

Summary
During the aging process, an accumulation of non-heme iron disrupts cellular homeostasis and contributes to the mitochondrial dysfunction typical of various neuromuscular degenerative diseases. Few studies have investigated the effects of iron accumulation on mitochondrial integrity and function in skeletal muscle and liver tissue. Thus, we isolated liver mitochondria (LM), as well as quadriceps-derived subsarcolemmal mitochondria (SSM) and interfibrillar mitochondria (IFM), from male Fischer 344× Brown Norway rats at 8, 18, 29 and 37 months of age. Non-heme iron content in SSM, IFM and LM was significantly higher with age, reaching a maximum at 37 months of age. The mitochondrial permeability transition pore (mPTP) was more susceptible to the opening in aged mitochondria containing high levels of iron (i.e. SSM and LM) compared to IFM. Furthermore, mitochondrial RNA oxidation increased significantly with age in SSM and LM, but not in IFM. Levels of mitochondrial RNA oxidation in SSM and LM correlated positively with levels of mitochondrial iron, whereas a significant negative correlation was observed between the maximum Ca2+ amounts needed to induce mPTP opening and iron contents in SSM, IFM and LM. Overall, our data suggest that age-dependent accumulation of mitochondrial iron may increase mitochondrial dysfunction and oxidative damage, thereby enhancing the susceptibility to apoptosis.
Keywords: mitochondrial aging, mitochondrial iron homeostasis, mitochondrial permeability transition pore, mitochondrial RNA, oxidative stress, skeletal muscle subsarcolemmal and interfibrillar mitochondria
Introduction
Iron-mediated redox chemistry is critical to mitochondrial oxidative phosphorylation and other life-sustaining functions, such as ATP production and redox signaling (Atamna et al., 2002). Perturbation of iron homeostasis causes a decline in mitochondrial function and plays a significant role in various neuromuscular degenerative diseases, possibly including age-related tissue dysfunction (Killilea et al., 2004). Recent studies have found that iron accumulates in various tissues and species with age (Cook & Yu, 1998; Killilea et al., 2003; Reverter-Branch et al., 2004). Age-related accumulation of iron increases the potential for free redox-active iron, which can promote oxidative stress and mitochondrial damage (Shigenaga et al., 1994; Halliwell & Gutteridge, 1999).
In contrast to heme iron, non-heme iron is believed to coexist with iron-binding proteins, such as ferritin, hemosiderin and neuromelanin (Schenck & Zimmerman, 2004). Most cellular non-heme iron is taken up by mitochondria and used for the biosynthesis of iron-based cofactors (e.g. heme and iron–sulfur clusters), which are essential components of the mitochondrial electron transfer chain (Steffens et al., 1987). Iron-based cofactors are also necessary for protein synthesis, signaling and regulation of tissue oxygen levels (Beinert & Kiley, 1999; Atamna et al., 2002). Therefore, well-maintained mitochondrial iron homeostasis may be critical for the functional fidelity of cells and tissues with age. Cellular iron can be imported into mitochondria by mitoferrin, a transmembrane protein residing in the inner mitochondrial membrane (Shaw et al., 2006). It is also possible that iron is directly transferred from endosomes to mitochondria (Sheftel et al., 2007). Several mitochondrial transporters (e.g. ABCB7, FLVCR, ABCG2 and ABC-me) are known to export iron in the form of heme or iron–sulfur clusters (Shirihai et al., 2000; Quigley et al., 2004; Cavadini et al., 2007) to maintain mitochondrial iron homeostasis. In addition, mitochondrial iron homeostasis requires frataxin, a 17-kDa protein whose deficiency causes iron accumulation in mitochondria (Babcock et al., 1997). Although the precise mechanisms of iron homeostasis in mitochondria remain unknown, defective mitochondrial iron homeostasis seems to play a major role in mitochondrial dysfunction, increased oxidative stress and abnormal cell death with age (Pandolfo, 2002). In fact, a recent study revealed that overexpression of frataxin increases resistance to oxidative stress and extends lifespan in Drosophila (Runko et al., 2008).
The mitochondrial permeability transition pore (mPTP) is a voltage-dependent, high-conductance, non-specific passive pore that spans the mitochondrial matrix and the outer and inner mitochondrial membranes. The opening of mPTP compromises mitochondrial membrane potential and leads to a transition in membrane permeability (Di Lisa & Bernardi, 2006). The permeability transition modifies the functional and structural features of mitochondria and may cause the release of various signaling transduction molecules, such as calcium (Ca2+ ) and apoptosis mediators (Halestrap et al., 1998). This event can be potentially cytotoxic in that the released mitochondrial apoptosis signals can activate various cell death pathways. Moreover, faulty Ca2+ cycling increases the generation of mitochondrial reactive oxygen species and further enhances other mitochondrion’s PTP opening within a cell (Kroemer et al., 2007; Deniaud et al., 2008). Thus, the integrity of the pore is important for maintaining mitochondrial function as well as cellular viability during the aging process. Recently, mitochondrial dysfunction, oxidative stress and apoptotic cell death are increasingly recognized among the fundamental mechanisms that drive the process of aging (Dirks & Leeuwenburgh, 2005; Leeuwenburgh & Prolla, 2006; Judge & Leeuwenburgh, 2007; Marzetti et al., 2008a). Past studies have indicated that advanced age renders mPTP more susceptible to oxidative stress and reduces the mitochondrial Ca2+-handling capacity (Kristal & Yu, 1998; Di Lisa & Bernardi, 2005). Similarly, iron-mediated oxidative stress has been shown to increase the susceptibility of mPTP against Ca2+ stimuli (Gogvadze et al., 2002, 2003). Therefore, it is possible that age-related accumulation of non-heme iron in mitochondria modulates mitochondrial oxidative stress and enhances permeability transition, which can potentially lead to cell death and tissue degeneration.
Unlike liver mitochondria, skeletal muscle mitochondria can be categorized into two subpopulations based on their subcellular localization: subsarcolemmal mitochondria (SSM) are located beneath the plasma membrane and interfibrillar mitochondria (IFM) are positioned in parallel rows between myofibrils (Palmer et al., 1977). Recent evidence suggests that age-related alterations in mitochondrial structure and bioenergetics differ in SSM and IFM (Riva et al., 2006; Chabi et al., 2008). In addition, past studies have determined that SSM and IFM respond differently to oxidative stress and apoptotic stimuli (Suh et al., 2003; Adhihetty et al., 2005; Judge et al., 2005a). For example, although IFM are more resistant to Ca2+-induced PTP opening than SSM in the rat skeletal muscle, the PTP in IFM opens faster than in SSM. In addition, IFM release larger amounts of pro-apoptotic proteins in response to exogenous iron-mediated oxidative stress than SSM (Adhihetty et al., 2005). Hence, it is possible that these two subpopulations also differ in their ability to deal with iron and Ca2+, oxidant production and induction of apoptotic cell death as skeletal muscles age.
Based on these premises, we studied the effects of age on mitochondrial non-heme iron levels, mitochondrial Ca2+-handling and oxidative damage in mitochondrial RNA (mtRNA) as an oxidative marker in rat skeletal muscle or liver. Evidence suggests that RNA damage alters the structural and functional fidelity of cells; thus, it is worthwhile to investigate alterations in mtRNA integrity during the aging process (Wallace, 1999; Honda et al., 2005; Seo et al., 2006). Our hypothesis is that age-related iron accumulation in the mitochondria contributes to mitochondrial dysfunction and increases the susceptibility of mitochondrial-mediated apoptosis which contributes to the aging process. Overall, our data suggest that age-dependent mitochondrial accumulation of non-heme iron increases oxidative damage and mitochondrial dysfunction, thereby enhancing susceptibility to cell death.
Results
Body and tissue weights
Body and liver wet weights increased with age ( p < 0.0001, Table 1) up to 29 months and decreased at 37 month of age. Quadriceps wet weight decreased after 18 months of age, with the greatest loss (~40%) occurring after 29 months of age ( p < 0.0001, Table 1). The ratio of quadriceps weight (mg) to body weight (g) progressively decreased among the age cohorts, from 17.0 ± 0.3 at 8 months to 8.0 ± 0.3 at 37 months. In contrast, liver weight to body weight ratio did not change among the age cohorts (Table 1).
Table 1.
Physical characteristics of 8-, 18-, 29- and 37-month-old male Brown Norway Fischer cross rats
| 8 months | 18 months | 29 months | 37 months | |
|---|---|---|---|---|
| BW (g) | (376.9)± 14.8a | (478.8)± 6.0b | (537.7)± 7.4c | (486.0)± 10.3b |
| MW (g) | (6.3)± 0.2b | (6.7)± 0.6b | (6.5)± 0.1b | (3.7)± 0.1a |
| LW (g) | (10.2)± 0.4a | (13.3)± 0.4b | (14.9)± 0.5b | (14.4)± 0.5b |
| MW/BW (mg g−1) | (17.0)± 0.3c | (14.0)± 1.1b | (12.0)± 0.2b | (8.0)± 0.3a |
| LW/BW (mg g−1) | (27.0)± 0.3 | (28.0)± 0.5 | (28.0)± 0.6 | (30.0)± 1.0 |
MW, quadriceps muscle weights; LW, liver weights; MW/BW, muscle weight to body weight ratio; LW/BW, liver weight to body weight ratio. Values are expressed as means ± SEM (n = 9).
Different letters indicate values are significantly different (p < 0.05).
Mitochondrial non-heme iron levels and Ca2+-retention capacity
To determine the extent of age-related changes in mitochondrial iron accumulation and mitochondrial Ca2+-handling ability, we measured non-heme iron levels and the amount of Ca2+ needed to induce mPTP opening in quadriceps and liver mitochondria. Iron content increased with age in SSM, IFM and LM and showed the greatest accumulation after 29 months of age (p < 0.0001, Fig. 1A–C). Moreover, in each age group, iron levels were higher in SSM and LM as compared with IFM. In SSM, the increases in mitochondrial iron levels during aging coincided with progressive decreases in mitochondrial Ca2+-retention capacity (p < 0.0001, Fig. 1D). Similar trends were observed in IFM (p = 0.068, Fig. 1E) and LM (p < 0.001, Fig. 1F). Interestingly, in each age group, IFM were more resistant to Ca2+-induced mPTP opening (i.e. 10-fold) compared with SSM and LM. To examine if the amount of mitochondrial protein differed among groups, we measured levels of porin, an abundant mitochondrial outer membrane protein (Mannella, 1998), in the isolated mitochondria. Western blot analysis revealed no significant differences among age groups (data not shown). Co-incubation with cyclosporin A significantly increased mitochondrial Ca2+-retention capacity in all mitochondrial populations, indicating that Ca2+ release results from mPTP opening (Fig. 2).
Fig. 1.
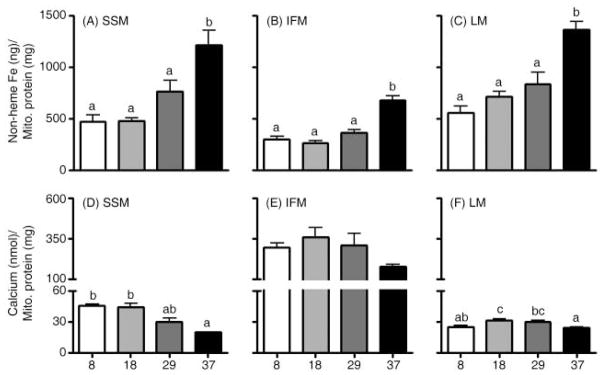
Effects of aging on mitochondrial non-heme iron levels and permeability transition pore opening (i.e. Ca2+-retention capacity) in skeletal muscle and liver. Non-heme iron levels increased with age in quadriceps (A) subsarcolemmal mitochondria (SSM), (B) interfibrillar mitochondria (IFM) and (C) liver mitochondria (LM) and showed the greatest accumulation after 29 months of age. Ca2+-retention capacity significantly decreased with age in (D) SSM, but not in (E) IFM (p = 0.068). (F) LM showed significantly decreased Ca2+-retention capacity in 37-month-old rats compared to 18-month-old rats. Data are expressed as mean ± SEM (n = 7–9). a,b,c Different letters indicate values are significantly different (p < 0.05).
Fig. 2.
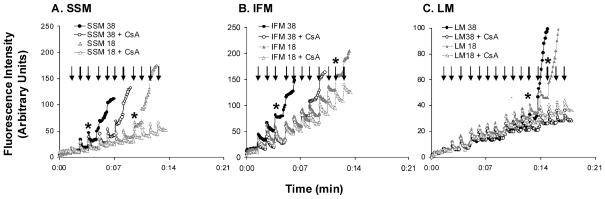
Representative experiments of calcium-induced mitochondrial permeability transition pore (mPTP) opening. (A) subsarcolemmal mitochondria (SSM; 0.75 mg mL−1), (B) interfibrillar mitochondria (IFM; 0.1 mg mL−1) and (C) liver mitochondria (LM; 1.0 mg mL−1) were energized with glutamate/malate. 1.25 nmol (SSM and IFM) and 0.65 nmol (LM) of CaCl2 were added to mitochondria with a 1-min interval between injections. During this time, extra-mitochondrial Ca2+ pulses were recorded in the presence of 1 μM calcium green-5 N. Arrows indicate Ca2+ injections. Asterisks (*) denote the injections which open mPTP, leading to mitochondrial Ca2+ release. Incubation with 0.5 μM of cyclosporin A (CsA) could significantly inhibit mPTP opening.
Mitochondrial RNA oxidation levels
To investigate whether redox-active iron accumulation was associated with oxidative damage in mitochondria, we measured total mtRNA oxidation by quantifying 8-oxoGuo using high-performance liquid chromatography coupled with electrochemical and UV detection. Oxidized mtRNA levels in SSM were significantly increased in 29- and 37-month-old animals relative to 8-month-old animals (p < 0.05, Fig. 3A). LM from 37-month-old rats also showed significantly elevated levels of oxidized mtRNA (p < 0.05, Fig. 3C). Despite the fact that our method was able to detect DNA and RNA oxidation simultaneously, the amount of mtDNA was insufficient to obtain a detectible signal.
Fig. 3.
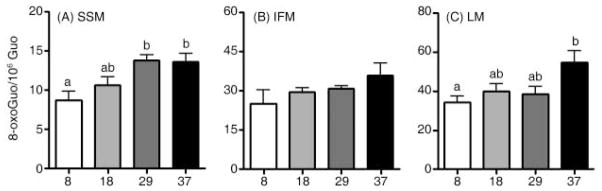
Effects of aging on mitochondrial RNA oxidation in skeletal muscle and liver. Levels of mtRNA increased significantly with age in (A) subsarcolemmal mitochondria (SSM), whereas age did not affect RNA oxidation in (B) interfibrillar mitochondria (IFM). With age, RNA oxidation was significantly increased in (C) liver mitochondria. Data are expressed as mean ± SEM (n = 4–7). a,bDifferent letters indicate values are significantly different (p < 0.05).
We further examined specific correlations between mitochondrial non-heme iron content and Ca2+-retention capacity, as well as between non-heme iron and levels of oxidized mtRNA (Fig. 4). We found a significant negative correlation between iron levels and the maximum amounts of Ca2+ needed to induce mPTP opening (Ca2+-retention capacity) in SSM (r = −0.74; p < 0.0001, Fig. 4A), IFM (r = −0.42; p < 0.05, Fig. 4B), and LM (r = −0.46; p < 0.05, Fig. 4C). The negative relationship between Ca2+-retention capacity and iron contents implies that higher iron content renders the mitochondria more susceptible to Ca2+-induced mPTP opening. This supports the notion that mPTP opening occurred with lesser amount of Ca2+ stimuli in the presence of higher levels of mitochondrial iron. With respect to oxidative damage, iron levels correlated with oxidized mtRNA levels in SSM (r = 0.74; p < 0.01; Fig. 4D), but not in IFM (Fig. 4E). Similar to SSM, there was a positive correlation between iron levels and oxidized mtRNA in LM (r = 0.60; p < 0.001; Fig. 4F).
Fig. 4.

Correlation between mitochondrial non-heme iron level, Ca2+-retention capacity and mtRNA oxidation. Correlation analyses were performed to determine how mitochondrial iron levels relate to mPTP opening and mtRNA oxidation. There was a significant negative correlation between iron levels and mPTP opening: (A) subsarcolemmal mitochondria (SSM; n = 32), (B) interfibrillar mitochondria (IFM; n = 29), and (C) liver mitochondria (LM; n = 30). In (D) SSM (n = 14), RNA oxidation levels was correlated with iron contents, but not in (E) IFM (p = 0.08, n = 16). A positive correlation between iron contents and RNA oxidation levels was detected in (F) LM (n = 30).
Caspase-9 and caspase-3 activities
To further characterize a possible role of mitochondrial iron accumulation in skeletal muscle cell degeneration during the aging process, we measured cytosolic caspase-9 and caspase-3 activities (key mediators of apoptotic cell death signal pathway) in the rat quadriceps muscle. Caspase-9 activity was not changed among the age cohorts (Fig. 5A), but caspase-3 activity was significantly increased with advancing age (p < 0.05, Fig. 5B). We also performed Pearson’s tests to determine whether mitochondrial iron levels are related with caspase-9 and caspase-3 activities. Interestingly, there was a significant positive correlation between iron levels and caspase-9 activity (r = 0.42; p < 0.05, Fig. 6A), as well as caspase-3 activity (r = 0.73; p < 0.0001, Fig. 6B) in SSM. Iron levels also positively correlated with caspase-3 activity in IFM (r = 0.41; p < 0.05, Fig. 6D), but not with caspase-9 activity.
Fig. 5.
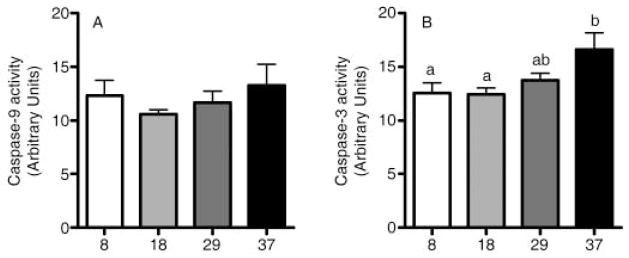
Cytosolic caspase-9 and caspase-3 activities. (A) Caspase-9 activity was not changed with age, whereas (B) caspase-3 activity was significantly increased in the aged rat quadriceps muscle (p < 0.05). Data are expressed as mean ± SEM (n = 4–8). a,bDifferent letters indicate values are significantly different (p < 0.05).
Fig. 6.

Correlation between mitochondrial non-heme iron level and caspase-9 and caspase-3 activities. Pearson’s tests were performed to determine whether mitochondrial iron levels correlate with caspase-9 and caspase-3 activities. Iron levels in subsarcolemmal mitochondria (SSM) significantly correlated with (A) caspase-9 activity (n = 26) and (B) caspase-3 activity (n = 26). There was no correlation between interfibrillar mitochondria’s (IFM) iron contents and (C) caspase-9 activity, whereas a positive correlation between iron levels and (D) caspase-3 activity was found in IFM (n = 25).
Discussion
Previous studies have suggested that accumulation of mitochondrial iron contributes to the decay of mitochondria and decreases life-sustaining functions, such as ATP production, intracellular Ca2+ buffering, regulation of cellular redox balance and apoptosis (Rötig et al., 1997; Pandolfo, 2006). Schipper and colleagues used histochemical analyses to visualize the age-associated sequestration of mitochondrial iron in human subcortical brain and in rat substantia nigra (Schipper & Cissé, 1995; Schipper et al., 1998). Several other studies have suggested that mitochondrial damage via excessive cellular iron overload may be an intrinsic factor in mitochondrial permeability transition and the functional decline associated with aging (Gogvadze et al., 2002, 2003; Walter et al., 2002; Llorens et al., 2007). However, no studies have investigated the accumulation of non-heme iron in mitochondria with age and the effects on oxidative stress.
The present study is the first to show that non-heme iron increases in mitochondria, especially at very old age. Non-heme iron accumulation correlates with age-related increases in mtRNA oxidative damage and diminished mitochondrial Ca2+-handling capacity in rat skeletal muscle and liver mitochondria. Previously, iron deposition in mitochondria was assessed in tissue section specimens using immuno-histochemical methods (Schipper & Cissé, 1995; Schipper et al., 1998). However, our study quantified the levels of non-heme iron in freshly fractionated mitochondria, thereby providing a mitochondrial-specific analysis of the biochemical changes that occur with age. IFM levels of non-heme iron remained relatively stable (i.e. ~300 ng mg−1) until 29 months of age, while a substantial increase in iron (~ 85%) was observed at 37 months of age. In contrast, non-heme iron gradually increased in SSM and LM with age, suggesting that age modulates non-heme iron differently in SSM vs. IFM. Indeed, following the Hoppel group’s finding that mitochondrial subpopulations exist in cardiac myocytes, various studies from Hood’s group and others found that mitochondrial subpopulations in skeletal muscle (i.e. SSM and IFM) differ in structure, bioenergetics and function (Palmer et al., 1977; Servais et al., 2003; Judge et al., 2005a,b; Riva et al., 2006; Chabi et al., 2008). Interestingly, our data also revealed that the levels of iron per milligram of protein were ~100% higher in SSM than in IFM, further suggesting that the two subpopulations differ in biochemical composition. The current study provides important initial results concerning the impact of iron accumulation in mitochondria with age, but more studies are warranted. Future research needs to study potential mechanisms of mitochondrial iron abnormality during the aging process (e.g. altered iron transport mechanisms and impaired iron metabolism). In addition, it is worthwhile to explore whether additional Fenton reactive metals (e.g. copper) accumulate in the aged mitochondria. This is because a pool of Fenton reactive metals can increase oxidative stress, and therein a combination of this reactive metal pool can damage mitochondria, subsequently leading to mitochondrial dysfunction and mitochondrial-mediated apoptosis (Valko et al., 2005; Marzetti et al., 2008a).
Unlike non-heme iron, which increased with age, mitochondrial Ca2+-buffering ability was significantly lower in SSM (~125%), IFM (~100%) and LM (~50%) from 37-month-old rats compared with mitochondria from younger (i.e. 8- and 18-month-old) animals. Furthermore, IFM were shown to have a much higher Ca2+-retention capacity than SSM, as demonstrated by the finding that mPTP opening occurred with larger amount of Ca2+ stimuli in IFM. This is in agreement with the result of Adhihetty et al. (2005), which showed that rat skeletal muscle IFM need more time to open the PTP than SSM in response to Ca2+ and oxidative stress. The study also found that once IFM opening is triggered by Ca2+ overload and/or exogenous iron-mediated oxidative stress, IFM open faster and release larger amounts of pro-apoptotic molecules than SSM. Although our current method was not able to measure the velocity of the pore opening, it is important to test whether age alters the PTP opening kinetics. This is because increased velocity of the pore opening against Ca2+ and oxidative stress can play an important role in the degree of mitochondrial-mediated apoptosis and cell degeneration during the aging process. Mitochondrial permeability transition by mPTP opening may cause mitochondrial swelling, inadequate mitochondrial Ca2+ buffering and ATP depletion (Javadov & Karmazyn, 2007). In addition, mPTP opening can increase cytosolic cycling of Ca2+, potentially increasing oxidative damage in mitochondria (Malis & Bonventre, 1986) and activating cellular Ca2+-dependent proteolytic pathways (Dargelos et al., 2008). Furthermore, as opening of the pore reduces mitochondrial membrane potential and disassociates various mitochondrial apoptotic proteins (e.g. endonuclease G, apoptosis inducing factor and cytochrome c), the increased vulnerability of the pore is thought to promote progressive subcellular deterioration and apoptotic or necrotic cell death (Halestrap et al., 1998; Kroemer et al., 2007; Deniaud et al., 2008). Supportively, our results indicate that the activity of caspase-3 (a key executioner in apoptotic cell death) was significantly increased in the aged rat quadriceps and positively correlated with iron levels in SSM and IFM. Caspase-9 activity was also positively related with iron concentrations in SSM (Fig. 6). In addition, according to our recently published study conducted on the gastrocnemius muscle from the same animal groups, we found that both cytosolic and nuclear levels of endonuclease G and apoptosis inducing factor increased in the aged muscle, indicating that advanced age was associated with muscle cell death (Marzetti et al., 2008b). Moreover, in the same muscle, iron levels were significantly increased in aged rats (p < 0.0001) and correlated with the extent of muscle cell death (r = 0.67; p < 0.0001; Fig. 7). Thus, these findings strongly imply that mitochondrial-mediated apoptosis is increased in aged rat skeletal muscle and that age-associated iron accumulation may play a role in tissue degeneration, possibly via enhanced mitochondrial permeability transition. Indeed, a recent study demonstrated that age-dependent skeletal muscle loss and functional declines are associated with increased oxidative stress and enhanced susceptibility to mitochondrial-mediated apoptosis (Chabi et al., 2008).
Fig. 7.
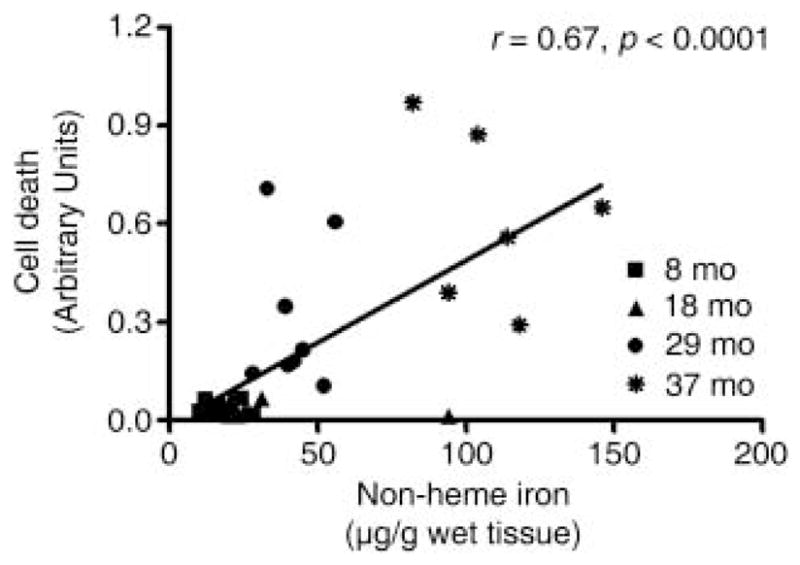
Correlation between non-heme iron level and cell death in rat gastrocnemius muscle at 8-, 18-, 29- and 37-month ages. Pearson’s tests were performed to determine whether non-heme iron levels correlate with muscle cell death. The correlation between non-heme iron and cell death was highly significant (n = 30).
Although various physiological and electrochemical factors are involved in the mitochondrial permeability transition, oxidative stress and Ca2+ overload are well-known triggers for mPTP opening and play important roles in the cell degeneration encountered with aging and age-related disorders (Shigenaga et al., 1994; Deniaud et al., 2008). Similarly, certain physiological conditions that increase oxidative stress and Ca2+ cycling (e.g. aging, ischemia-reperfusion injury and iron accumulation) can elicit mitochondrial permeability transition. For example, Martin et al. (2007) reported that skeletal muscle mitochondria become more susceptible to the permeability transition against Ca2+ overload with advancing age. In addition, ischemia-reperfusion, which increases oxidative and Ca2+ stress, dissociates mPTP and can trigger apoptotic or necrotic cell death during a cardiac infarction (Pepe, 2000; Halestrap, 2006; Kroemer et al., 2007). Furthermore, Gogvadze et al. (2003) demonstrated that pre-incubation with Fe2+ oxidizes mitochondrial lipid components and increases susceptibility to the permeability transition against Ca2+ stimuli. Therefore, age-associated accumulation of mitochondrial iron can alter the susceptibility of mPTP with age and elicit cell degeneration via enhanced mitochondrial permeability transition (Rötig et al., 1997; Puccio et al., 2001; Pandolfo, 2002; Fontenay et al., 2006). Consistent with these ideas, our correlation analysis suggested that non-heme iron levels in mitochondria are negatively related to the Ca2+-retention capacities of SSM, IFM and LM (Fig. 4).
Excess non-heme iron in aged mitochondria may increase the amount of oxidative stress from mitochondrial O2•− and H2O2, presumably via Fe2+ reduction of O2, and may further result in the decay of mitochondrial structural components, such as lipid, protein, and nucleic acids. Shigenaga et al. (1994) thoroughly reviewed the role of iron in mitochondrial lipids, proteins and mtDNA. Several studies provide additional evidence that abnormal iron results in the significant decay of mitochondria (Hanstein et al., 1975; Walter et al., 2002). Similarly, excessive iron may degrade mtRNA, leading to cumulative mtRNA damage that could play a role in mitochondrial senescence. In fact, emerging evidence suggests that RNA-specific oxidative damage is an excellent indicator of oxidative stress with age (Hofer et al., 2006; Seo et al., 2006; Cui et al., 2007) because, unlike DNA damage, there are no damage-specific repair mechanisms for oxidized RNA lesions. Therefore, the extent of RNA damage may represent the overall level of oxidative stress in the cell, relative to cellular antioxidant and turnover capacity. By contrast, DNA oxidation is governed by specific repair mechanisms and is independently regulated by the overall cellular redox status. Interestingly, rRNA has been found to have a high binding affinity for labile iron and is oxidized more easily by exposure to iron and reactive oxygen species than DNA (Honda et al., 2005; Hofer et al., 2005, 2006). This indicates that mitochondrial iron overload would not only disrupt the mitochondrial redox balance and increase free radical damage, but would also increase mtRNA oxidation. Recent findings, indeed, show that iron overloads in aged tissues and atrophied skeletal muscle are associated with oxidative damage to RNA and can be related to functional decline (Hofer et al., 2008; Xu et al., 2008). Furthermore, the present study demonstrates a positive correlation between iron levels and extent of mtRNA oxidation in SSM and LM, indicating that excessive non-heme iron increases mtRNA oxidation and oxidative stress in mitochondria from the skeletal muscle and liver of rats. However, further studies, particularly focusing on non-heme iron chelation, are required for conclusively determining the causal relationship between excessive iron and age-related oxidative damage in mitochondrial structural components.
In summary, our study identifies a potential role for mitochondrial iron abnormality with age and supports the hypothesis that age-dependent iron accumulation in mitochondria enhances the permeability transition, resulting in cell degeneration via oxidative damage (Fig. 8). Furthermore, our findings underline that biochemical and physiological differences exist among mitochondrial subpopulations in skeletal muscle. Thus, our study provides new insight into the contribution of mitochondrial iron homeostasis to aging and suggests that mitochondrial non-heme iron represents a novel target for delaying the aging process.
Fig. 8.

Potential role of mitochondrial iron accumulation in the aging process. Accumulation of mitochondrial iron, possibly due to altered mechanisms of mitochondrial iron transport increases oxidative stress via Fenton chemistry and the decay of mitochondrial structural components, such as proteins, lipids and nucleic acids. With age, an increase in the susceptibility of the permeability transition pore (PTP) may cause mitochondrial dysfunction and cellular degeneration via apoptosis or necrosis.
Experimental procedures
Animals
Thirty-six male Fischer 344× Brown Norway rats, ages 8, 18, 29 and 37 months, were purchased from the National Institute on Aging colony at Harlan Industries (Indianapolis, IN, USA). Rats were individually housed and maintained on a 12-h light/dark cycle at constant temperature and humidity in a facility approved by the Association for Assessment and Accreditation of Laboratory Animal Care. Rats had ad libitum access to standard rat chow and tap water. Body weights were recorded biweekly. Two rats were randomly selected for euthanasia each day. As anesthetic agents have been shown to influence mitochondrial function and gene expression (Brunner et al., 1975; Stowe & Kevin, 2004), rapid decapitation was used to avoid these confounding influences. All procedures were approved by the University of Florida’s animal care and use committee prior to the study.
Isolation of skeletal mitochondrial subpopulations
Quadriceps and liver were quickly removed and tissue weights were recorded. Isolation of quadriceps IFM and SSM was achieved as previously described (Servais et al., 2003). Briefly, after removing of excess fat and tendons, quadriceps were minced in ice-cold isolation buffer (50 mM Tris-HCl, 75 mM KCl, 150 mM sucrose, 5 mM MgCl2, 1 mM KH2PO4, 1 mM EGTA and 0.4% fatty acid-free BSA, pH 7.4), followed by homogenization in 10 mL buffer per gram of tissue on ice, using five full strokes with a mechanically driven Potter-Elvehjem glass-Teflon homogenizer. Homogenates were centrifuged at 800 g (10 min, 4 °C) to pellet large organelles and filaments containing IFM. The SSM-containing supernatant was filtered through synthetic cheese cloth to avoid contamination with cell debris and was centrifuged at 8000 g (10 min, 4 °C) to obtain the SSM fraction. The initial IFM-containing pellet was immediately resuspended in 10 mL isolation buffer. IFM were released from myofibrils via incubation on ice with freshly prepared protease (0.8 U g−1 tissue; Sigma-Aldrich Co., St. Louis, MO, USA) for 1 min with agitation. After five strokes of homogenization, large organelles and nuclei were removed by centrifugation at 800 g (10 min, 4 °C). The IFM-containing supernatant was filtered through cheese cloth and centrifuged at 8000 g (10 min, 4 °C) to collect the IFM pellet. The IFM and SSM pellets were immediately and gently resuspended in BSA-free buffer, centrifuged twice and kept on ice for the mPTP assay.
Isolation of liver mitochondria
After dissection, liver was immediately rinsed with ice-cold isolation buffer (20 mM Tris-HCl, 250 mM sucrose, 1 mM EGTA and 0.2% fatty acid-free BSA at pH 7.4) and minced in 10 mL isolation buffer per gram of tissue. Homogenization was performed on ice, using five strokes with a mechanically driven Potter-Elvehjem glass-Teflon homogenizer. The homogenate was centrifuged at 1000 g (10 min, 4 °C) and the supernatant was recovered. After repeating the previous step, the LM-containing supernatant was centrifuged at 8000 g (10 min, 4 °C). The resulting pellet, containing the mitochondrial fraction, was immediately and gently resuspended in BSA-free buffer, centrifuged and kept on ice for the mPTP assay. For non-heme iron measurement and mtRNA oxidation assay, mitochondria were further purified using the gradient ultra-centrifugation method described by Strømhaug et al. (1998). Isolated LM were diluted in 0.25 M sucrose (~9.3 mL) and placed on top of a freshly prepared 26.3 mL ultracentrifugation tube containing two gradients (i.e. 5 mL 22.5% and 12 mL 9.5% Nycodenz) (Gentaur, Brussels, Belgium). LM were then centrifuged using a Beckman Optima LE-80K (Beckman Coulter, Inc., Fullerton, CA, USA) ultracentrifuge at 141 000 g (34 100 r.p.m. for the 50.2Ti rotor) at 4 °C for 60 min. The LM layer was carefully collected, resuspended in 0.25 M sucrose buffer and centrifuged at 8000 g (10 min, 4 °C). The resulting LM pellet was frozen at −80 °C for biochemical analysis.
Measurement of mitochondrial non-heme iron levels
The mitochondrial non-heme iron content in muscle and liver was measured as described by Rebouche et al. (2004) with minor modifications. Briefly, liver mitochondria homogenate were diluted fourfold in de-ionized water. Forty microliters of an iron-releasing and protein-precipitating solution [i.e. 1 N HCl and 10% (v/v) trichloroacetic acid] was added to 40 μL of either mitochondrial homogenate, water (i.e. blank) or iron standards. Samples were then incubated at 95 °C for 60 min and subsequently cooled to room temperature. After centrifugation (i.e. 10 000 g, 10 min, room temperature), 40 μL of de-ionized water were added to the reaction mixtures to remove heme-containing proteins. Forty microliters of supernatant were mixed with equal volumes of sample blank solution [1.5 M sodium acetate and 0.1% (v/v) thioglycolic acid] or chromogen solution [0.508 mM ferrozine, 1.5 M sodium acetate, 0.1% (v/v) thioglycolic acid]. Samples were incubated at room temperature for 30 min to allow color development. Absorbance was read at 562 nm in a quartz cuvette, using a Beckman DU 640 spectrophotometer. A commercially available iron standard (High-Purity Standards, Charleston, SC, USA) was diluted to 2, 4, 6, 8 and 10 μg iron per milliliter in de-ionized water and used to construct a standard curve. Non-heme iron content was calculated as micrograms of iron per gram of mitochondrial protein.
Measurement of mitochondrial calcium retention capacity
To determine the maximum amount of Ca2+ required for mPTP opening, freshly isolated mitochondria were monitored using the membrane-impermeable fluorescent probe calcium green-5 N (Molecular Probes, Eugene, OR, USA), as modified from Ichas et al. (1997). IFM (0.1 mg mL−1) and SSM (0.75 mg mL−1) were incubated with 250 μL of a reaction buffer containing 250 mM sucrose, 10 mM Tris and 10 mM KH2PO4 at pH 7.4. LM (1.0 mg mL−1) was incubated with 250 μL of a similar buffer containing a reduced concentration of KH2PO4 (i.e. 1 mM). All reactions were maintained at 37 °C and energized with 5 μL of respiratory substrate (5 mM glutamate and 2.5 mM malate). A Synergy HT multidetection microplate reader (Bio-TeK Instruments, Vinooski, VT, USA) with automatic shaking and a programmable injector was used to inject 1.25 nmol CaCl2 (for reactions containing IFM and SSM) and 0.625 nmol CaCl2 (for reactions containing LM) into each well, with a 1-min interval between injections. During this time, extra-mitochondrial Ca2+ pulses were recorded in the presence of 1 μM calcium green-5 N, with excitation and emission wave lengths set at 506 and 532 nm, respectively. Ca2+ injection was continued until mPTP completely opened. In a parallel assay, 0.5 μM of cyclosporin A (Sigma, St. Louis, MO, USA) was used to confirm the release of Ca2+ in response to mPTP opening (Halestrap, 2006).
Measurement of mtRNA oxidation
Nucleic acids were extracted from isolated mitochondria as previously described, using a high-salt phenol–chloroform method in the presence of the metal chelator deferoxamine mesylate [DFOM; affinity constant for Fe(III): log K = 30.8] (Hofer et al., 2006). Oxidized RNA and DNA was quantified as 8-hydroxy-guanosine/106 guanosine (RNA) and 8-hydroxy-2′-deoxyguanosine/106 2′-deoxyguanosine (DNA), respectively. Briefly, isolated IFM (~3 mg), SSM (~3 mg) and LM (~5 mg) were resuspended in freshly prepared guanidine thiocyanate buffer [3 M GTC, 0.2% (w/v) N-lauroylsarcosinate, 20 mM Tris, 10 mM DFOM, pH 7.5] and vortexed on ice. After transferring the homogenates into phase-lock gel tubes (5 Prime Inc., Hamburg, Germany), an equal volume of phenol/chloroform/isoamyl alcohol (25 : 24 : 1, pH 6.7) was added and the mixtures were briefly vortexed on ice several times for a total of 10 min. After centrifugation (4500 g, 5 min, 0 °C), the upper aqueous phase was transferred to a new phase-lock gel tube, gently mixed with an equal volume of chloroform:isoamyl alcohol (24 : 1) and centrifuged at 4500 g for 5 min at 0 °C. Upon collection of the aqueous phase, nucleic acids were mixed with an equal amount of isopropanol and allowed to precipitate overnight at −80 °C. After centrifugation (i.e. 10 000 g, 10 min, 0 °C), nucleic acid pellets were washed with 70% (v/v) ice-cold ethanol and quickly dried under N2 gas. Nucleic acid pellets were dissolved in deoxygenated water containing 30 μM DFOM by degassing with N2 gas. The nucleic acids were then hydrolyzed using 4 U nuclease P1 and 5 U alkaline phosphatase in buffer (i.e. 30 mM sodium acetate, 20 μM ZnCl2, pH 5.3, final volume 100 μL) at 50 °C for 60 min. After filtration, levels of mtRNA oxidation were evaluated by selectively analyzing the levels of 8-oxo-7,8-dihydroguanosine (8-oxo Guo) and guanosine (Guo) using high-performance liquid chromatography coupled with electrochemical and UV detection.
Measurement of cytosolic caspase-9 and caspase-3 activities
Quadriceps cytosolic fractions were used to measure caspase-9 and caspase-3 activities, as modified from the caspase-9 and caspase-3 fluorometirc assay kits (BioVision Research Products, Mountain View, CA, USA). Samples (50 μg of proteins) were incubated with either caspase-9-specific substrate (LEHD-AFC; catalog #K118) or caspase-3-specific substrate (DEVD-AFC; catalog #K105) at 37 °C for 90 min. The level of free AFC (7-amino-4-trifluoromethyl coumarin) produced by cleavage activity of caspase-9 and caspase-3 was detected with excitation (400 nm) and emission (505 nm) wave lengths by using a SpectraMax Gemini XS microplate fluorometer.
Statistics
All statistical analyses were conducted using GraphPad Prism 4 (GraphPad Software Inc., San Diego, CA, USA). For normally distributed data, differences among experimental groups were tested by one-way analysis of variance with Tukey’s post-hoc analysis when indicated. In the case of not normally distributed variables, the Kruskal–Wallis H, with Dunn’s post-test when applicable, was used. Correlations between variables were explored using Pearson’s test (or Spearman’s when appropriate). All tests were two-sided with statistical significance defined as p < 0.05. Data are presented as mean ± SEM.
Acknowledgments
This research was supported by grants from the National Institute of Health to C.L. (AG17994 and AG21042) and a University of Florida Claude D. Pepper Older Americans Independence Center NIH grant (1 P30 AG028740) and a fellowship grant from the American Heart Association to A.Y.S. (0615256B) and to T.H. (0525346B).
References
- Adhihetty PJ, Ljubicic V, Menzies KJ, Hood DA. Differential susceptibility of subsarcolemmal and intermyofibrillar mitochondria to apoptotic stimuli. Am J Physiol Cell Physiol. 2005;289:C994–C1001. doi: 10.1152/ajpcell.00031.2005. [DOI] [PubMed] [Google Scholar]
- Atamna H, Walter PB, Ames BN. The role of heme and iron-sulfur clusters in mitochondrial biogenesis, maintenance, and decay with age. Arch Biochem Biophys. 2002;397:345–353. doi: 10.1006/abbi.2001.2671. [DOI] [PubMed] [Google Scholar]
- Babcock M, de SD, Oaks R, vis-Kaplan S, Jiralerspong S, Montermini L, Pandolfo M, Kaplan J. Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science. 1997;276:1709–1712. doi: 10.1126/science.276.5319.1709. [DOI] [PubMed] [Google Scholar]
- Beinert H, Kiley PJ. Fe-S proteins in sensing and regulatory functions. Curr Opin Chem Biol. 1999;3:152–157. doi: 10.1016/S1367-5931(99)80027-1. [DOI] [PubMed] [Google Scholar]
- Brunner EA, Cheng SC, Berman ML. Effects of anesthesia on intermediary metabolism. Annu Rev Med. 1975;26:391–401. doi: 10.1146/annurev.me.26.020175.002135. [DOI] [PubMed] [Google Scholar]
- Cavadini P, Biasiotto G, Poli M, Levi S, Verardi R, Zanella I, Derosas M, Ingrassia R, Corrado M, Arosio P. RNA silencing of the mitochondrial ABCB7 transporter in HeLa cells causes an iron-deficient phenotype with mitochondrial iron overload. Blood. 2007;109:3552–3559. doi: 10.1182/blood-2006-08-041632. [DOI] [PubMed] [Google Scholar]
- Chabi B, Ljubicic V, Menzies KJ, Huang JH, Saleem A, Hood DA. Mitochondrial function and apoptotic susceptibility in aging skeletal muscle. Aging Cell. 2008;7:2–12. doi: 10.1111/j.1474-9726.2007.00347.x. [DOI] [PubMed] [Google Scholar]
- Cook CI, Yu BP. Iron accumulation in aging: modulation by dietary restriction. Mech Ageing Dev. 1998;102:1–13. doi: 10.1016/s0047-6374(98)00005-0. [DOI] [PubMed] [Google Scholar]
- Cui L, Hofer T, Rani A, Leeuwenburgh C, Foster TC. Comparison of lifelong and late life exercise on oxidative stress in the cerebellum. Neurobiol Aging. 2007 doi: 10.1016/j.neurobiolaging.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dargelos E, Poussard S, Brule C, Daury L, Cottin P. Calcium-dependent proteolytic system and muscle dysfunctions: a possible role of calpains in sarcopenia. Biochimie. 2008;90:359–368. doi: 10.1016/j.biochi.2007.07.018. [DOI] [PubMed] [Google Scholar]
- Deniaud A, Sharaf el dO, Maillier E, Poncet D, Kroemer G, Lemaire C, Brenner C. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene. 2008;27:285–299. doi: 10.1038/sj.onc.1210638. [DOI] [PubMed] [Google Scholar]
- Di Lisa F, Bernardi P. Mitochondrial function and myocardial aging. A critical analysis of the role of permeability transition. Cardiovasc Res. 2005;66:222–232. doi: 10.1016/j.cardiores.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Di Lisa LF, Bernardi P. Mitochondria and ischemia-reperfusion injury of the heart: fixing a hole. Cardiovasc Res. 2006;70:191–199. doi: 10.1016/j.cardiores.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Dirks AJ, Leeuwenburgh C. The role of apoptosis in age-related skeletal muscle atrophy. Sports Med. 2005;35:473–483. doi: 10.2165/00007256-200535060-00002. [DOI] [PubMed] [Google Scholar]
- Fontenay M, Cathelin S, Amiot M, Gyan E, Solary E. Mitochondria in hematopoiesis and hematological diseases. Oncogene. 2006;25:4757–4767. doi: 10.1038/sj.onc.1209606. [DOI] [PubMed] [Google Scholar]
- Gogvadze V, Walter PB, Ames BN. Fe2+ induces a transient Ca2+ release from rat liver mitochondria. Arch Biochem Biophys. 2002;398:198–202. doi: 10.1006/abbi.2001.2721. [DOI] [PubMed] [Google Scholar]
- Gogvadze V, Walter PB, Ames BN. The role of Fe2+-induced lipid peroxidation in the initiation of the mitochondrial permeability transition. Arch Biochem Biophys. 2003;414:255–260. doi: 10.1016/s0003-9861(02)00750-6. [DOI] [PubMed] [Google Scholar]
- Halestrap AP. Calcium, mitochondria and reperfusion injury: a pore way to die. Biochem Soc Trans. 2006;34:232–237. doi: 10.1042/BST20060232. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Kerr PM, Javadov S, Woodfield KY. Elucidating the molecular mechanism of the permeability transition pore and its role in reperfusion injury of the heart. Biochim Biophys Acta. 1998;1366:79–94. doi: 10.1016/s0005-2728(98)00122-4. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. Oxford, UK: Oxford University Press; 1999. [Google Scholar]
- Hanstein WG, Sacks PV, Muller-Eberhard U. Properties of liver mitochondria from iron-loaded rats. Biochem Biophys Res Commun. 1975;67:1175–1184. doi: 10.1016/0006-291x(75)90797-4. [DOI] [PubMed] [Google Scholar]
- Hofer T, Badouard C, Bajak E, Ravanat JL, Mattsson A, Cotgreave IA. Hydrogen peroxide causes greater oxidation in cellular RNA than in DNA. Biol Chem. 2005;386:333–337. doi: 10.1515/BC.2005.040. [DOI] [PubMed] [Google Scholar]
- Hofer T, Marzetti E, Xu J, Seo AY, Gulec S, Knutson MD, Leeuwenburgh C, Dupont-Versteegden EE. Increased iron content and RNA oxidative damage in skeletal muscle with aging and disuse atrophy. Exp Gerontol. 2008;43:563–570. doi: 10.1016/j.exger.2008.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofer T, Seo AY, Prudencio M, Leeuwenburgh C. A method to determine RNA and DNA oxidation simultaneously by HPLC-ECD: greater RNA than DNA oxidation in rat liver after doxorubicin administration. Biol Chem. 2006;387:103–111. doi: 10.1515/BC.2006.014. [DOI] [PubMed] [Google Scholar]
- Honda K, Smith MA, Zhu X, Baus D, Merrick WC, Tartakoff AM, Hattier T, Harris PL, Siedlak SL, Fujioka H, Liu Q, Moreira PI, Miller FP, Nunomura A, Shimohama S, Perry G. Ribosomal RNA in Alzheimer disease is oxidized by bound redox-active iron. J Biol Chem. 2005;280:20978–20986. doi: 10.1074/jbc.M500526200. [DOI] [PubMed] [Google Scholar]
- Ichas F, Jouaville LS, Mazat JP. Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell. 1997;89:1145–1153. doi: 10.1016/s0092-8674(00)80301-3. [DOI] [PubMed] [Google Scholar]
- Javadov S, Karmazyn M. Mitochondrial permeability transition pore opening as an endpoint to initiate cell death and as a putative target for cardioprotection. Cell Physiol Biochem. 2007;20:1–22. doi: 10.1159/000103747. [DOI] [PubMed] [Google Scholar]
- Judge S, Jang YM, Smith A, Hagen T, Leeuwenburgh C. Age-associated increases in oxidative stress and antioxidant enzyme activities in cardiac interfibrillar mitochondria: implications for the mitochondrial theory of aging. FASEB J. 2005a;19:419–421. doi: 10.1096/fj.04-2622fje. [DOI] [PubMed] [Google Scholar]
- Judge S, Jang YM, Smith A, Selman C, Phillips T, Speakman JR, Hagen T, Leeuwenburgh C. Exercise by lifelong voluntary wheel running reduces subsarcolemmal and interfibrillar mitochondrial hydrogen peroxide production in the heart. Am J Physiol Regul Integr Comp Physiol. 2005b;289:R1564–R1572. doi: 10.1152/ajpregu.00396.2005. [DOI] [PubMed] [Google Scholar]
- Judge S, Leeuwenburgh C. Cardiac mitochondrial bioenergetics, oxidative stress, and aging. Am J Physiol Cell Physiol. 2007;292:C1983–C1992. doi: 10.1152/ajpcell.00285.2006. [DOI] [PubMed] [Google Scholar]
- Killilea DW, Atamna H, Liao C, Ames BN. Iron accumulation during cellular senescence in human fibroblasts in vitro. Antioxid Redox Signal. 2003;5:507–516. doi: 10.1089/152308603770310158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killilea DW, Wong SL, Cahaya HS, Atamna H, Ames BN. Iron accumulation during cellular senescence. Ann N Y Acad Sci. 2004;1019:365–367. doi: 10.1196/annals.1297.063. [DOI] [PubMed] [Google Scholar]
- Kristal BS, Yu BP. Dietary restriction augments protection against induction of the mitochondrial permeability transition. Free Radic Biol Med. 1998;24:1269–1277. doi: 10.1016/s0891-5849(97)00444-9. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- Leeuwenburgh C, Prolla TA. Genetics, redox signaling, oxidative stress, and apoptosis in mammalian aging. Antioxid Redox Signal. 2006;8:503–505. doi: 10.1089/ars.2006.8.503. [DOI] [PubMed] [Google Scholar]
- Llorens JV, Navarro JA, Martinez-Sebastian MJ, Baylies MK, Schneuwly S, Botella JA, Molto MD. Causative role of oxidative stress in a Drosophila model of Friedreich ataxia. FASEB J. 2007;21:333–344. doi: 10.1096/fj.05-5709com. [DOI] [PubMed] [Google Scholar]
- Malis CD, Bonventre JV. Mechanism of calcium potentiation of oxygen free radical injury to renal mitochondria. A model for post-ischemic and toxic mitochondrial damage. J Biol Chem. 1986;261:14201–14208. [PubMed] [Google Scholar]
- Mannella CA. Conformational changes in the mitochondrial channel protein, VDAC, and their functional implications. J Struct Biol. 1998;121:207–218. doi: 10.1006/jsbi.1997.3954. [DOI] [PubMed] [Google Scholar]
- Martin C, Dubouchaud H, Mosoni L, Chardigny JM, Oudot A, Fontaine E, Vergely C, Keriel C, Rochette L, Leverve X, Demaison L. Abnormalities of mitochondrial functioning can partly explain the metabolic disorders encountered in sarcopenic gastrocnemius. Aging Cell. 2007;6:165–177. doi: 10.1111/j.1474-9726.2007.00271.x. [DOI] [PubMed] [Google Scholar]
- Marzetti E, Lawler JM, Hiona A, Manini T, Seo AY, Leeuwenburgh C. Modulation of age-induced apoptotic signaling and cellular remodeling by exercise and calorie restriction in skeletal muscle. Free Radic Biol Med. 2008a;44:160–168. doi: 10.1016/j.freeradbiomed.2007.05.028. [DOI] [PubMed] [Google Scholar]
- Marzetti E, Wohlgemuth SE, Lees HA, Chung HY, Giovannini S, Leeuwenburgh C. Age-related activation of mitochondrial caspase-independent apoptotic signaling in rat gastrocnemius muscle. Mech Ageing Dev. 2008b;129:542–549. doi: 10.1016/j.mad.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer JW, Tandler B, Hoppel CL. Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J Biol Chem. 1977;252:8731–8739. [PubMed] [Google Scholar]
- Pandolfo M. Iron metabolism and mitochondrial abnormalities in Friedreich ataxia. Blood Cells Mol Dis. 2002;29:536–547. doi: 10.1006/bcmd.2002.0591. [DOI] [PubMed] [Google Scholar]
- Pandolfo M. Iron and Friedreich ataxia. J Neural Transm Suppl. 2006;70:143–146. doi: 10.1007/978-3-211-45295-0_22. [DOI] [PubMed] [Google Scholar]
- Pepe S. Mitochondrial function in ischaemia and reperfusion of the ageing heart. Clin Exp Pharmacol Physiol. 2000;27:745–750. doi: 10.1046/j.1440-1681.2000.03326.x. [DOI] [PubMed] [Google Scholar]
- Puccio H, Simon D, Cossee M, Criqui-Filipe P, Tiziano F, Melki J, Hindelang C, Matyas R, Rustin P, Koenig M. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat Genet. 2001;27:181–186. doi: 10.1038/84818. [DOI] [PubMed] [Google Scholar]
- Quigley JG, Yang Z, Worthington MT, Phillips JD, Sabo KM, Sabath DE, Berg CL, Sassa S, Wood BL, Abkowitz JL. Identification of a human heme exporter that is essential for erythropoiesis. Cell. 2004;118:757–766. doi: 10.1016/j.cell.2004.08.014. [DOI] [PubMed] [Google Scholar]
- Rebouche CJ, Wilcox CL, Widness JA. Microanalysis of non-heme iron in animal tissues. J Biochem Biophys Methods. 2004;58:239–251. doi: 10.1016/j.jbbm.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Reverter-Branchat G, Cabiscol E, Tamarit J, Ros J. Oxidative damage to specific proteins in replicative and chronological-aged Saccharomyces cerevisiae: common targets and prevention by calorie restriction. J Biol Chem. 2004;279:31983–31989. doi: 10.1074/jbc.M404849200. [DOI] [PubMed] [Google Scholar]
- Riva A, Tandler B, Lesnefsky EJ, Conti G, Loffredo F, Vazquez E, Hoppel CL. Structure of cristae in cardiac mitochondria of aged rat. Mech Ageing Dev. 2006;127:917–921. doi: 10.1016/j.mad.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rötig A, deLonlay P, Chretien D, Foury F, Koenig M, Sidi D, Munnich A, Rustin P. Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat Genet. 1997;17:215–217. doi: 10.1038/ng1097-215. [DOI] [PubMed] [Google Scholar]
- Runko AP, Griswold AJ, Min KT. Overexpression of frataxin in the mitochondria increases resistance to oxidative stress and extends lifespan in Drosophila. FEBS Lett. 2008;582:715–719. doi: 10.1016/j.febslet.2008.01.046. [DOI] [PubMed] [Google Scholar]
- Schenck JF, Zimmerman EA. High-field magnetic resonance imaging of brain iron: birth of a biomarker? NMR Biomed. 2004;17:433–445. doi: 10.1002/nbm.922. [DOI] [PubMed] [Google Scholar]
- Schipper HM, Cissé S. Mitochondrial constituents of corpora amylacea and autofluorescent astrocytic inclusions in senescent human brain. Glia. 1995;14:55–64. doi: 10.1002/glia.440140108. [DOI] [PubMed] [Google Scholar]
- Schipper HM, Vininsky R, Brull R, Small L, Brawer JR. Astrocyte mitochondria: a substrate for iron deposition in the aging rat substantia nigra. Exp Neurol. 1998;152:188–196. doi: 10.1006/exnr.1998.6854. [DOI] [PubMed] [Google Scholar]
- Seo AY, Hofer T, Sung B, Judge S, Chung HY, Leeuwenburgh C. Hepatic oxidative stress during aging: effects of 8% long-term calorie restriction and lifelong exercise. Antioxid Redox Signal. 2006;8:529–538. doi: 10.1089/ars.2006.8.529. [DOI] [PubMed] [Google Scholar]
- Servais S, Couturier K, Koubi H, Rouanet JL, Desplanches D, Sornay-Mayet MH, Sempore B, Lavoie JM, Favier R. Effect of voluntary exercise on H2O2 release by subsarcolemmal and intermyofibrillar mitochondria. Free Radic Biol Med. 2003;35:24–32. doi: 10.1016/s0891-5849(03)00177-1. [DOI] [PubMed] [Google Scholar]
- Shaw GC, Cope JJ, Li L, Corson K, Hersey C, Ackermann GE, Gwynn B, Lambert AJ, Wingert RA, Traver D, Trede NS, Barut BA, Zhou Y, Minet E, Donovan A, Brownlie A, Balzan R, Weiss MJ, Peters LL, Kaplan J, Zon LI, Paw BH. Mitoferrin is essential for erythroid iron assimilation. Nature. 2006;440:96–100. doi: 10.1038/nature04512. [DOI] [PubMed] [Google Scholar]
- Sheftel AD, Zhang AS, Brown C, Shirihai OS, Ponka P. Direct interorganellar transfer of iron from endosome to mitochondrion. Blood. 2007;110:125–132. doi: 10.1182/blood-2007-01-068148. [DOI] [PubMed] [Google Scholar]
- Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci USA. 1994;91:10 771–10 778. doi: 10.1073/pnas.91.23.10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirihai OS, Gregory T, Yu C, Orkin SH, Weiss MJ. ABC-me: a novel mitochondrial transporter induced by GATA-1 during erythroid differentiation. EMBO J. 2000;19:2492–2502. doi: 10.1093/emboj/19.11.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffens GC, Biewald R, Buse G. Cytochrome c oxidase is a threecopper, two-heme-A protein. Eur J Biochem. 1987;164:295–300. doi: 10.1111/j.1432-1033.1987.tb11057.x. [DOI] [PubMed] [Google Scholar]
- Stowe DF, Kevin LG. Cardiac preconditioning by volatile anesthetic agents: a defining role for altered mitochondrial bioenergetics. Antioxid Redox Signal. 2004;6:439–448. doi: 10.1089/152308604322899512. [DOI] [PubMed] [Google Scholar]
- Strømhaug PE, Berg TO, Fengsrud M, Seglen PO. Purification and characterization of autophagosomes from rat hepatocytes. Biochem J. 1998;335:217–224. doi: 10.1042/bj3350217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh JH, Heath SH, Hagen TM. Two subpopulations of mitochondria in the aging rat heart display heterogenous levels of oxidative stress. Free Radic Biol Med. 2003;35:1064–1072. doi: 10.1016/s0891-5849(03)00468-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valko M, Morris H, Cronin MT. Metals, toxicity and oxidative stress. Curr Med Chem. 2005;12:1161–1208. doi: 10.2174/0929867053764635. [DOI] [PubMed] [Google Scholar]
- Wallace DC. Mitochondrial diseases in man and mouse. Science. 1999;283:1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- Walter PB, Knutson MD, Paler-Martinez A, Lee S, Xu Y, Viteri FE, Ames BN. Iron deficiency and iron excess damage mitochondria and mitochondrial DNA in rats. Proc Natl Acad Sci USA. 2002;99:2264–2269. doi: 10.1073/pnas.261708798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Knutson MD, Carter CS, Leeuwenburgh C. Iron accumulation with age, oxidative stress and functional decline. PLoS. 2008;3:e2865. doi: 10.1371/journal.pone.0002865. [DOI] [PMC free article] [PubMed] [Google Scholar]