OX2/OX1 |
| 69 | n-Propyl | 2-Pyridyl | 20.6±5.5 | >10000 | >485 |
| 70 | n-Propyl | 3-Pyridyl | 41.9±16 | >10000 | >239 |
| 71 | CF3CH2 | 2-Pyridyl | 22.5±2.7 | >10000 | >444 |
| 72 | CF3CH2 | 3-Pyridyl | 8.5±1.0 | >10000 | >1180 |
| 73 | Ethyl | 2-Pyridyl | 87.4±12 | >10000 | >114 |
| 74 | Ethyl | 3-Pyridyl | 15.7±1.7 | >10000 | >637 |
Values are the mean of at least three independent experiments in duplicate
Values are the mean of at least three independent experiments in duplicate.
Computational Analysis
A comprehensive 3D-pharmacophore for the orexin antagonists evaluated in this study was developed. The pharmacophore was initiated by generating conformational libraries for each analog in this class. This included computation of the 3D-distance metrics between 4-candidate pharmacophore points (variable substituents off the 7-position, the two centroids for each of the aromatic ring substituents and the nitrogen on the tetrahydroisoquinoline core), and determination of the conformations of each ligand giving rise to a common 3D display (Figure 2A and B). Superposition of all ligands in the training set with these four pharmacophore points allowed us to investigate a series of substituent properties that proved important for OX1 potency. Twenty-one properties, including stereochemical (Verloop parameters,44, 45 volume, area, globularity), electrostatic (positions of electrostatic minima and maxima), polar (polar surface area), electronic (E-HOMO, E-LUMO, polarizability), thermodynamic (vibrational and rotational heat capacity and entropies at 300K) and atom based hydrophobicity (ClogP), were evaluated for ligand conformations that complied with a 3D-pharmacophore overlap rule.46 In addition, the same subset of properties was computed for substituents (fragments) alone without the tetrahydroisoquinoline core compliant pharmacophoric region. This dual approach was chosen to allow the addition of substituent independent properties to the QSAR analysis.
Figure 2.

A) Ligands superimposed at 3D-pharmacophore points defined by B) the abstract pharmacophore representation computed from 21 ligands in Table 1. C) a plot of the predicted lnKe(OX1) vs. experimental for a fragment (2D) QSAR illustrating that contributions from the width (Verloop Sterimol “B1”), polar surface area, ClogP and lowest unoccupied (LUMO) energy of the substituent give a robust analysis (r2=0.7/F=8.2 (n1=4,n2=18), p=0.001/ 2.8B1+0.7*E(LUMO)-0.13logP-5.5)). D) 3D-COMFA/QSAR illustration of the increased steric bulk contributions region of the substituent variation highlighted by green correlate with low Ke values with minor modulation due to electrostatic contributions. E) a depiction of the location of the LUMO density on compound 6.
Three properties were found to correlate structural modification at the 7-position with alterations in OX1 potency using a three variable QSAR analysis. The Sterimol parameters including spatial extent of the substituent at position 7 and particularly the width (Verloop B1) relative to the long-axis, logP of the substituent, and energy of the lowest unoccupied molecular orbital (E-LUMO) demonstrated statistically significant correlations with changes in OX1 potency (r2 = 0.69) (Figure 2C). It is apparent from the 3-D QSAR model (Figure 2D) and the functional activity data that size of the substituent plays a considerable role in determining potency at the 7-position. OX1 potency increases monotonically from 1530 nM to 17 nM for compounds 11 (−OH), 6 (−OCH3), 12 (−OCH2CH3), and 13 (−OCH2CH2CH3) respectively at which point the trend begins to reverse. This indicates that substituents of a particular size are required for activity.
Although steric properties play a main role in determining potency at the 7-position, the QSAR analysis also highlights the importance of other non-observable factors such as LUMO energy on antagonist potency. Figure 2E depicts the spatial distribution of the LUMO density on compound 6. Position 7 is on the aromatic ring most associated with LUMO electron density. Naturally, substitutions at that position would modulate the energy of the LUMO orbital and charge density transfer, a facet plausibly having a role in stabilization of the receptor bound ligand configuration.
Figure 3 depicts a similar 3D-QSAR and COMFA analysis for substituent variation corresponding to the N-alkylation variations summarized in Table 2. Figure 3B illustrates results from a COMFA analysis of the training set superposition shown in Figure 3A. Both steric and electrostatic variations in the ligand substituents contributed relatively equally to the underlying rationale for the Ke(OX1) variations. This was independently verified in the 2D-QSAR analysis shown in Figure 3D, where coefficients of the B1-Verloop parameter and the polar surface area made significant contributions, along with the energy of the HOMO, and led to a model predictive of lnKe(OX1). Analogous to our identification of the E(LUMO) term for position 7 variations, the atomic centers containing the highest occupied molecular orbital were found on the ring system directly attached to the position 2 substitutions. The results from these studies highlight the manner in which substitutions can be rationalized in terms of underlying physical properties keyed to the molecular interactions and binding free energies and provide a basis for a comprehensive 3D-pharmacophore.
Figure 3.

Depiction of COMFA steric (left) and electrostatic fields (right) about compound 6 derived from the analysis of 6, 20, 21, 23, 30, 41, 44, 47, 50, 51, 52, 55 and 60. The analysis (R2=0.761/F[n1=1,n2=11)] 35.108, SE=0.61) for lnKe(OX1) using the 3D-pharmacophore quaternion least squares superposition of pharmacophore points illustrated in Figure 2.
Through the establishment of this 3D-pharmacophore model a basic understanding of the impact specific substitutions have on receptor potency and selectivity has been achieved. This information will be important to guide the selection of scaffold modifications to favorably alter receptor potency, subtype selectivity, or ADM. Alternatively, establishing quantitative pharmacophore models allows us to transfer information of the 3D-display of physiochemical properties to new scaffolds with an understanding of those that are critical for activity.
Conditioned Place Preference
Given the well-established activity of OX1 antagonists in attenuating the rewarding and reinforcing effects of drugs, compound 72 was studied in conditioned place preference (CPP). As shown in Figure 4, vehicle did not produce significant CPP. Cocaine at 10 mg/kg induced robust and significant CPP. Compound 72 at 20 mg/kg did not produce CPP or place aversion but significantly attenuated the development of place preference induced by cocaine.
Figure 4.

Effects of compound 72 on cocaine-induced conditioned place preference in rats (n=8-9). Data presented as mean ± S.E.M. * P < 0.05 as compared to vehicle treated rats. $ P < 0.05 as compared to 10 mg/kg cocaine treated rats.
Conclusions
In an attempt to develop OX1 selective antagonists, we synthesized and evaluated a series of tetrahydroisoquinolines, a core structure that is present in both the dual OX1/OX2 antagonist 2 and the OX2 selective antagonist 5. SAR studies suggested a preference for steric bulk at the 7-position; however, this tolerance is limited, as the potency decreases with the further size increase of the substituents. Electron deficient groups are also well tolerated at this position. At the acetamide position, an aromatic system was generally preferred, and the pyridylmethyl groups gave the best potency. The pharmacophore model obtained through computational analysis confirmed the steric and electronic requirement at the 7-position. This model also suggests that steric and electrostatic variations in the ligand substituents contributed equally to the underlying rationale for the Ke (OX1) variations at the acetamide position. In the CPP paradigm, compound 72, which had excellent OX1 potency and selectivity over OX2 in the calcium assay, significantly attenuated cocaine CPP. In summary, several compounds with excellent potency at and selectivity for the OX1 receptor have been identified, and they will serve as probes to further investigate the pharmacology and function of the OX1 receptor and the orexin system. Modifications at other positions are ongoing and will be reported in due course.
Experimental
Chemistry
All solvents and chemicals were reagent grade. Unless otherwise mentioned, all were purchased from commercial vendors and used as received. Flash column chromatography was done on a Teledyne ISCO CombiFlash Rf system using prepacked columns. Solvents used were hexane, ethyl acetate (EtOAc), dichloromethane, methanol and chloroform:methanol:ammonium hydroxide (80:18:2) (CMA-80). Purity and characterization of compounds was established by a combination of high pressure liquid chromatography (HPLC), thin layer chromatography (TLC), mass spectrometry (MS) and nuclear magnetic resonance (NMR) analysis. 1H and 13C NMR spectra were recorded on a Bruker Avance DPX-300 (300 MHz) spectrometer and were determined in chloroform-d or methanol-d4 with tetramethylsilane (TMS) (0.00 ppm) or solvent peaks as the internal reference. Chemical shifts are reported in ppm relative to the reference signal, and coupling constant (J) values are reported in Hz. TLC was done on EMD precoated silica gel 60 F254 plates, and spots were visualized with UV light or iodine staining. Low resolution mass spectra were obtained using a Waters Alliance HT/Micromass ZQ system (ESI). All test compounds were greater than 95% pure (except where noted) as determined by HPLC on an Agilent 1100 system using an Agilent Zorbax SB-Phenyl, 2.1 mm x 150 mm, 5 μm column with gradient elution using the mobile phases (A) H2O containing 0.1% CF3COOH and (B) MeCN, with a flow rate of 1.0 mL/min.
2-(3,4-dimethoxyphenyl)-N-[2-(4-hydroxy-3-methoxyphenyl)ethyl]acetamide (9)
3,4-Dimethoxyphenylacetic acid (0.45 g, 2.29 mmol), 4-hydroxy-3-methoxyphenethylamine hydrochloride (0.47 g, 2.29 mmol) and HBTU (0.96 g, 2.52 mmol) were combined in dry dimethylformamide (20 mL) at RT under N2. Diisopropylethylamine (1.19 g, 1.6 mL, 9.17 mmol) was added and the reaction stirred at RT overnight. The reaction was diluted with ethyl acetate, washed with 2N hydrochloric acid, sodium bicarbonate solution and brine, dried over MgSO4 and the solvent removed under reduced pressure to give the desired amide as a yellow oil which solidified upon standing (0.72 g, 91%). 1H NMR (300 MHz, CHLOROFORM-d) δ ppm 2.67 (t, J=6.88 Hz, 2 H) 3.36 - 3.43 (m, 2 H) 3.45 (s, 2 H) 3.82 (s, 3 H) 3.83 (s, 3 H) 3.88 (s, 3 H) 6.04 (br. s., 1 H) 6.44 - 6.53 (m, 1 H) 6.61 (d, J=1.70 Hz, 1 H) 6.66 - 6.77 (m, 3 H) 6.78 - 6.86 (m, 1 H) 7.97 (s, 1 H).
General procedure for Bischler-Napieralski reaction/sodium borohydride reduction 1-[(3,4-dimethoxyphenyl)methyl]-6-methoxy-1,2,3,4-tetrahydroisoquinolin-7-ol (10)
Amide 9 (0.39 g, 0.91 mmol) was suspended in anhydrous toluene (5 mL) and phosphorous oxychloride (0.70 g, 0.4 mL, 4.56 mmol) added slowly. The reaction was heated to 90 °C for 2 hours. The reaction was cooled, then quenched by slow addition of the reaction mixture to water and stirred vigorously at room temperature for 15 min. Sodium hydroxide solution (2N) was added until pH was 8-9, then the solution was extracted 3 times with dichloromethane. The combined extracts were dried over MgSO4 and the solvent removed under reduced pressure.
The crude dihydroisoquinoline was dissolved in methanol (5 mL) and cooled in an ice bath under N2. Sodium borohydride (0.17 g, 4.52 mmol) was added portionwise and the reaction allowed to warm slowly to RT overnight. The reaction was quenched with water then the methanol removed under reduced pressure. The aqueous solution was extracted 3 times with dichloromethane and the combined extracts were dried over MgSO4 and the solvent removed under reduced pressure to give the desired tetrahydroisoquinoline 10 as a frothy solid (0.26 g, 67%). 1H NMR (300 MHz, CHLOROFORM-d) δ ppm 2.59 - 2.93 (m, 5 H) 3.11 - 3.24 (m, 1 H) 3.34 - 3.58 (m, 2 H) 3.79 - 3.92 (m, 9 H) 4.08 (dd, J=9.42, 3.20 Hz, 1 H) 6.57 (s, 1 H) 6.60 - 6.71 (m, 1 H) 6.72 - 6.88 (m, 3 H).
N-benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-7-hydroxy-6-methoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (11)
Amine 10 (3.65 g, 11.08 mmol), N-benzyl-2-bromoacetamide (3.77 g, 16.62 mmol) and tetrabutylammounium iodide (0.82 g, 2.21 mmol) were combined in dry DMF (110 mL) and diisopropylethylamine (3.58 g, 4.8 mL, 27.70 mmol) was added. The reaction was stirred at RT overnight under N2. The reaction was diluted with EtOAc, washed with NaHCO3 solution, water and brine (x2), then dried over MgSO4 and the solvent removed under reduced pressure. The crude was purified by chromatography on silica (0-100% EtOAc in hexane) to obtain the desired product as a frothy white solid (2.98 g, 56%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.15 - 7.36 (m, 3H), 7.07 (d, J = 7.06 Hz, 2H), 6.89 (br. s., 1H), 6.65 - 6.77 (m, 4H), 6.52 - 6.64 (m, 3H), 5.51 (s, 1H), 4.48 (dd, J = 8.24, 15.02 Hz, 1H), 3.88 (s, 3H), 3.80 (s, 3H), 3.73 (s, 3H), 3.33 - 3.68 (m, 3H), 3.06 - 3.32 (m, 2H), 2.78 - 2.99 (m, 4H), 2.46 (d, J = 15.82 Hz, 1H). m/z 477 (M+H).
General procedure for O-alkylation: N-benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-7-ethoxy-6-methoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (12)
Phenol 11 (25 mg, 0.025 mmol) and potassium carbonate (22 mg, 0.157 mmol) were combined in dry dimethylformamide, then 1-bromoethane (12 mg, 6 μL, 0.079 mmol) was added and the reaction stirred at RT under N2 overnight. It was diluted with ethyl acetate, washed with sodium bicarbonate solution and brine, dried over MgSO4 and the solvent was removed under reduced pressure. The compound was purified by chromatography on silica (0-75% EtOAc in hexane) to obtain the desired product as a pale yellow solid (19 mg, 73%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.19 - 7.34 (m, 3H), 7.07 - 7.14 (m, 1H), 6.94 - 7.01 (m, 1H), 6.56 - 6.74 (m, 5H), 6.48 (s, 1H), 4.49 (dd, J = 8.05, 14.93 Hz, 1H), 4.02 (q, J = 6.97 Hz, 2H), 3.85 (s, 3H), 3.81 (s, 3H), 3.74 (s, 3H), 3.57 - 3.70 (m, 2H), 3.34 - 3.48 (m, 1H), 3.12 - 3.34 (m, 2H), 2.79 - 2.99 (m, 4H), 2.41 - 2.54 (m, 1H), 1.45 (t, J = 6.97 Hz, 3H). m/z 505 (M+H).
N-benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (6)
Prepared in 51% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 - 7.35 (m, 2H), 7.07 - 7.14 (m, 2H), 6.98 (dd, J = 4.94, 7.58 Hz, 1H), 6.62 - 6.74 (m, 3H), 6.59 (s, 1H), 6.45 (s, 1H), 4.50 (dd, J = 8.05, 14.93 Hz, 1H), 3.84 - 3.89 (m, 3H), 3.81 (d, J = 1.88 Hz, 6H), 3.78 - 3.83 (m, 6H), 3.75 (s, 3H), 3.59 - 3.71 (m, 2H), 3.35 - 3.48 (m, 1H), 3.11 - 3.35 (m, 2H), 2.80 - 3.00 (m, 4H), 2.41 - 2.55 (m, 1H). m/z 491 (M+H).
N-benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-6-methoxy-7-propoxy-1,2,3,4-tetrahydroisoquinolin-2-.yl}acetamide (13)
Prepared in 48% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.20 - 7.34 (m, 3H), 7.07 - 7.14 (m, 2H), 6.98 (dd, J = 4.95, 7.77 Hz, 1H), 6.61 - 6.75 (m, 3H), 6.59 (s, 1H), 6.47 (s, 1H), 4.50 (dd, J = 8.10, 14.98 Hz, 1H), 3.89 (t, J = 6.88 Hz, 2H), 3.85 (s, 3H), 3.81 (s, 3H), 3.75 (s, 3H), 3.58 - 3.70 (m, 2H), 3.34 - 3.48 (m, 1H), 3.11 - 3.34 (m, 2H), 2.79 - 2.99 (m, 4H), 2.49 (d, J = 15.92 Hz, 1H), 1.77 - 1.92 (m, 2H), 1.00 - 1.09 (m, 3H). m/z 519 (M+H).
N-benzyl-2-{7-butoxy-1-[(3,4-dimethoxyphenyl)methyl]-6-methoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (14)
Prepared in 75% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.19 - 7.35 (m, 3H), 7.10 (d, J = 6.78 Hz, 2H), 6.93 - 7.02 (m, 1H), 6.61 - 6.75 (m, 3H), 6.58 (s, 1H), 6.47 (s, 1H), 4.50 (dd, J = 8.10, 14.98 Hz, 1H), 3.89 - 3.97 (m, 2H), 3.83 - 3.87 (m, 3H), 3.81 (s, 3H), 3.75 (s, 3H), 3.58 - 3.71 (m, 2H), 3.34 - 3.48 (m, 1H), 3.11 - 3.34 (m, 2H), 2.80 - 2.98 (m, 4H), 2.42 - 2.54 (m, 1H), 1.75 - 1.88 (m, 2H), 1.42 - 1.55 (m, 2H), 0.98 (t, J = 7.30 Hz, 3H). m/z 533 (M+H).
N-benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-7-(hexyloxy)-6-methoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (15)
Prepared in 64% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 - 7.35 (m, 3H), 7.10 (d, J = 6.69 Hz, 2H), 6.97 (dd, J = 4.99, 7.44 Hz, 1H), 6.61 - 6.76 (m, 3H), 6.58 (s, 1H), 6.48 (s, 1H), 4.50 (dd, J = 8.10, 14.98 Hz, 1H), 3.92 (t, J = 6.83 Hz, 2H), 3.82 - 3.87 (m, 3H), 3.81 (s, 3H), 3.74 (s, 3H), 3.58 - 3.69 (m, 2H), 3.34 - 3.49 (m, 1H), 3.11 - 3.34 (m, 2H), 2.80 - 2.99 (m, 4H), 2.41 - 2.54 (m, 1H), 1.75 - 1.89 (m, 2H), 1.30 - 1.53 (m, 6H), 0.91 (t, J = 6.90 Hz, 3H). m/z 561 (M+H).
N-benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-6-methoxy-7-(3-methylbutoxy)-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (16)
Prepared in 88% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.20 - 7.36 (m, 3H), 7.11 (d, J = 6.97 Hz, 2H), 6.95 - 7.04 (m, 1H), 6.62 - 6.76 (m, 3H), 6.59 (s, 1H), 6.48 (s, 1H), 4.51 (dd, J = 8.01, 14.98 Hz, 1H), 3.95 (t, J = 6.88 Hz, 2H), 3.85 (s, 3H), 3.81 (s, 3H), 3.75 (s, 3H), 3.60 - 3.71 (m, 2H), 3.35 - 3.49 (m, 1H), 3.13 - 3.34 (m, 2H), 2.78 - 2.99 (m, 4H), 2.49 (d, J = 15.73 Hz, 1H), 1.84 (td, J = 6.62, 13.33 Hz, 1H), 1.71 - 1.77 (m, 2H), 0.98 (d, J = 6.50 Hz, 6H). m/z 547 (M+H).
N-benzyl-2-[7-(cyclohexylmethoxy)-1-[(3,4-dimethoxyphenyl)methyl]-6-methoxy-1,2,3,4-tetrahydroisoquinolin-2-yl]acetamide (17)
Prepared in 33% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.19 - 7.34 (m, 3H), 7.10 (d, J = 6.78 Hz, 2H), 7.01 (d, J = 6.97 Hz, 1H), 6.61 - 6.76 (m, 3H), 6.58 (s, 1H), 6.45 (s, 1H), 4.49 (dd, J = 8.01, 14.98 Hz, 1H), 3.82 (s, 3H), 3.81 (s, 3H), 3.75 (s, 3H), 3.59 - 3.72 (m, 4H), 3.34 - 3.47 (m, 1H), 3.12 - 3.34 (m, 2H), 2.79 - 2.95 (m, 4H), 2.48 (d, J = 16.01 Hz, 1H), 1.61 - 1.97 (m, 6H), 0.92 - 1.40 (m, 5H). m/z 573 (M+H).
N-Benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-7-[2-(dimethylamino)ethoxy]-6-methoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (18)
Prepared in 46% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.19 - 7.34 (m, 3H), 7.07 - 7.14 (m, 2H), 6.95 (dd, J = 4.76, 7.77 Hz, 1H), 6.68 - 6.75 (m, 2H), 6.61 - 6.67 (m, 1H), 6.59 (s, 1H), 6.56 (s, 1H), 4.50 (dd, J = 8.19, 14.98 Hz, 1H), 4.01 - 4.12 (m, 2H), 3.84 (s, 3H), 3.82 (s, 3H), 3.75 (s, 3H), 3.57 - 3.68 (m, 2H), 3.36 - 3.50 (m, 1H), 3.12 - 3.34 (m, 2H), 2.83 - 2.99 (m, 4H), 2.75 - 2.82 (m, 2H), 2.44 - 2.54 (m, 1H), 2.38 (s, 6H). m/z 548 (M+H).
N-benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-7-[4-(dimethylamino)butoxy]-6-methoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (19)
Phenol 11 (0.30 g, 0.63 mmol) and potassium carbonate (0.35 g, 1.26 mmol) were combined in dimethylformamide (6 mL) and 1-bromo-4-chlorobutane (0.22 g, 0.15 mL, 1.26 mmol) was added. The reaction was heated to 50 °C overnight. The reaction was cooled, diluted with water and extracted twice with EtOAc. The combined extracts were washed with brine, dried over MgSO4 and the solvent removed under reduced pressure. It was then purified by chromatography on silica (0-80% EtOAc in hexane) to obtain the desired chloride (0.22 g, 61%) for use in the following reaction. 1H NMR (300 MHz, CHLOROFORM-d) δ ppm 7.20 - 7.35 (m, 3 H) 7.11 (d, J=7.16 Hz, 2 H) 6.93 - 7.01 (m, 1 H) 6.62 - 6.75 (m, 3 H) 6.60 (s, 1 H) 6.48 (s, 1 H) 4.50 (dd, J=14.93, 8.05 Hz, 1 H) 3.97 (t, J=5.51 Hz, 2 H) 3.85 (s, 3 H) 3.80 (s, 3 H) 3.75 (s, 3 H) 3.58 - 3.70 (m, 4 H) 3.36 - 3.55 (m, 1 H) 3.12 - 3.34 (m, 2 H) 2.79 - 2.98 (m, 4 H) 2.42 - 2.56 (m, 1 H) 1.94 - 2.03 (m, 4 H).
The chloride (30 mg, 0.053 mmol), dimethylamine hydrochloride (7 mg, 0.079 mmol), potassium carbonate (18 mg, 0.132 mmol) and tetrabutylammonium iodide (4 mg, 0.011 mmol) were combined in dimethylformamide (0.5 mL) and heated at 50 °C overnight. The reaction was cooled, diluted with water and extracted 3 times with EtOAc. The combined extracts were washed with brine, dried over MgSO4 and the solvent removed under reduced pressure. The compound was purified by chromatography on silica (0-50% CMA-80 in EtOAc) to give the desired product (6 mg, 19%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.19 - 7.35 (m, 3H), 7.07 - 7.15 (m, 2H), 6.91 - 7.00 (m, 1H), 6.62 - 6.76 (m, 3H), 6.59 (s, 1H), 6.51 (s, 1H), 4.50 (dd, J = 8.15, 15.02 Hz, 1H), 3.98 (t, J = 6.50 Hz, 2H), 3.84 (s, 3H), 3.82 (s, 3H), 3.75 (s, 3H), 3.57 - 3.68 (m, 2H), 3.36 - 3.49 (m, 1H), 3.11 - 3.34 (m, 2H), 2.81 - 2.99 (m, 4H), 2.43 - 2.54 (m, 1H), 2.33 - 2.42 (m, 2H), 2.27 (s, 6H), 1.79 - 1.92 (m, 2H), 1.62 - 1.76 (m, 2H). m/z 576 (M+H).
N-benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-6-methoxy-7-[2-(piperidin-1-yl)ethoxy]-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (20)
Prepared in 24% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.20 - 7.35 (m, 3H), 7.10 (d, J = 7.06 Hz, 2H), 6.93 (dd, J = 4.71, 7.82 Hz, 1H), 6.68 - 6.76 (m, 2H), 6.61 - 6.66 (m, 1H), 6.59 - 6.61 (m, 1H), 6.58 (s, 1H), 4.49 (dd, J = 8.24, 15.02 Hz, 1H), 4.11 (t, J = 6.45 Hz, 2H), 3.85 (s, 3H), 3.81 - 3.84 (m, 3H), 3.74 (s, 3H), 3.55 - 3.65 (m, 2H), 3.36 - 3.51 (m, 1H), 3.11 - 3.33 (m, 2H), 2.75 - 2.98 (m, 6H), 2.41 - 2.61 (m, 5H), 1.56 - 1.71 (m, 4H), 1.48 (d, J = 4.99 Hz, 2H). m/z 588 (M+H).
N-benzyl-2-[7-(benzyloxy)-1-[(3,4-dimethoxyphenyl)methyl]-6-methoxy-1,2,3,4-tetrahydroisoquinolin-2-yl]acetamide (21)
Prepared in 78% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.39 (td, J = 7.11, 15.16 Hz, 3H), 7.19 - 7.33 (m, 4H), 7.10 (d, J = 6.69 Hz, 2H), 6.91 - 7.01 (m, 1H), 6.57 - 6.70 (m, 5H), 6.49 (s, 1H), 5.06 (s, 2H), 4.48 (dd, J = 8.05, 14.93 Hz, 1H), 3.87 (s, 3H), 3.79 (s, 3H), 3.74 (s, 3H), 3.52 - 3.70 (m, 2H), 3.32 - 3.45 (m, 1H), 3.10 - 3.32 (m, 2H), 2.79 - 2.98 (m, 3H), 2.67 - 2.78 (m, 1H), 2.41 - 2.53 (m, 1H). m/z 567 (M+H).
N-benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-6-methoxy-7-(pyridin-2-ylmethoxy)-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (22)
Prepared in 61% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.60 (d, J = 4.71 Hz, 1H), 7.68 - 7.77 (m, 1H), 7.58 (d, J = 7.82 Hz, 1H), 7.17 - 7.35 (m, 5H), 7.08 (d, J = 7.06 Hz, 2H), 6.89 (br. s., 1H), 6.56 - 6.75 (m, 4H), 5.27 (s, 2H), 4.48 (dd, J = 8.19, 14.98 Hz, 1H), 3.90 (s, 3H), 3.82 (s, 3H), 3.74 (s, 3H), 3.50 - 3.62 (m, 2H), 3.33 - 3.48 (m, 1H), 3.08 - 3.31 (m, 2H), 2.68 - 3.00 (m, 4H), 2.48 (d, J = 15.64 Hz, 1H). m/z 568 (M+H).
N-benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-6-methoxy-7-(pyridin-3-ylmethoxy)-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (23)
Prepared in 47% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.68 (s, 1H), 8.58 (d, J = 4.71 Hz, 1H), 7.79 (d, J = 7.82 Hz, 1H), 7.18 - 7.37 (m, 4H), 7.11 (d, J = 7.06 Hz, 2H), 6.98 (br. s., 1H), 6.57 - 6.73 (m, 4H), 6.50 (s, 1H), 5.05 (s, 2H), 4.49 (dd, J = 8.01, 14.98 Hz, 1H), 3.87 (s, 3H), 3.81 (s, 3H), 3.75 (s, 3H), 3.54 - 3.71 (m, 2H), 3.34 - 3.48 (m, 1H), 3.10 - 3.33 (m, 2H), 2.72 - 3.01 (m, 4H), 2.43 - 2.57 (m, 1H). m/z 568 (M+H).
N-benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-6-methoxy-7-(4-phenoxybutoxy)-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (24)
Prepared in 82% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.20 - 7.36 (m, 5H), 7.11 (d, J = 6.78 Hz, 2H), 6.87 - 7.01 (m, 4H), 6.62 - 6.76 (m, 3H), 6.60 (s, 1H), 6.51 (s, 1H), 4.51 (dd, J = 8.05, 14.93 Hz, 1H), 3.98 - 4.11 (m, 4H), 3.84 (s, 3H), 3.81 (s, 3H), 3.75 (s, 3H), 3.58 - 3.70 (m, 2H), 3.35 - 3.51 (m, 1H), 3.11 - 3.34 (m, 2H), 2.81 - 3.02 (m, 4H), 2.42 - 2.56 (m, 1H), 1.94 - 2.11 (m, 4H). m/z 624 (M+H).
2-[(benzylcarbamoyl)methyl]-1-[(3,4-dimethoxyphenyl)methyl]-6-methoxy-1,2,3,4-tetrahydroisoquinolin-7-yl butanoate (25)
Phenol 11 (50 mg, 0.105 mmol), butyric acid (9 mg, 0.105 mmol) and BOP (46 mg, 0.105 mmol) were combined in dichloromethane (1 mL). Diisopropylethylamine (34 mg, 46 μL, 0.262 mmol) was added and the reaction stirred at RT under N2 overnight. The reaction was diluted with EtOAc, washed with 2M HCl, NaHCO3 solution and brine, dried over MgSO4 and the solvent removed under reduced pressure. The crude was purified by chromatography on silica (0-75% EtOAc in hexane) to give the desired product as a pale yellow solid (54 mg, 95%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 - 7.34 (m, 4H), 7.10 (d, J = 6.69 Hz, 2H), 6.92 (d, J = 6.97 Hz, 1H), 6.74 (s, 1H), 6.56 - 6.71 (m, 4H), 4.47 (dd, J = 8.01, 15.07 Hz, 1H), 3.80 (s, 3H), 3.79 (s, 3H), 3.74 (s, 3H), 3.59 - 3.70 (m, 2H), 3.11 - 3.44 (m, 3H), 2.77 - 3.01 (m, 4H), 2.55 (t, J = 7.00 Hz, 2H), 2.49 (d, J = 2.64 Hz, 1H), 1.72 - 1.87 (m, 2H), 1.05 (t, J = 7.39 Hz, 3H). m/z 547 (M+H).
General procedure for sulfonate formation: 2-[(benzylcarbamoyl)methyl]-1-[(3,4-dimethoxyphenyl)methyl]-6-methoxy-1,2,3,4-tetrahydroisoquinolin-7-yl methanesulfonate (26)
To a solution of phenol 11 (30 mg, 0.063 mmol) in dichloromethane (0.5 mL) cooled in ice under N2 was added triethylamine (16 mg, 22 μL, 0.157 mmol), and methanesulfonyl chloride (14 mg, 10 μL, 0.126 mmol). The reaction was allowed to warm slowly to RT overnight. Solvent was removed under reduced pressure and the crude purified by chromatography on silica (0-100% EtOAc in hexane) to give the desired product (35 mg, 54%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.20 - 7.36 (m, 3H), 7.03 - 7.12 (m, 3H), 6.86 (br. s., 1H), 6.67 - 6.76 (m, 3H), 6.60 - 6.65 (m, 1H), 4.48 (dd, J = 8.10, 14.98 Hz, 1H), 3.89 (s, 3H), 3.82 (s, 3H), 3.74 (s, 3H), 3.54 - 3.72 (m, 2H), 3.24 - 3.48 (m, 2H), 3.19 (s, 3H), 3.10 - 3.17 (m, 1H), 2.82 - 3.05 (m, 4H), 2.48 - 2.61 (m, 1H). m/z 555 (M+H).
2-[(benzylcarbamoyl)methyl]-1-[(3,4-dimethoxyphenyl)methyl]-6-methoxy-1,2,3,4-tetrahydroisoquinolin-7-yl trifluoromethanesulfonate (27)
Prepared in 18% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.21 - 7.36 (m, 4H), 7.10 (d, J = 8.01 Hz, 2H), 6.90 (s, 1H), 6.87 (br. s., 1H), 6.75 (s, 1H), 6.59 - 6.72 (m, 2H), 4.49 (dd, J = 8.05, 15.02 Hz, 1H), 3.90 (s, 3H), 3.80 (s, 3H), 3.74 (s, 3H), 3.60 - 3.73 (m, 2H), 3.33 - 3.45 (m, 1H), 3.11 - 3.33 (m, 2H), 2.77 - 3.04 (m, 4H), 2.56 (d, J = 18.46 Hz, 1H). 19F NMR (282 MHz, CHLOROFORM-d) δ -73.7. m/z 609 (M+H).
N-benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-6-methoxy-7-(2,2,2-trifluoroethoxy)-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (28)
Phenol 11 (100 mg, 0.24 mmol) and cesium carbonate (235 mg, 0.72 mmol) were combined in dry dimethylformamide (1.5 mL) and 2,2,2-trifluoroiodoethane (101 mg. 47 μL, 0.48 mmol) was added. The reaction was heated to 50 °C overnight until TLC (4:1 EtOAc/hexane) showed all starting material gone. The reaction was cooled, diluted with ethyl acetate and then washed with sodium bicarbonate solution and brine. The solution was then dried over MgSO4 and the solvent removed under reduced pressure. The compound was purified by chromatography on silica (0-50% EtOAc in hexane) to obtain the desired product as a pale yellow solid (86 mg, 72%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.19 - 7.34 (m, 3H), 7.09 (d, J = 6.69 Hz, 2H), 6.94 (d, J = 5.09 Hz, 1H), 6.59 - 6.75 (m, 5H), 4.48 (dd, J = 8.15, 15.02 Hz, 1H), 4.23 - 4.41 (m, 2H), 3.87 (s, 3H), 3.81 (s, 3H), 3.74 (s, 3H), 3.57 - 3.68 (m, 2H), 3.34 - 3.50 (m, 1H), 3.09 - 3.34 (m, 2H), 2.76 - 3.00 (m, 4H), 2.44 - 2.57 (m, 1H). m/z 559 (M+H).
2-[(benzylcarbamoyl)methyl]-1-[(3,4-dimethoxyphenyl)methyl]-6-methoxy-1,2,3,4-tetrahydroisoquinolin-7-yl benzenesulfonate (29)
Prepared in 64% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.87 - 7.94 (m, 2H), 7.62 - 7.71 (m, 1H), 7.48 - 7.58 (m, 2H), 7.19 - 7.35 (m, 3H), 7.09 (d, J = 6.88 Hz, 2H), 6.96 (s, 1H), 6.83 (d, J = 2.83 Hz, 1H), 6.58 - 6.73 (m, 3H), 6.55 (s, 1H), 4.48 (dd, J = 8.24, 15.02 Hz, 1H), 3.82 (s, 3H), 3.74 (s, 3H), 3.54 - 3.67 (m, 2H), 3.51 (s, 3H), 3.32 - 3.47 (m, 1H), 3.07 - 3.31 (m, 2H), 2.73 - 2.99 (m, 4H), 2.41 - 2.56 (m, 1H). m/z 617 (M+H).
2-[(benzylcarbamoyl)methyl]-1-[(3,4-dimethoxyphenyl)methyl]-6-methoxy-1,2,3,4-tetrahydroisoquinolin-7-yl 4-methylbenzene-1-sulfonate (30)
Prepared in 63% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.78 (d, J = 8.19 Hz, 2H), 7.19 - 7.37 (m, 5H), 7.08 (d, J = 6.97 Hz, 2H), 6.94 (s, 1H), 6.79 - 6.86 (m, 1H), 6.59 - 6.72 (m, 3H), 6.56 (s, 1H), 4.49 (dd, J = 8.19, 14.98 Hz, 1H), 3.82 (s, 3H), 3.74 (s, 3H), 3.58 - 3.67 (m, 2H), 3.54 (s, 3H), 3.32 - 3.45 (m, 1H), 3.07 - 3.31 (m, 2H), 2.73 - 2.98 (m, 4H), 2.47 -2.54 (m, 1H), 2.45 (s, 3H). m/z 631 (M+H).
General procedure for N-alkylation with acetate: 2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetate (32)
Tetrahydroisoquinoline 31 (0.26 g, 0.63 mmol), ethyl bromoacetate (0.16 g, 0.11 mL, 0.95 mmol) and tetrabutylammonium iodide (47 mg, 0.13 mmol) were combined in anhydrous dimethylformamide (6 mL) and diisopropylethylamine (0.20 g, 0.28 mL, 1.58 mmol) was added. The reaction was stirred at RT under N2 overnight. The reaction was diluted with ethyl acetate, washed with sodium bicarbonate solution and brine, dried over MgSO4 and solvent removed under reduced pressure. The compound was purified by chromatography on silica (0-50% EtOAc/hexane) to give the desired product as a frothy solid (0.17 g, 55%). 1H NMR (300 MHz, CHLOROFORM-d) δ ppm 6.73 - 6.79 (m, 1 H) 6.60 - 6.68 (m, 2 H) 6.55 (s, 1 H) 6.01 (s, 1 H) 4.11 - 4.22 (m, 2 H) 3.93 (dd, J=7.39, 5.23 Hz, 1 H) 3.85 (s, 3 H) 3.83 (s, 3 H) 3.80 (s, 3 H) 3.57 (s, 3 H) 3.39 - 3.55 (m, 2 H) 3.28 (ddd, J=12.88, 9.68, 5.04 Hz, 1 H) 3.14 (dd, J=13.37, 4.99 Hz, 1 H) 2.98 (dt, J=12.60, 4.77 Hz, 1 H) 2.73 - 2.87 (m, 2 H) 2.45 - 2.59 (m, 1 H) 1.26 (t, J=7.11 Hz, 3 H).
General procedure for ester hydrolysis: 2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetic acid
Ester (80 mg, 0.16 mmol) was dissolved in ethanol (2 mL) and aqueous sodium hydroxide solution (2N, 0.3 mL, 0.64 mmol) was added. The reaction was stirred at RT for 2 hours. The pH of the solution was adjusted to between 8 and 9 with 2N HCl, then all solvents were removed under reduced pressure. The crude solid was redissolved as far as possible in methanol, the remaining solids removed by filtration and the filtrate concentrated. The desired acid was obtained as a white solid (75 mg, 100%). 1H NMR (300 MHz, METHANOL-d4) δ ppm 6.92 (d, J=8.10 Hz, 1 H) 6.82 (s, 1 H) 6.68 - 6.78 (m, 2 H) 5.96 (s, 1 H) 4.67 (dd, J=9.04, 5.09 Hz, 1 H) 3.81 - 3.82 (m, 3 H) 3.76 (s, 3 H) 3.71 - 3.85 (m, 6 H) 3.46 (s, 3 H) 3.38 - 3.66 (m, 2 H) 2.95 - 3.18 (m, 3 H).
General procedure for final coupling: 2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-(2-phenylethyl)acetamide (33)
Acid (30 mg, 0.075 mmol), phenethylamine (9 mg, 9 μL, 0.075 mmol) and BOP (33 mg, 0.075 mmol) were combined in anhydrous dimethylformamide (1 mL) and diisopropylethylamine (24 mg, 33 μL, 0.187 mmol) was added. The reaction was stirred at RT under N2 overnight. The reaction was diluted with ethyl acetate, washed with sodium bicarbonate solution and brine, dried over MgSO4 and the solvent removed under reduced pressure. The compound was purified by chromatography on silica (0-75% EtOAc/hexane) to give the desired product as a pale yellow solid (10 mg, 26%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 - 7.33 (m, 3H), 7.11 - 7.18 (m, 2H), 6.66 - 6.83 (m, 4H), 6.58 (s, 1H), 6.41 (s, 1H), 3.87 (s, 6H), 3.85 (s, 3H), 3.80 (s, 3H), 3.59 (dd, J = 5.56, 9.23 Hz, 1H), 3.28 - 3.47 (m, 2H), 3.03 - 3.28 (m, 2H), 2.73 - 2.99 (m, 5H), 2.39 - 2.67 (m, 3H). m/z 505 (M+H).
2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-(3-phenylpropyl)acetamide (34)
Prepared in 64% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.23 - 7.33 (m, 2H), 7.11 - 7.22 (m, 3H), 6.69 - 6.85 (m, 3H), 6.61 - 6.68 (m, 1H), 6.59 (s, 1H), 6.42 (s, 1H), 3.86 (s, 3H), 3.85 (s, 3H), 3.81 (s, 3H), 3.80 (s, 3H), 3.60 (dd, J = 5.37, 9.23 Hz, 1H), 3.01 - 3.50 (m, 4H), 2.78 - 3.00 (m, 4H), 2.60 - 2.78 (m, 1H), 2.42 - 2.57 (m, 3H), 1.45 - 1.69 (m, 2H). m/z 519 (M+H).
2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-1-(1,2,3,4-tetrahydroisoquinolin-2-yl)ethan-1-one (35)
Prepared in 32% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.01 - 7.24 (m, 3H), 6.53 - 6.90 (m, 5H), 6.27 (d, J = 11.87 Hz, 1H), 4.24 - 4.89 (m, 2H), 3.65 - 4.01 (m, 12H), 3.22 - 3.64 (m, 5H), 2.73 - 3.10 (m, 6H), 2.35 - 2.72 (m, 2H). m/z 517.4 (M+H).
2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-1-(3-phenylpiperidin-1-yl)ethan-1-one (36)
Prepared in 42% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.13 - 7.41 (m, 6H), 6.88 - 7.10 (m, 1H), 6.52 - 6.83 (m, 3H), 4.64 (d, J = 7.72 Hz, 1H), 3.76 - 3.95 (m, 12H), 3.53 - 3.75 (m, 4H), 3.20 - 3.52 (m, 4H), 2.71 - 3.15 (m, 5H), 2.30 - 2.66 (m, 4H). m/z 545 (M+H).
2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-1-(4-phenylpiperidin-1-yl)ethan-1-one (37)
Prepared in 52% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.02 - 7.40 (m, 5H), 6.63 - 6.83 (m, 3H), 6.59 (s, 1H), 6.31 (s, 1H), 4.53 - 4.85 (m, 1H), 3.77 - 3.91 (m, 9H), 3.72 (s, 3H), 3.62 (s, 1H), 3.21 - 3.55 (m, 4H), 2.75 - 3.18 (m, 5H), 2.36 - 2.72 (m, 4H), 1.86 (br. s., 1H), 1.33 - 1.59 (m, 2H). m/z 545 (M+H).
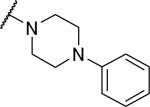
2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-1-(4-phenylpiperazin-1-yl)ethan-1-one (38)
Prepared in 42% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 - 7.39 (m, 3H), 6.89 (s, 2H), 6.71 (s, 3H), 6.59 (s, 1H), 6.33 (s, 1H), 3.77 - 3.94 (m, 9H), 3.73 (s, 3H), 3.47 (d, J = 12.81 Hz, 3H), 3.34 (d, J = 12.81 Hz, 5H), 2.70 - 3.07 (m, 8H), 2.47 (dd, J = 3.86, 16.48 Hz, 1H). m/z 546 (M+H).
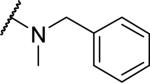
N-benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-methylacetamide (39)
Prepared in 47% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.17 - 7.37 (m, 4H), 7.01 (d, J = 7.16 Hz, 1H), 6.64 - 6.80 (m, 3H), 6.58 (d, J = 4.90 Hz, 1H), 6.18 - 6.34 (m, 1H), 4.40 - 4.74 (m, 1H), 3.89 - 4.33 (m, 2H), 3.78 - 3.88 (m, 9H), 3.65 - 3.76 (m, 3H), 3.28 - 3.58 (m, 3H), 2.83 - 3.15 (m, 4H), 2.70 - 2.82 (m, 3H), 2.39 - 2.60 (m, 1H). m/z 505 (M+H).
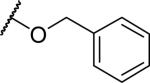
2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetate (40)
Acid (30 mg, 0.075 mmol), potassium carbonate (26 mg, 0.187 mmol) and tetrabutylammonium iodide (6 mg, 0.015 mmol) were combined in dimethylformamide (1 mL) and benzyl bromide (19 mg, 13 μL, 0.112 mmol) was added. The reaction was heated to 50 °C under N2 overnight. The reaction was cooled and diluted with EtOAc. It was washed with NaHCO3 solution and brine, dried over MgSO4 and the solvent was removed under reduced pressure. The material was purified by chromatography on silica (0-50% EtOAc in hexane) to give the desired ester (9 mg, 24%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.35 (s, 5H), 6.73 (d, J = 8.01 Hz, 1H), 6.57 - 6.66 (m, 2H), 6.55 (s, 1H), 6.00 (s, 1H), 5.16 (s, 2H), 3.90 - 4.03 (m, 1H), 3.84 (s, 6H), 3.78 (s, 3H), 3.45 - 3.65 (m, 5H), 3.22 - 3.36 (m, 1H), 3.09 - 3.20 (m, 1H), 2.94 - 3.08 (m, 1H), 2.71 - 2.90 (m, 2H), 2.53 (d, J = 16.39 Hz, 1H). m/z 492 (M+H). HPLC purity 91.0%.
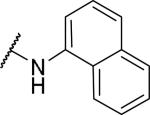
2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-(naphthalen-1-yl)acetamide (41)
Prepared in 64% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 9.25 (s, 1H), 7.84 (d, J = 7.63 Hz, 2H), 7.66 (d, J = 8.10 Hz, 1H), 7.33 - 7.54 (m, 4H), 6.63 - 6.72 (m, 2H), 6.59 (s, 1H), 6.46 (s, 1H), 6.30 (d, J = 8.10 Hz, 1H), 3.87 - 3.92 (m, 3H), 3.90 (s, 1H), 3.80 (s, 3H), 3.65 (s, 3H), 3.36 - 3.61 (m, 3H), 3.27 (s, 3H), 2.92 - 3.15 (m, 4H), 2.64 (d, J = 15.82 Hz, 1H). m/z 527 (M+H).
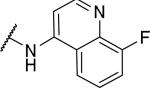
2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-(8-fluoroquinolin-4-yl)acetamide (42)
Prepared in 27% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.88 (d, J = 4.99 Hz, 1H), 8.31 (d, J = 4.99 Hz, 1H), 7.54 (dd, J = 1.60, 8.10 Hz, 1H), 7.28 - 7.46 (m, 2H), 6.87 (d, J = 8.38 Hz, 1H), 6.61 - 6.71 (m, 2H), 6.53 (d, J = 1.79 Hz, 1H), 6.43 (s, 1H), 6.37 (d, J = 8.10 Hz, 1H), 4.01 (t, J = 6.73 Hz, 1H), 3.90 (s, 3H), 3.75 - 3.81 (m, 3H), 3.68 (s, 3H), 3.39 - 3.55 (m, 6H), 2.89 - 3.20 (m, 4H), 2.62 - 2.76 (m, 1H). m/z 546 (M+H). HPLC purity 93.0%.
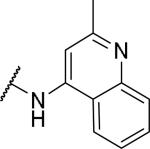
2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-(2-methylquinolin-4-yl)acetamide (43)
Prepared in 28% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 9.89 (s, 1H), 8.13 (s, 1H), 8.00 (d, J = 8.48 Hz, 1H), 7.66 (t, J = 7.30 Hz, 1H), 7.33 (t, J = 7.35 Hz, 1H), 7.11 (d, J = 8.38 Hz, 1H), 6.61 - 6.69 (m, 2H), 6.47 - 6.54 (m, 1H), 6.40 (s, 1H), 6.36 (d, J = 8.19 Hz, 1H), 3.99 (t, J = 6.64 Hz, 1H), 3.90 (s, 3H), 3.79 - 3.87 (m, 2H), 3.76 (s, 3H), 3.62 - 3.69 (m, 3H), 3.43 - 3.54 (m, 2H), 3.39 (s, 3H), 2.87 - 3.21 (m, 4H), 2.73 (s, 3H). m/z 542 (M+H). HPLC purity 93.0%.
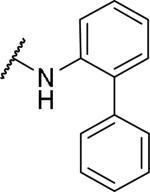
2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-(2-phenylphenyl)acetamide (44)
Prepared in 40% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 9.41 (s, 1H), 8.35 (d, J = 8.29 Hz, 1H), 7.35 - 7.45 (m, 1H), 7.15 - 7.32 (m, 8H), 6.63 (d, J = 8.29 Hz, 1H), 6.51 (s, 1H), 6.36 - 6.45 (m, 2H), 5.85 (s, 1H), 3.87 (s, 3H), 3.73 (d, J = 11.68 Hz, 6H), 3.57 - 3.66 (m, 1H), 3.54 (s, 3H), 3.30 - 3.45 (m, 1H), 3.17 - 3.29 (m, 1H), 2.93 - 3.08 (m, 1H), 2.60 (d, J = 8.29 Hz, 4H), 2.27 - 2.39 (m, 1H). m/z 553 (M+H).
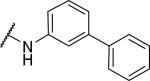
2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-(3-phenylphenyl)acetamide (45)
Prepared in 27% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.68 (s, 1H), 7.57 (d, J = 7.91 Hz, 2H), 7.39 - 7.53 (m, 3H), 7.29 - 7.37 (m, 1H), 7.23 (d, J = 8.29 Hz, 3H), 6.71 (d, J = 1.51 Hz, 3H), 6.63 (s, 1H), 6.48 (s, 1H), 3.89 (s, 3H), 3.81 (d, J = 6.40 Hz, 6H), 3.76 (s, 3H), 3.45 - 3.57 (m, 1H), 3.24 - 3.45 (m, 2H), 2.89 - 3.09 (m, 4H), 2.52 - 2.64 (m, 1H). m/z 553 (M+H)
2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-(4-phenylphenyl)acetamide (46)
Prepared in 16% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.74 (br. s., 1H), 7.59 (d, J = 6.78 Hz, 3H), 7.28 - 7.51 (m, 4H), 7.06 (br. s., 1H), 6.58 - 6.92 (m, 4H), 6.45 (s, 1H), 4.12 (q, J = 7.16 Hz, 1H), 3.77 - 3.97 (m, 6H), 3.56 - 3.76 (m, 6H), 3.23 - 3.54 (m, 4H), 2.85 - 3.13 (m, 3H), 2.58 (d, J = 15.82 Hz, 1H). m/z 553 (M+H).
2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-(1,2,3,4-tetrahydronaphthalen-1-yl)acetamide (47)
Prepared in 63% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.06 - 7.22 (m, 4H), 6.53 - 6.63 (m, 2H), 6.18 - 6.49 (m, 3H), 5.00 - 5.21 (m, 1H), 3.79 - 3.88 (m, 3H), 3.74 (d, J = 6.03 Hz, 6H), 3.67 (s, 3H), 3.11 - 3.49 (m, 3H), 2.62 - 3.05 (m, 7H), 2.42 - 2.56 (m, 1H), 1.66 - 2.02 (m, 4H). m/z 531 (M+H).
2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-(2-methyl-1,2,3,4-tetrahydroquinolin-4-yl)acetamide (48)
Quinoline 43 (20 mg, 0.037 mmol) and 10% palladium on carbon (20 mg) were combined in ethanol (5 mL) and chloroform (1 mL) and hydrogenated at 50 psi of H2 in a Parr shaker for 3 days. The reaction was filtered through Celite and the solvent was removed under reduced pressure. The crude was purified by chromatography on silica (0-20% MMA-80 in EtOAc) to give the tetrahydroisoquinoline derivative (3 mg, 15%). 1H NMR (300 MHz, CHLOROFORM-d) δ 9.09 (s, 1H), 7.78 (s, 1H), 7.26 (s, 3H), 6.57 - 6.67 (m, 3H), 6.51 (s, 1H), 6.28 (s, 1H), 3.82 - 3.99 (m, 5H), 3.59 - 3.81 (m, 10H), 3.27 - 3.54 (m, 4H), 3.04 - 3.19 (m, 1H), 2.79 - 3.03 (m, 5H), 2.58 - 2.70 (m, 1H), 2.48 (s, 3H). m/z 546 (M+H).
2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-[(1S)-1-phenylethyl]acetamide (49 and 50)
Prepared from tetrahydroisoquinoline 11 and 2-bromo-N-[(1S)-1-phenylethyl]acetamide as above as a mixture of diastereomers in 54% yield. Diastereomers were separated by reverse phase HPLC; the first to elute was 49 and the second was 50. 49: 1H NMR (300 MHz, CHLOROFORM-d) δ ppm 8.24 (br. s., 1 H) 7.20 - 7.37 (m, 5 H) 6.74 (d, J=8.10 Hz, 1 H) 6.62 (s, 1 H) 6.58 (s, 1 H) 6.49 (d, J=8.19 Hz, 1 H) 5.86 (br. s., 1 H) 4.97 - 5.09 (m, 1 H) 4.60 - 4.70 (m, 1 H) 3.88 - 4.03 (m, 2 H) 3.86 (s, 3 H) 3.83 - 3.85 (m, 3 H) 3.75 (s, 3 H) 3.61 (dd, J=11.73, 6.64 Hz, 2 H) 3.53 (s, 3 H) 3.45 - 3.50 (m, 1 H) 3.19 (d, J=10.83 Hz, 1 H) 2.83 - 3.04 (m, 2 H) 1.48 (d, J=6.97 Hz, 3 H). m/z 505 (M+H). 50: 1H NMR (300 MHz, CHLOROFORM-d) δ ppm 8.49 (d, J=7.63 Hz, 1 H) 7.21 - 7.39 (m, 5 H) 6.73 (d, J=8.19 Hz, 1 H) 6.62 (s, 1 H) 6.54 (d, J=1.70 Hz, 1 H) 6.43 (d, J=8.10 Hz, 1 H) 5.78 (s, 1 H) 5.05 (s, 1 H) 4.52 (d, J=6.31 Hz, 1 H) 3.86 (s, 3 H) 3.84 (s, 3 H) 3.78 - 3.95 (m, 2 H) 3.75 (s, 3 H) 3.56 - 3.69 (m, 2 H) 3.51 - 3.54 (m, 1 H) 3.49 (s, 3 H) 3.10 - 3.33 (m, 1 H) 2.83 - 3.04 (m, 2 H) 1.50 (d, J=6.97 Hz, 3 H). m/z 505 (M+H).
N-[(4-chlorophenyl)methyl]-2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (51)
Prepared in 60% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.21 - 7.29 (m, 2H), 7.03 (d, J = 8.38 Hz, 2H), 6.91 (t, J = 6.31 Hz, 1H), 6.63 - 6.78 (m, 3H), 6.59 (s, 1H), 6.48 (s, 1H), 4.41 (dd, J = 8.05, 15.02 Hz, 1H), 3.87 (s, 3H), 3.84 (s, 3H), 3.82 (s, 3H), 3.77 (s, 3H), 3.52 - 3.65 (m, 2H), 3.36 - 3.51 (m, 1H), 3.09 - 3.34 (m, 2H), 2.80 - 3.01 (m, 4H), 2.42 - 2.55 (m, 1H). m/z 527, 525 (M+H).
2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-[(4-fluorophenyl)methyl]acetamide (52)
Prepared in 64% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.02 - 7.11 (m, 2H), 6.87 - 7.02 (m, 3H), 6.64 - 6.78 (m, 3H), 6.59 (s, 1H), 6.47 (s, 1H), 4.42 (dd, J = 8.10, 14.79 Hz, 1H), 3.87 (s, 3H), 3.83 (s, 3H), 3.82 (s, 3H), 3.78 (s, 3H), 3.52 - 3.66 (m, 2H), 3.36 - 3.51 (m, 1H), 3.09 - 3.34 (m, 2H), 2.80 - 3.00 (m, 4H), 2.42 - 2.56 (m, 1H). m/z 509 (M+H).
2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-(pyridin-2-ylmethyl)acetamide (53)
Prepared in 57 % yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.52 (d, J = 4.52 Hz, 1H), 7.53 - 7.69 (m, 2H), 7.16 (dd, J = 5.18, 7.16 Hz, 1H), 7.09 (d, J = 7.82 Hz, 1H), 6.62 - 6.72 (m, 3H), 6.60 (s, 1H), 6.29 (s, 1H), 4.53 (dd, J = 6.55, 16.25 Hz, 1H), 4.17 (dd, J = 5.32, 16.25 Hz, 1H), 3.86 (s, 3H), 3.77 (s, 3H), 3.67 - 3.75 (m, 7H), 3.17 - 3.51 (m, 3H), 2.82 - 3.11 (m, 4H), 2.45 - 2.60 (m, 1H). m/z 492 (M+H).
2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-(pyridin-3-ylmethyl)acetamide (54)
Prepared in 49% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.50 (dd, J = 1.37, 4.76 Hz, 1H), 8.33 (d, J = 1.70 Hz, 1H), 7.43 (d, J = 7.82 Hz, 1H), 7.22 (dd, J = 4.80, 7.72 Hz, 1H), 6.89 - 6.98 (m, 1H), 6.64 - 6.79 (m, 3H), 6.60 (s, 1H), 6.51 (s, 1H), 4.41 (dd, J = 8.01, 15.26 Hz, 1H), 3.87 (s, 3H), 3.85 (s, 3H), 3.84 (s, 3H), 3.77 (s, 3H), 3.56 - 3.70 (m, 2H), 3.38 - 3.54 (m, 1H), 3.09 - 3.34 (m, 2H), 2.82 - 3.02 (m, 4H), 2.43 - 2.57 (m, 1H). m/z 492 (M+H).
2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-(pyridin-4-ylmethyl)acetamide (55)
Prepared in 89% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.48 - 8.56 (m, 2H), 6.96 - 7.02 (m, 2H), 6.92 (dd, J = 5.13, 7.68 Hz, 1H), 6.71 - 6.80 (m, 2H), 6.58 - 6.68 (m, 2H), 6.53 (s, 1H), 4.40 (dd, J = 8.10, 15.92 Hz, 1H), 3.88 (s, 3H), 3.86 (s, 3H), 3.83 (s, 3H), 3.69 (s, 3H), 3.55 - 3.66 (m, 2H), 3.41 - 3.54 (m, 1H), 3.10 - 3.37 (m, 2H), 2.82 - 3.04 (m, 4H), 2.53 (d, J = 16.29 Hz, 1H). m/z 492 (M+H).
2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-{[4-(dimethylamino)phenyl]methyl}acetamide (56)
Prepared in 7% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.03 (d, J = 8.57 Hz, 2H), 6.92 - 6.99 (m, 1H), 6.62 - 6.73 (m, 5H), 6.58 (s, 1H), 6.38 (s, 1H), 4.38 - 4.49 (m, 1H), 3.86 (s, 3H), 3.83 (s, 3H), 3.82 (s, 3H), 3.78 (s, 3H), 3.52 - 3.69 (m, 2H), 3.31 - 3.45 (m, 1H), 3.21 (q, J = 16.80 Hz, 2H), 2.90 - 2.96 (m, 6H), 2.77 - 2.90 (m, 4H), 2.41 - 2.53 (m, 1H). m/z 534 (M+H).
2-bromo-N-[(4-nitrophenyl)methyl]acetamide
To a solution of 4-nitrobenzylamine hydrochloride (1.89 g, 10 mmol) and triethylamine (1.01 g, 1.39 mL, 10 mmol) in dichloromethane (10 mL) cooled in an ice bath was added dropwise bromoacetyl bromide (1.01 g, 0.44 mL, 5 mmol). A precipitate formed immediately. The ice bath was removed and the reaction stirred at RT for 2 hr. The precipitate was removed by filtration and the solution washed twice with 2N HCl, dried over MgSO4 and the solvent removed under reduced pressure to give the bromide as a white solid (1.4 g, 100%). 1H NMR (300 MHz, CHLOROFORM-d) δ 8.22 (d, J = 8.67 Hz, 2H), 7.47 (d, J = 8.48 Hz, 2H), 4.57 - 4.64 (m, 2H), 4.15 (s, 2H).
2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-[(4-nitrophenyl)methyl]acetamide
Prepared as per general alkylation procedure using 2-bromo-N-[(4-nitrophenyl)methyl]acetamide in 63% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.15 (d, J = 8.76 Hz, 2H), 7.23 (d, J = 8.67 Hz, 2H), 6.90 - 7.00 (m, 1H), 6.65 - 6.81 (m, 3H), 6.61 (s, 1H), 6.53 (s, 1H), 4.47 (dd, J = 8.01, 15.54 Hz, 1H), 3.87 (s, 3H), 3.87 (s, 3H), 3.83 (s, 3H), 3.70 - 3.75 (m, 3H), 3.42 - 3.71 (m, 3H), 3.11 - 3.35 (m, 2H), 2.83 - 3.03 (m, 4H), 2.45 - 2.57 (m, 1H).
N-[(4-aminophenyl)methyl]-2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide
To a solution of nitrophenyl (197 mg, 0.37 mmol) and hydrazine monohydrate (0.15 mL) in ethanol (5 mL) warmed to 50 °C was added Raney nickel (2800, as a slurry in water, 25 mg) and the reaction heated at 50 °C for 1 hr. It was cooled and filtered through Celite and the solvent removed under reduced pressure to give the aniline (155 mg, 83%). 1H NMR (300 MHz, CHLOROFORM-d) δ 6.92 (d, J = 8.29 Hz, 2H), 6.70 (s, 2H), 6.56 - 6.68 (m, 4H), 6.40 (s, 1H), 4.39 (dd, J = 7.91, 14.51 Hz, 1H), 3.86 (s, 3H), 3.82 (s, 6H), 3.79 (s, 3H), 3.51 - 3.67 (m, 3H), 3.32 - 3.44 (m, 1H), 3.10 - 3.31 (m, 2H), 2.78 - 2.98 (m, 4H), 2.42 - 2.53 (m, 1H).
2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-[(4-acetamidophenyl)methyl]acetamide (57)
To the aniline (30 mg, 0.059 mmol) in dichloromethane (1 mL) was added acetic anhydride (12 mg, 11 μL, 0.119 mmol) and diisopropylethylamine (19 mg, 26 μL, 0.148 mmol) and the reaction stirred at RT overnight. The reaction was diluted with EtOAc, washed with NaHCO3 solution. The aqueous was extracted once with EtOAc and the combined organics were washed with brine, dried over MgSO4 and the solvent removed under reduced pressure. Purified by chromatography on silica (0-100% EtOAc in hexane) to give the amide (34 mg, 100%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.41 (d, J = 8.38 Hz, 2H), 7.23 (s, 1H), 7.05 (d, J = 8.38 Hz, 2H), 6.89 - 6.99 (m, 1H), 6.64 - 6.79 (m, 3H), 6.59 (s, 1H), 6.46 (s, 1H), 4.41 (dd, J = 7.91, 15.07 Hz, 1H), 3.87 (s, 3H), 3.81 - 3.84 (m, 6H), 3.78 (s, 3H), 3.54 - 3.67 (m, 2H), 3.35 - 3.50 (m, 1H), 3.09 - 3.33 (m, 2H), 2.78 - 2.99 (m, 4H), 2.42 - 2.56 (m, 1H), 2.17 (s, 3H). m/z 548 (M+H).
N-{4-[(2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamido)methyl]phenyl}hexanamide (58)
Aniline (23 mg, 0.046 mmol), hexanoic acid (5 mg, 0.046 mmol) and BOP (20 mg, 0.046 mmol) were combined in dichloromethane (1 mL) and diisopropylethylamine (12 mg, 16 μL, 0.091 mmol) was added. The reaction was stirred at RT overnight and diluted with EtOAc. It was washed with NaHCO3 solution and brine, dried over MgSO4 and the solvent was removed under reduced pressure. It was purified by chromatography on silica (0-100% EtOAc in hexane) to give the amide (30 mg, 100%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.43 (d, J = 8.29 Hz, 2H), 7.21 - 7.28 (m, 1H), 7.04 (d, J = 8.38 Hz, 2H), 6.94 (dd, J = 5.09, 7.72 Hz, 1H), 6.65 - 6.75 (m, 3H), 6.59 (s, 1H), 6.46 (s, 1H), 4.41 (dd, J = 8.05, 14.83 Hz, 1H), 3.86 (s, 3H), 3.81 - 3.84 (m, J = 0.80 Hz, 6H), 3.78 (s, 3H), 3.54 - 3.66 (m, 2H), 3.35 - 3.53 (m, 1H), 3.09 - 3.33 (m, 2H), 2.79 - 2.99 (m, 4H), 2.43 - 2.56 (m, 1H), 2.34 (t, J = 7.54 Hz, 2H), 1.64 - 1.79 (m, 2H), 1.31 - 1.39 (m, 4H), 0.87 - 0.95 (m, 3H). m/z 604 (M+H).
2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-({4-[(hexylcarbamoyl)amino]phenyl}methyl)acetamide (59)
To the aniline (40 mg, 0.079 mmol) in toluene (1 mL) was added n-hexyl isocyanate (11 mg, 13 μL, 0.087 mmol) and the mixture heated to 75 °C for 2 hr. The reaction was cooled, the solvent was removed under reduced pressure and the crude was purified by chromatography on silica (0-100% EtOAc in hexane) to give the urea (38 mg, 76%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.12 (d, J = 8.48 Hz, 2H), 7.00 (dd, J = 5.27, 7.72 Hz, 1H), 6.93 (d, J = 8.48 Hz, 2H), 6.87 (s, 1H), 6.66 - 6.77 (m, 3H), 6.60 (s, 1H), 6.49 (s, 1H), 5.29 (t, J = 5.56 Hz, 1H), 4.33 (dd, J = 7.82, 14.98 Hz, 1H), 3.87 (s, 3H), 3.84 (s, 3H), 3.82 (s, 3H), 3.75 (s, 3H), 3.54 - 3.66 (m, 2H), 3.36 - 3.51 (m, 1H), 3.07 - 3.32 (m, 4H), 2.79 - 3.01 (m, 4H), 2.43 - 2.56 (m, 1H), 1.39 - 1.54 (m, 2H), 1.21 - 1.36 (m, 6H), 0.82 - 0.90 (m, 3H). m/z 633 (M+H).
2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-heptylacetamide (60)
Prepared in 63% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 6.67 - 6.91 (m, 3H), 6.59 (m, 2H), 6.42 (m, 1H), 3.74 - 3.96 (m, 12H), 3.62 (m, 1H), 3.33 - 3.49 (m, 1H), 3.03 - 3.30 (m, 3H), 2.78 - 3.02 (m, 4H), 2.62 (m, 1H), 2.51 (d, J = 16.29 Hz, 1H), 1.08 - 1.38 (m, 10H), 0.89 (m, 3H). m/z 499 (M+H).
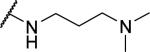
2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-[3-(dimethylamino)propyl]acetamide (61)
Prepared in 57% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 6.86 (d, J = 8.10 Hz, 2H), 6.72 - 6.79 (m, 1H), 6.70 (d, J = 1.79 Hz, 1H), 6.59 (s, 1H), 6.35 (s, 1H), 3.89 (s, 3H), 3.87 (s, 3H), 3.86 (s, 3H), 3.77 (s, 3H), 3.58 - 3.65 (m, 1H), 3.32 - 3.46 (m, 1H), 3.05 - 3.31 (m, 3H), 2.78 - 3.03 (m, 4H), 2.59 - 2.74 (m, 1H), 2.44 - 2.58 (m, 1H), 2.12 - 2.22 (m, 6H), 2.10 - 2.24 (m, 2H), 1.35 - 1.53 (m, 2H). m/z 486 (M+H).
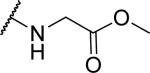
2-(2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamido)acetate (62)
Prepared in 55% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.27 (d, J = 2.35 Hz, 1H), 6.94 (br. s., 1H), 6.73 - 6.86 (m, 2H), 6.59 (d, J = 2.17 Hz, 1H), 6.41 (d, J = 2.07 Hz, 1H), 4.06 (s, 1H), 3.84 - 3.91 (m, 8H), 3.75 - 3.83 (m, 5H), 3.68 - 3.75 (m, 3H), 3.38 - 3.57 (m, 2H), 3.11 - 3.33 (m, 2H), 2.82 - 3.00 (m, 4H), 2.46 - 2.59 (m, 1H). m/z 473 (M+H).
2-(2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamido)acetic acid
Ester (60 mg, 0.127 mmol) was dissolved in methanol (5 mL) and 2N sodium hydroxide solution (0.25 mL, 0.508 mmol) was added. The reaction was stirred at RT overnight. The pH was adjusted to 7-8 with 2N HCl, then all solvent was removed under reduced pressure. The solids were redissolved as far as possible in methanol, the solids were removed by decantation, then the solvents were removed under reduced pressure to give the acid as a white solid (93 mg, 100%). 1H NMR (300 MHz, METHANOL-d4) δ 6.78 - 6.96 (m, 3H), 6.69 (s, 1H), 6.51 (s, 1H), 3.77 - 3.86 (m, 9H), 3.63 - 3.77 (m, 6H), 3.46 (d, J = 5.18 Hz, 1H), 3.17 - 3.28 (m, 2H), 2.83 - 3.04 (m, 4H), 2.47 - 2.61 (m, 1H).
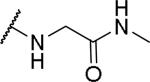
2-(2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamido)-N-methylacetamide (63)
The acid (30 mg, 0.065 mmol), methylamine hydrochloride (9 mg, 0.131 mmol) and BOP (38 mg, 0.085 mmol) were combined in dry DMF (1 mL) and diisopropylethylamine (42 mg, 57 μL, 0.327 mmol) was added. The reaction was stirred at RT overnight and then diluted with EtOAc. It was washed with NaHCO3 solution and brine, dried over MgSO4 and the solvents were removed under reduced pressure. The crude was purified by chromatography on silica (0-50% CMA-80 in EtOAc) to give the desired amide (13 mg, 42 %). 1H NMR (300 MHz, METHANOL-d4) δ 6.80 - 6.96 (m, 3H), 6.69 (s, 1H), 6.62 (s, 1H), 3.78 - 3.85 (m, 10H), 3.75 (s, 3H), 3.60 - 3.73 (m, 3H), 3.42 - 3.59 (m, 1H), 3.01 - 3.09 (m, 1H), 2.84 - 2.99 (m, 6H), 2.72 (s, 3H), 2.53 (dd, J = 5.09, 15.73 Hz, 1H). m/z 472 (M+H).
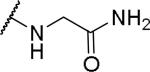
N-(carbamoylmethyl)-2-{1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (64)
Prepared from acid using ammonium chloride in 23% yield. 1H NMR (300 MHz, METHANOL-d4) δ 6.79 - 6.95 (m, 3H), 6.69 (s, 1H), 6.57 - 6.61 (m, 1H), 3.83 (s, 3H), 3.82 (s, 3H), 3.80 (s, 3H), 3.73 (s, 3H), 3.65 - 3.76 (m, 2H), 3.43 - 3.57 (m, 1H), 3.26 - 3.37 (m, 2H), 3.01 - 3.11 (m, 1H), 2.84 - 3.00 (m, 4H), 2.53 (dd, J = 5.04, 15.49 Hz, 1H). m/z 458 (M+H).
2-{1-[(3,4-dimethoxyphenyl)methyl]-6-methoxy-7-propoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-(pyridin-2-ylmethyl)acetamide (69)
Prepared in 52% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.52 (d, J = 4.80 Hz, 1H), 7.52 - 7.69 (m, 2H), 7.13 - 7.21 (m, 1H), 7.08 (d, J = 7.82 Hz, 1H), 6.62 - 6.74 (m, 3H), 6.59 (s, 1H), 6.33 (s, 1H), 4.53 (dd, J = 6.69, 16.20 Hz, 1H), 4.16 (dd, J = 5.27, 16.20 Hz, 1H), 3.85 (s, 3H), 3.77 (s, 3H), 3.72 (s, 3H), 3.66 - 3.83 (m, 3H), 3.17 - 3.51 (m, 3H), 2.81 - 3.09 (m, 4H), 2.46 - 2.59 (m, 1H), 1.73 - 1.88 (m, 2H), 1.01 (t, J = 7.39 Hz, 3H). m/z 520 (M+H).
2-{1-[(3,4-dimethoxyphenyl)methyl]-6-methoxy-7-propoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-(pyridin-3-ylmethyl)acetamide (70)
Prepared in 43% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.45 - 8.52 (m, 1H), 8.33 (s, 1H), 7.43 (d, J = 7.82 Hz, 1H), 7.22 (dd, J = 4.85, 7.77 Hz, 1H), 6.89 - 6.99 (m, 1H), 6.63 - 6.78 (m, 3H), 6.59 (s, 1H), 6.53 (s, 1H), 4.40 (dd, J = 8.01, 15.26 Hz, 1H), 3.93 (t, J = 6.88 Hz, 2H), 3.85 (s, 3H), 3.83 (s, 3H), 3.76 (s, 3H), 3.54 - 3.70 (m, 2H), 3.37 - 3.54 (m, 1H), 3.09 - 3.33 (m, 2H), 2.80 - 2.99 (m, 4H), 2.42 - 2.55 (m, 1H), 1.79 - 1.93 (m, 2H), 1.05 (t, J = 7.44 Hz, 3H). m/z 520 (M+H).
2-{1-[(3,4-dimethoxyphenyl)methyl]-6-methoxy-7-(2,2,2-trifluoroethoxy)-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-(pyridin-2-ylmethyl)acetamide (71)
Prepared in 31% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.52 (d, J = 3.96 Hz, 1H), 7.63 (t, J = 7.63 Hz, 1H), 7.51 (br. s., 1H), 7.13 - 7.22 (m, 1H), 7.07 (d, J = 7.63 Hz, 1H), 6.55 - 6.73 (m, 4H), 6.50 (s, 1H), 4.50 (dd, J = 6.55, 15.97 Hz, 1H), 4.08 - 4.35 (m, 3H), 3.86 (s, 3H), 3.77 (s, 3H), 3.72 (s, 3H), 3.66 - 3.75 (m, 1H), 3.14 - 3.49 (m, 3H), 2.78 - 3.09 (m, 4H), 2.47 - 2.62 (m, 1H). m/z 560 (M+H).
2-{1-[(3,4-dimethoxyphenyl)methyl]-6-methoxy-7-(2,2,2-trifluoroethoxy)-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-(pyridin-3-ylmethyl)acetamide (72)
Prepared in 29% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.49 (d, J = 4.71 Hz, 1H), 8.32 (s, 1H), 7.42 (d, J = 7.82 Hz, 1H), 7.22 (dd, J = 4.85, 7.68 Hz, 1H), 6.81 - 6.92 (m, 1H), 6.62 - 6.77 (m, 5H), 4.24 - 4.49 (m, 3H), 3.86 (s, 3H), 3.84 (s, 3H), 3.76 (s, 3H), 3.54 - 3.66 (m, 2H), 3.38 - 3.53 (m, 1H), 3.07 - 3.34 (m, 2H), 2.77 - 3.02 (m, 4H), 2.45 - 2.61 (m, 1H). m/z 560 (M+H).
2-{1-[(3,4-dimethoxyphenyl)methyl]-7-ethoxy-6-methoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-(pyridin-2-ylmethyl)acetamide (73)
Prepared in 36% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.49 - 8.55 (m, 1H), 7.63 (dt, J = 1.84, 7.70 Hz, 1H), 7.55 (t, J = 5.79 Hz, 1H), 7.16 (dd, J = 5.32, 7.02 Hz, 1H), 7.08 (d, J = 7.82 Hz, 1H), 6.61 - 6.72 (m, 3H), 6.59 (s, 1H), 6.34 (s, 1H), 4.53 (dd, J = 6.69, 16.20 Hz, 1H), 4.14 (dd, J = 5.27, 16.20 Hz, 1H), 3.88 - 4.00 (m, 2H), 3.85 (s, 3H), 3.77 (s, 3H), 3.72 (s, 3H), 3.65 - 3.70 (m, 1H), 3.17 - 3.48 (m, 3H), 2.81 - 3.08 (m, 4H), 2.46 - 2.57 (m, 1H), 1.41 (t, J = 6.97 Hz, 2H). m/z 506 (M+H).
2-{1-[(3,4-dimethoxyphenyl)methyl]-7-ethoxy-6-methoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-(pyridin-3-ylmethyl)acetamide (74)
Prepared in 41% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.49 (dd, J = 1.55, 4.76 Hz, 1H), 8.33 (d, J = 1.70 Hz, 1H), 7.43 (td, J = 1.93, 7.82 Hz, 1H), 7.18 - 7.25 (m, 1H), 6.93 (dd, J = 5.23, 7.58 Hz, 1H), 6.71 - 6.78 (m, 2H), 6.64 - 6.70 (m, 1H), 6.60 (s, 1H), 6.54 (s, 1H), 4.40 (dd, J = 8.01, 15.26 Hz, 1H), 4.06 (q, J = 7.00 Hz, 2H), 3.86 (s, 3H), 3.83 (s, 3H), 3.76 (s, 3H), 3.55 - 3.68 (m, 2H), 3.38 - 3.54 (m, 1H), 3.10 - 3.33 (m, 2H), 2.77 - 3.01 (m, 4H), 2.43 - 2.56 (m, 1H), 1.47 (t, J = 6.97 Hz, 3H). m/z 506 (M+H).
Functional determinations
Activity of the target compounds at the OX1 and OX2 receptors was assayed using RD-HGA16 cells (Molecular Devices), a CHO cell line stably over-expressing the promiscuous Gq-protein Ga16. Two individual cell lines were used that stably express either OX1 or OX2 receptors. Cells were cultured overnight in black, clear bottom, 96-well tissue culture plates at 25,000 cells per well in 100 μL/well Ham's F12 medium supplemented with 10% FBS, 1x pen/strep, 0.1 μg/mL Normocin, 400 μg/mL Geneticin, and 200 μg/mL Hygromycin. Medium was then removed and cells were loaded with 200 μL/well of a calcium sensitive fluorescent dye (calcium 5, Molecular Devices) in assay buffer (1x HBSS, 20 μM HEPES plus 2.7 mM probenecid) as per manufacturer's instructions for 45 min. at 37 °C. Cells were then pretreated by addition of 25 μL/well 10% DMSO (for intrinsic activity measurements) or 25 μL 10% DMSO including 10x final concentration of test compound (for antagonist assays) for 15 min. at 37 °C. Activity was then assayed on a FlexStation II at 37 °C. Baseline fluorescence was measured every 1.52 seconds for 19 seconds at which time 25 μl of 100 μM test compound (for intrinsic activity) or an 8-point half-log concentration curve of Orexin A (for antagonist activity) was added by the FlexStation II. Fluorescence was measured every 1.52 seconds for another 41 seconds. Activity was measured as change in fluorescence intensity by setting the average baseline value of each well to zero and measuring peak fluorescence intensity following compound addition. EC50 values were calculated for orexin A (EC50−) and orexin A + test compound (EC50+), and these were used to calculate the test compound Ke. The concentration/response data were fit to a three-parameter logistic equation using GraphPad Prism (v5 for Windows; GraphPad Software, San Diego, CA) to calculate the EC50 values. For OX1, at least two different concentrations of test compound were used for these experiments, and these were chosen such that they caused a 4-fold or greater rightward shift in the orexin A EC50. For OX2, test compounds were run at 10 μM due to the low potency of these compounds at OX2. The Ke was calculated from the formula Ke = [L]/ ((EC50-/EC50+)-1), where [L] equals the test compound concentration, EC50– equals the EC50 of orexin A alone, and EC50+ equals the EC50 of orexin A in the presence of test compound. The data represent the mean from at least three independent experiments.
Conformational Libraries and 3D-Pharmacophores
Initial 3D geometries of the ligands were generated from SMILES strings using CORINA (Molecular Networks, Erlangen, Germany). Conformational libraries were generated using 12 Genetic Algorithm (GA) and Energy Minimization runs, each based on a distinct random number seed, employing BALLOON as the GA-engine.47 An initial population size of 200 was employed for every GA run, each consisting of 200 generations. The energy (fitness) function was evaluated using the full MMFF94 forcefield and a continuum dielectric of 80 for dielectric response screening electrostatics. The GA runs for each ligand were then pooled and unique conformers retained. For the purposes of this study, a unique conformer was defined as any structure differing by less than 30 degrees for any torsion (excluding those terminating in hydrogens).
3D-pharmacophores were developed using MOLMOD.48, 49 This code takes as input 1) pharmacophore hypotheses, 2) conformational libraries for each of the ligands, 3) distance and tolerance criteria for each set of pharmacophore points defining pharmacophore metric compliance and 4) an energy window for consideration of conformations from each library. Given the relative homogeneity of the present study chemotype, the objective of pharmacophore analysis was primarily the development of an overlap rule for those structural commonalities in the scaffold common to all ligands, thus enabling property evaluation and multivariate statistical analysis (2D and 3D QSAR) of the pharmacophore compliant conformations. Should one or more pharmacophores be found, MOLMOD outputs both distance metrics for each and a quaternion least squares superposition of conformations of each of the ligands complying with the pharmacophore.
Property Evaluation: Properties of both whole ligand and fragments (substituents) were evaluated for each of the conformations complying with the pharmacophore(s) using semiempirical QM (MOPAC) models. Post-computation analysis of results was done with GRAPHA, a utility which extracts QM-energetic, electrostatic, hydrophobic, frontier-orbital energetic, Sterimol, shape, polar-non-polar surface area, volume and thermodynamic descriptors.
Fragment/2D/3D QSAR: Properties compiled were analyzed to develop quantitative structure relationships in an effort to ascertain the key physiochemical properties modulating orexin Ke values as a function of substitution. Multivariate least squares approaches embodied in the R PROJECT software for Statistical Computing (http://www.r-project.org/) and Tripos Sybyl were employed.
Conditioned Place Preference
Subjects
Adult Sprague-Dawley rats (Harlan, Indianapolis, IN) were housed individually on a 12/12-h light/dark cycle with free access to water and food except during experimental sessions. Testing occurred during the light period. Animal maintenance and experiments were done in accordance with the Institutional Animal Care and Use Committee, University at Buffalo, and the 2010 Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources on Life Sciences, National Research Council, National Academy of Sciences, Washington DC).
Apparatus
The standard experimental chamber for automated assessment of CPP (LE890, Harvard Apparatus, Holliston, MA) consisted of two Perspex compartments of the same size (30 cm × 30 cm × 34 cm) interconnected by a central corridor (8 cm × 10 cm × 34 cm). Both side compartments were equipped with a grey Perspex guillotine door that separated the end compartment from the central grey corridor. Each compartment had distinct, yet neutral, visual and tactile cues: One had black walls and a white rough floor, the other had white walls and a black smooth floor. The corridor had grey walls and grey floor. Time spent in each compartment was monitored by a video camera located on the ceiling and controlled by video-tracking software (Smart Junior, Harvard Apparatus, Holliston, MA).
Procedure
Details were described previously (Thorn et al., 2012). Briefly, the experiment started with a 30-min pretest session during which rats had free access to both compartments to verify the absence of preference for either. Only the first 15 min (900 sec) of the pretest session was recorded and analyzed. Rats spending more than 75 % (> 675 sec) or less than 25 % ( < 225 sec) of the total time in a compartment were eliminated from further testing (only 1 rat was excluded from the studies). While this was an unbiased setup, individual rats somewhat preferred one side over the other. On average individual rats spent 58% of total pretest time in one chamber and 38% in the other chamber with the remaining 4% being spent in the central corridor. Following the pre-test phase, rats underwent place conditioning across 8 days, with alternating treatment-compartment pairings. Rats received vehicle-compartment pairing on odd days and drug (cocaine, compound 72 or cocaine + compound 72)-compartment pairing on even days. For a conditioning session, vehicle or drug was administered, and the rat was placed immediately in the paired compartment with no access to the other compartment for 30 min. This conditioning time was chosen because it was used in other studies and was established in our laboratory.50 Drug and compartment pairing were counterbalanced. On the test day (24 hours after last conditioning session), rats were randomly placed into one compartment without treatment and had access to both compartments during the 15-min observation period, and time spent in each compartment was recorded. For the combination group, compound 72 was administered 10 min before cocaine. This pretreatment time was chosen because initial studies found that 10 min was adequate for compound 72 to be behaviorally active in a drug discrimination assay (data not shown).
Data Analyses
The magnitude of place preference was presented as the preference score, which was defined as time spent in the drug-associated compartment on the test day minus time spent in the drug-associated compartment on pretest day. Data were submitted to one-way analysis of variance with treatment (four groups) as the factor, followed by Bonferroni's post hoc test. An effect was considered significant if p < .05.
Drugs
Cocaine hydrochloride (Research Technology Branch, National Institute on Drug Abuse, Rockville, MD) and compound 72 were dissolved in 0.9 % physiological saline and administered i.p. Dose was expressed as milligrams of the form indicated above per kilogram of body weight (mg/kg). Injection volume was 1 mL/kg.
Supplementary Material
Figure 1.

Orexin antagonists.
Acknowledgements
We are grateful to the National Institute of Health/National Institute on Drug Abuse (grants DA032837 and DA026582 to YZ, DA034806 to JXL) for the financial support of this research. We thank Ms. Tiffany Langston, Mr. Keith Warner, Ms. Allyson Smith, Dr. Angela Giddings and Dr. Ann Decker for their valuable technical assistance. We also thank Dr. Scott Runyon and Dr. Gerald Pollard for their critical reading and editing of the manuscript.
ABBREVIATIONS
- ADME
Absorption Distribution Metabolism Excretion
- BOP
(Benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate
- COMFA
comparative molecular field analysis
- CPP
conditioned place preference
- HBTU
O-Benzotriazole-N,N,N’,N’-tetramethyl-uronium-hexafluoro-phosphate
- HOMO
highest occupied molecular orbital
- HPLC
high performance liquid chromatography
- LUMO
lowest unoccupied molecular orbital
- OX1
orexin 1 receptor
- OX2
orexin 2 receptor
- QSAR
quantitative structure activity relationship
- SAR
structure-activity relationship
- TLC
thin layer chromatography
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Supporting Information. HPLC analysis of target compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.de Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, Fukuhara C, Battenberg EL, Gautvik VT, Bartlett FS, 2nd, Frankel WN, van den Pol AN, Bloom FE, Gautvik KM, Sutcliffe JG. The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci U S A. 1998;95:322–327. doi: 10.1073/pnas.95.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richardson JA, Kozlowski GP, Wilson S, Arch JR, Buckingham RE, Haynes AC, Carr SA, Annan RS, McNulty DE, Liu WS, Terrett JA, Elshourbagy NA, Bergsma DJ, Yanagisawa M. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92:573–585. doi: 10.1016/s0092-8674(00)80949-6. [DOI] [PubMed] [Google Scholar]
- 3.Gotter AL, Webber AL, Coleman PJ, Renger JJ, Winrow CJ. International Union of Basic and Clinical Pharmacology. LXXXVI. Orexin receptor function, nomenclature and pharmacology. Pharmacol Rev. 2012;64:389–420. doi: 10.1124/pr.111.005546. [DOI] [PubMed] [Google Scholar]
- 4.Kukkonen JP. Physiology of the orexinergic/hypocretinergic system: a revisit in 2012. Am J Physiol Cell Physiol. 2013;304:C2–32. doi: 10.1152/ajpcell.00227.2012. [DOI] [PubMed] [Google Scholar]
- 5.Date Y, Ueta Y, Yamashita H, Yamaguchi H, Matsukura S, Kangawa K, Sakurai T, Yanagisawa M, Nakazato M. Orexins, orexigenic hypothalamic peptides, interact with autonomic, neuroendocrine and neuroregulatory systems. Proc Natl Acad Sci U S A. 1999;96:748–753. doi: 10.1073/pnas.96.2.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nambu T, Sakurai T, Mizukami K, Hosoya Y, Yanagisawa M, Goto K. Distribution of orexin neurons in the adult rat brain. Brain Res. 1999;827:243–260. doi: 10.1016/s0006-8993(99)01336-0. [DOI] [PubMed] [Google Scholar]
- 7.Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, Kilduff TS. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci. 1998;18:9996–10015. doi: 10.1523/JNEUROSCI.18-23-09996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sakurai T. Roles of orexin/hypocretin in regulation of sleep/wakefulness and energy homeostasis. Sleep Med Rev. 2005;9:231–241. doi: 10.1016/j.smrv.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 9.Sutcliffe JG, de Lecea L. The hypocretins: setting the arousal threshold. Nat Rev Neurosci. 2002;3:339–349. doi: 10.1038/nrn808. [DOI] [PubMed] [Google Scholar]
- 10.Marcus JN, Aschkenasi CJ, Lee CE, Chemelli RM, Saper CB, Yanagisawa M, Elmquist JK. Differential expression of orexin receptors 1 and 2 in the rat brain. J Comp Neurol. 2001;435:6–25. doi: 10.1002/cne.1190. [DOI] [PubMed] [Google Scholar]
- 11.Trivedi P, Yu H, MacNeil DJ, Van der Ploeg LH, Guan XM. Distribution of orexin receptor mRNA in the rat brain. FEBS Lett. 1998;438:71–75. doi: 10.1016/s0014-5793(98)01266-6. [DOI] [PubMed] [Google Scholar]
- 12.Scammell TE, Winrow CJ. Orexin receptors: pharmacology and therapeutic opportunities. Annu Rev Pharmacol Toxicol. 2011;51:243–266. doi: 10.1146/annurev-pharmtox-010510-100528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kilduff TS, Peyron C. The hypocretin/orexin ligand-receptor system: implications for sleep and sleep disorders. Trends Neurosci. 2000;23:359–365. doi: 10.1016/s0166-2236(00)01594-0. [DOI] [PubMed] [Google Scholar]
- 14.Ohno K, Sakurai T. Orexin neuronal circuitry: role in the regulation of sleep and wakefulness. Front Neuroendocrinol. 2008;29:70–87. doi: 10.1016/j.yfrne.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 15.Boutrel B, de Lecea L. Addiction and arousal: the hypocretin connection. Physiol Behav. 2008;93:947–951. doi: 10.1016/j.physbeh.2007.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harris GC, Wimmer M, Aston-Jones G. A role for lateral hypothalamic orexin neurons in reward seeking. Nature. 2005;437:556–559. doi: 10.1038/nature04071. [DOI] [PubMed] [Google Scholar]
- 17.Tsujino N, Sakurai T. Orexin/hypocretin: a neuropeptide at the interface of sleep, energy homeostasis, and reward system. Pharmacol Rev. 2009;61:162–176. doi: 10.1124/pr.109.001321. [DOI] [PubMed] [Google Scholar]
- 18.Harris GC, Aston-Jones G. Arousal and reward: a dichotomy in orexin function. Trends Neurosci. 2006;29:571–577. doi: 10.1016/j.tins.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 19.Boutrel B, Kenny PJ, Specio SE, Martin-Fardon R, Markou A, Koob GF, de Lecea L. Role for hypocretin in mediating stress-induced reinstatement of cocaine-seeking behavior. Proc Natl Acad Sci U S A. 2005;102:19168–19173. doi: 10.1073/pnas.0507480102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Narita M, Nagumo Y, Hashimoto S, Narita M, Khotib J, Miyatake M, Sakurai T, Yanagisawa M, Nakamachi T, Shioda S, Suzuki T. Direct involvement of orexinergic systems in the activation of the mesolimbic dopamine pathway and related behaviors induced by morphine. J Neurosci. 2006;26:398–405. doi: 10.1523/JNEUROSCI.2761-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hara J, Yanagisawa M, Sakurai T. Difference in obesity phenotype between orexin-knockout mice and orexin neuron-deficient mice with same genetic background and environmental conditions. Neurosci Lett. 2005;380:239–242. doi: 10.1016/j.neulet.2005.01.046. [DOI] [PubMed] [Google Scholar]
- 22.Lawrence AJ, Cowen MS, Yang HJ, Chen F, Oldfield B. The orexin system regulates alcohol-seeking in rats. Br J Pharmacol. 2006;148:752–759. doi: 10.1038/sj.bjp.0706789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aston-Jones G, Smith RJ, Moorman DE, Richardson KA. Role of lateral hypothalamic orexin neurons in reward processing and addiction. Neuropharmacology. 2009;56(Suppl 1):112–121. doi: 10.1016/j.neuropharm.2008.06.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Richards JK, Simms JA, Steensland P, Taha SA, Borgland SL, Bonci A, Bartlett SE. Inhibition of orexin-1/hypocretin-1 receptors inhibits yohimbine-induced reinstatement of ethanol and sucrose seeking in Long-Evans rats. Psychopharmacology (Berl) 2008;199:109–117. doi: 10.1007/s00213-008-1136-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hollander JA, Lu Q, Cameron MD, Kamenecka TM, Kenny PJ. Insular hypocretin transmission regulates nicotine reward. Proc Natl Acad Sci U S A. 2008;105:19480–19485. doi: 10.1073/pnas.0808023105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boss C, Brisbare-Roch C, Jenck F. Biomedical application of orexin/hypocretin receptor ligands in neuroscience. J Med Chem. 2009;52:891–903. doi: 10.1021/jm801296d. [DOI] [PubMed] [Google Scholar]
- 27.Roecker AJ, Coleman PJ. Orexin receptor antagonists: medicinal chemistry and therapeutic potential. Curr Top Med Chem. 2008;8:977–987. doi: 10.2174/156802608784936746. [DOI] [PubMed] [Google Scholar]
- 28.Gatfield J, Brisbare-Roch C, Jenck F, Boss C. Orexin receptor antagonists: a new concept in CNS disorders? ChemMedChem. 2010;5:1197–1214. doi: 10.1002/cmdc.201000132. [DOI] [PubMed] [Google Scholar]
- 29.Kodadek T, Cai D. Chemistry and biology of orexin signaling. Mol Biosyst. 2010;6:1366–1375. doi: 10.1039/c003468a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smart D, Sabido-David C, Brough SJ, Jewitt F, Johns A, Porter RA, Jerman JC. SB-334867-A: the first selective orexin-1 receptor antagonist. Br J Pharmacol. 2001;132:1179–1182. doi: 10.1038/sj.bjp.0703953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Porter RA, Chan WN, Coulton S, Johns A, Hadley MS, Widdowson K, Jerman JC, Brough SJ, Coldwell M, Smart D, Jewitt F, Jeffrey P, Austin N. 1,3-Biarylureas as selective non-peptide antagonists of the orexin-1 receptor. Bioorg Med Chem Lett. 2001;11:1907–1910. doi: 10.1016/s0960-894x(01)00343-2. [DOI] [PubMed] [Google Scholar]
- 32.Perrey DA, Gilmour BP, Runyon SP, Thomas BF, Zhang Y. Diaryl urea analogues of SB-334867 as orexin-1 receptor antagonists. Bioorg Med Chem Lett. 2011;21:2980–2985. doi: 10.1016/j.bmcl.2011.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McElhinny CJ, Jr., Lewin AH, Mascarella SW, Runyon S, Brieaddy L, Carroll FI. Hydrolytic instability of the important orexin 1 receptor antagonist SB-334867: possible confounding effects on in vivo and in vitro studies. Bioorg Med Chem Lett. 2012;22:6661–6664. doi: 10.1016/j.bmcl.2012.08.109. [DOI] [PubMed] [Google Scholar]
- 34.Langmead CJ, Jerman JC, Brough SJ, Scott C, Porter RA, Herdon HJ. Characterisation of the binding of [3H]-SB-674042, a novel nonpeptide antagonist, to the human orexin-1 receptor. Br J Pharmacol. 2004;141:340–346. doi: 10.1038/sj.bjp.0705610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang R, Song X, Bali P, Smith A, Bayona CR, Lin L, Cameron MD, McDonald PH, Kenny PJ, Kamenecka TM. Disubstituted piperidines as potent orexin (hypocretin) receptor antagonists. Bioorg Med Chem Lett. 2012;22:3890–3894. doi: 10.1016/j.bmcl.2012.04.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stasi PL, Rovati L. Spiro amino compounds suitable for the treatment of inter alia sleep disorders and drug addiction. 2011 WO2011006960.
- 37.Steiner MA, Gatfield J, Brisbare-Roch C, Dietrich H, Treiber A, Jenck F, Boss C. Discovery and Characterization of ACT-335827, an Orally Available, Brain Penetrant Orexin Receptor Type 1 Selective Antagonist. ChemMedChem. 2013;8:898–903. doi: 10.1002/cmdc.201300003. [DOI] [PubMed] [Google Scholar]
- 38.Hirose M, Egashira S, Goto Y, Hashihayata T, Ohtake N, Iwaasa H, Hata M, Fukami T, Kanatani A, Yamada K. N-acyl 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline: the first orexin-2 receptor selective non-peptidic antagonist. Bioorg Med Chem Lett. 2003;13:4497–4499. doi: 10.1016/j.bmcl.2003.08.038. [DOI] [PubMed] [Google Scholar]
- 39.Koberstein R, Aissaoui H, Bur D, Clozel M, Fischli W, Jenck F, Mueller C, Nayler O, Sifferlen T, Treiber A, Weller T. Tetrahydroisoquinolines as orexin receptor antagonists: Strategies for lead optimization by solution-phase chemistry. Chimia. 2003;57:270–275. [Google Scholar]
- 40.Rice KC, Brossi A. Expedient Synthesis of Racemic and Optically-Active NNorreticuline and N-Substituted and 6'-Bromo-N-Norreticulines. Journal of Organic Chemistry. 1980;45:592–601. [Google Scholar]
- 41.Nilsson BL, Kiessling LL, Raines RT. Staudinger ligation: A peptide from a thioester and azide. Organic Letters. 2000;2:1939–1941. doi: 10.1021/ol0060174. [DOI] [PubMed] [Google Scholar]
- 42.Caddick S, Afonso CAM, Candeias SX, Hitchcock PB, Jenkins K, Murtagh L, Pardoe D, Santos AG, Treweeke NR, Weaving R. Synthesis of alpha-amino esters by dynamic kinetic resolution of alpha-haloacyl imidazolidinones. Tetrahedron. 2001;57:6589–6605. [Google Scholar]
- 43.Neubauer DN. Almorexant, a dual orexin receptor antagonist for the treatment of insomnia. Curr Opin Investig Drugs. 2010;11:101–110. [PubMed] [Google Scholar]
- 44.Verloop A, Tipker J. QSAR in Drug Design and Toxicology. Elsevier. 97:1987. [Google Scholar]
- 45.Verloop A. The STERIMOL Approach to Drug Design. Marcel Dekker; New York: 1987. [Google Scholar]
- 46.Kantola A, Villar HO, Loew GH. Atom Based Parametrization for a Conformationally Dependent Hydrophobic Index. Journal of Computational Chemistry. 1991;12:681–689. [Google Scholar]
- 47.Vainio MJ, Johnson MS. Generating conformer ensembles using a multiobjective genetic algorithm. J Chem Inf Model. 2007;47:2462–2474. doi: 10.1021/ci6005646. [DOI] [PubMed] [Google Scholar]
- 48.Harris D, Clayton T, Cook J, Sahbaie P, Halliwell RF, Furtmüller R, Huck S, Sieghart W, DeLorey TM. Selective influence on contextual memory: Physiochemical properties associated with selectivity of benzodiazepine ligands at GABAA receptors containing the a5 subunit. J. Med. Chem. 2008;51:3788–3803. doi: 10.1021/jm701433b. [DOI] [PubMed] [Google Scholar]
- 49.Harris DL, DeLorey TM, He X, Cook JM, Loew GH. Determinants of recognition of ligands binding to benzodiazepine receptor/GABA(A) receptors initiating sedation. Eur J Pharmacol. 2000;401:271–287. doi: 10.1016/s0014-2999(00)00462-3. [DOI] [PubMed] [Google Scholar]
- 50.Thorn DA, An XF, Zhang Y, Pigini M, Li JX. Characterization of the hypothermic effects of imidazoline I(2) receptor agonists in rats. Br J Pharmacol. 2012;166:1936–1945. doi: 10.1111/j.1476-5381.2012.01894.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.