

Abstract
Introduction: Prednisolone is a class II substance according to the Biopharmaceutics Classification System. It is a poorly water soluble agent. The aim of the present study was to improve dissolution rate of a poorly water-soluble drug, prednisolone, by a solid dispersion technique. Methods: Solid dispersion of prednisolone was prepared with PEG 6000 or different carbohydrates such as lactose and dextrin with various ratios of the drug to carrier i.e., 1:10, 1:20 and 1:40. Solid dispersions were prepared by coevaporation method. The evaluation of the properties of the dispersions was performed using dissolution studies, Fourier-transform infrared spectroscopy and x-ray powder diffractometery. Results: The results indicated that lactose is suitable carriers to enhance the in vitro dissolution rate of prednisolone. The data from the x-ray diffraction showed that the drug was still detectable in its solid state in all solid dispersions except solid dispersions prepared by dextrin as carrier. The results from infrared spectroscopy showed no well-defined drug–carrier interactions for coevaporates. Conclusion: Solid dispersion of a poorly water-soluble drug, prednisolone may alleviate the problems of delayed and inconsistent rate of dissolution of the drug.
Keywords: Prednisolone, Solid dispersion, Coevaporate, X-ray diffraction, FT-IR spectroscopy, Dissolution
Introduction
Improvement of the dissolution rates of water-insoluble drugs is one of the most challenging issues of drug development, because the enhanced dissolution rates can enhance drug oral bioavailability. 1 The solid dispersion technique for water-insoluble drugs developed by Chiou and Reigelman2 supplies an effective way to increase the dissolution rates of drugs. 3,4 Details of different types of solid dispersion systems and the mechanisms of drug release from these systems have been reviewed in details.2 Solid dispersion can be prepared by formulating supersaturated systems of the drug employing various types of carriers, ranging widely from water-soluble to amphiphilic and lipid-soluble ones.4–7 Drugs molecularly dispersed in proper carriers may reach the maximum levels of particle size reduction, therefore the surface area increase, which results in enhanced dissolution rates.8
Numerous attempts 9-16 have been made to improve the dissolution rate of prednisolone, a very slightly water soluble glucocorticoid to obtain more rapid and complete absorption. One of the limitations of common solvent method is the need for a solvent that dissolves hydrophilic carrier and hydrophobic drug at the same time which may not be applicable for some carriers such as carbohydrates. In our previous investigations we have demonstrated that co-evaporates with hydrophilic carriers could be prepared by using two different but mutually soluble solvents.17,18 Therefore the need for a common solvent of drug and carrier is not considered as an absolute requisite for preparation of co-evaporates. This broadens the application of the co-evaporation technique for more extensive range of drugs and carriers.
In the present study, efforts were made to improve the dissolution behavior and consequently absorption, of prednisolone by applying the solid dispersion technique using lactose, dextrin, and PEG 6000 as hydrophilic carriers.
Materials and Methods
Materials
For preparation of solid dispersions the following materials were used: lactose monohydrate (DMV, Netherland); mannitol, dextrin, PEG 2000, PEG 4000, PEG 6000 (Merck, Germany); prednisolone (Abureihan, Iran); Chemicals used for buffer preparation were reagent grade. All other materials used were of analytical or high performance liquid chromatography (HPLC) grade and were purchased from Merck (Germany).
Preparation of solid dispersions, physical mixtures and treated physical mixtures
Physical mixtures were prepared by mixing prednisolone with the hydrophilic carriers for 5 min in 100 mL bottles, until a homogenous mixture was obtained. The resulting mixture was sieved and the 105–250 micron particle size fraction was obtained using 60-140 mesh screens. The powders were stored in a screw-cap vial at room temperature.
Treated physical mixtures were prepared by mixing treated drug and treated carriers in 100-mL bottles. Treated drug was prepared by mixing alcoholic solution of prednisolone with distilled water until a homogenous mixture was obtained. Treated carrier was prepared by dissolving it in water and adding alcohol 96% until a homogenous mixture was obtained.
The objective of preparation of treated physical mixtures is to investigate if evaporation of the drug and carrier separately may affect release of the drug from solid dispersions.
Coevaporates were prepared in the following prednisolone-carrier ratios: 1:10, 1:20, and 1:40 (w/w). Coevaporates were prepared by dissolving the components separately in a minimum volume of ethanol or distilled water, respectively. The alcoholic solution of prednisolone was then poured into the aqueous solution of the carrier under continuous stirring. The mixture was then heated in a water bath (70 oC) under vacuum and vigorous stirring.
Initially, a transparent to a translucent viscous mass was observed and finally a pale yellow coevaporate was formed. The moist mass was transferred to a vacuum desicator and kept at 50oC overnight. The solid mass was ground and the particle size fraction of 105–250 micron was separated using 60-140 mesh screens and kept in a screw-capped glass vial until use.
Dissolution studies
Hand–filled, hard gelatin capsules of the formulations, containing 5 mg of the drug, were used for the dissolution studies. The test was conducted using USP 30 Apparatus I at 50 rpm. The dissolution medium consisted of 900 ml distilled water which was maintained at 37 (±.5) °C. The duration of the test was 30 minutes. A 5-ml aliquots were withdrawn at appropriate time intervals, and replaced with a 5 ml of distilled water and assayed by UV spectrophotometry at 246.8 nm. Cumulative percentages of the drug dissolved from the preparations were calculated. Three replicates of each dissolution test were carried out.
Mean percent dissolution (MPD) and dissolution efficiency (DE)19, were used to compare dissolution profiles of binary systems Eq. (1):
(1).
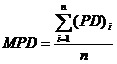
wherei is the sample number, n is the number of dissolution sample times and PD is the percent drug dissolved at time intevals Eq. (2):
(2).
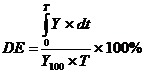
Where Y is percent drug released as a function of time, T is the total time of drug release and Y100 is 100 percent drug released.
Powder X-ray diffraction
The powder X-ray diffraction (PXRD) pattern of all ingredients and all binary systems were recorded using an automated siemens X-ray diffractometer (Siemens D5000, Munich, Germany). Cross sections of the ingredients and all binary systems were taken and held in place on a quartz plate to Cu Kα radiation of wave length 1.5406 A°. The samples were analyzed at room temperature over a range of 5-70° 2θ with sampling intervals of 0.02° 2θ and scanning rate of 6°/min.
Fourier-transform infrared spectroscopy
Fourier-transform infrared Spectroscopy (FT-IR) were obtained on a Bomem 2000 FT-IR system (Bomem Quebec, Canada) using the KBr disk method. Samples were mixed with KBr powder and compressed to 10-mm discs by hydraulic press at pressure of 10 tons for 30 seconds. The scanning range was 450-4000 cm-1 and resolution was 2 cm-1.
Results and discussion
Dissolution studies
The release of prednisolne from solid dispersions, physical mixtures and treated physical mixtures was determined in distilled water. DE and MPD values are represented in Table 1.
Table 1. DE and MPD of prednisolone-carrier binary systems (standard deviations were all less than 10%).
| Sample |
Pure drug |
Solid dispersion |
Treated Physical mixture |
Physical mixture | |||||
| Drug Carrier Ratio | - | 1:10 | 1:20 | 1:40 | 1:40 | 1:10 | 1:20 | 1:40 | |
|
Without carrier |
DE | 20.65 | - | - | - | - | - | - | - |
| MPD | 18.32 | - | - | - | - | - | - | - | |
| Lactose | DE | - | 80.98 | 83.18 | 80.71 | 71.24 | 65.39 | 73.73 | 70.24 |
| MPD | - | 73.05 | 75.12 | 72.95 | 63.77 | 57.80 | 66.60 | 62.45 | |
| Dextrin | DE | - | 50.62 | 62.49 | 68.82 | 65.28 | 31.42 | 38.11 | 39.51 |
| MPD | - | 45.10 | 56.00 | 61.31 | 58.37 | 27.47 | 33.68 | 34.41 | |
| PEG 6000 | DE | - | 60.93 | 60.78 | 53.74 | 55.18 | 42.36 | 51.61 | 43.25 |
| MPD | - | 53.95 | 52.90 | 46.05 | 48.42 | 35.76 | 44.14 | 37.17 | |
It is evident that the dissolution rate of the solid dispersions of prednisolone prepared with lactose, dextrin and PEG 6000, are higher than that of pure drug and physical mixtures of ingredients. Moreover the dissolution rate of treated physical mixtures was higher compared to related physical mixtures.
Possible explanations of the increased dissolution rate of solid dispersions have been proposed by Craig and Ford 20,21 , which include: reduction of drug crystallite size, a solubilization effect of the carrier, absence of aggregation of drug crystallites, improved wettability and dispersibility of the drug, dissolution of the drug in the hydrophilic carrier, conversion of the drug to the amorphous state and finally the combination of the above mentioned mechanisms.
Dry mixing of prednisolone with carrier brings the drug in close contact with the hydrophilic carrier. The increased dissolution rate observed in these cases can be contributed to several factors such as a solubilization effect of the carrier, improved wettability of the drug and inhibition of particle aggregation.
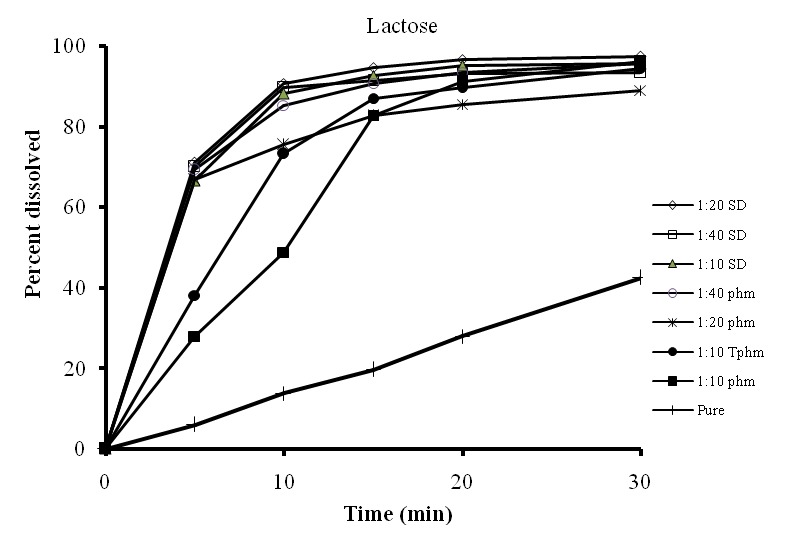
Dissolution profiles of solid dispersions with lactose showed an increase in the dissolution rate of prednisolone with respect to the drug by itself which could be due to high hyrophilicity of lactose (Figure 1). Also, increasing the weight fraction of prednisolone in the solid dispersions did not affect noticeably the dissolution rate of the dispersions prepared with lactose( Figure 2).
Figure 1. Dissolution profiles of physical mixtures (PM), treated physical mixtures (TPM) and solid dispersions (SD) of prednisolone with lactose and Dextrin.
Lactose.
Dextrin.
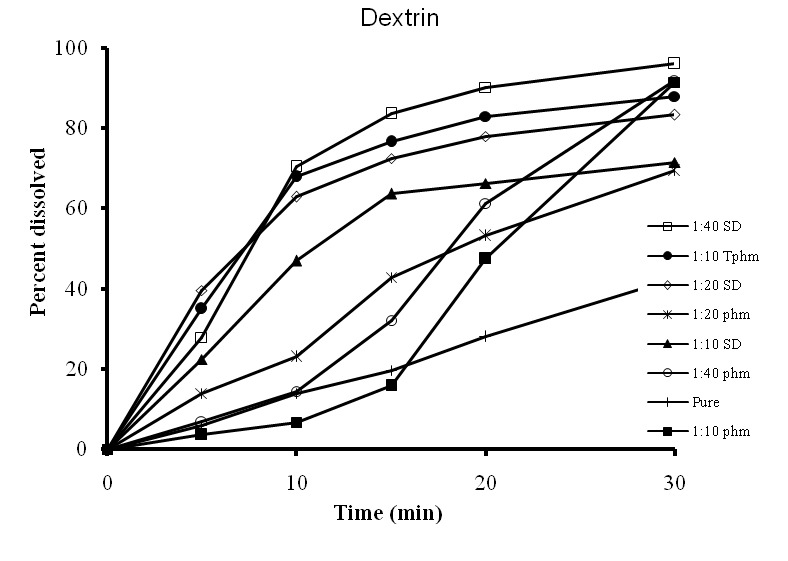
Figure 2.

FT-IR spectra of pure prednisolone, lactose, 1:40 physical mixture (PM) of prednisolone with lactose, and 1:40 solid dispersion (SD) of prednisolone with lactose
This is not unexpected, as it has been previously shown that an increase in the weight fraction of the drug does not necessarily decrease the dissolution rate. 17,22Hence utilizing lactose in a ratio of 1:10 should be enough for reaching higher dissolution rate.
As it can be seen from Figure 1, the solid dispersion of the drug with dextrin showed enhanced dissolution rate.17,18
Compared to the drug alone, an improvement in the dissolution rate was achieved for the formulations containing PEG 6000. Dissolution rate was decreased increasing the ratio of PEG 6000. This phenomenon may be attributed to formation of viscous boundary layer at the surface of the polymer at higher carrier ratios. However, the release of prednisolone from treated physical mixtures in the ratio of (1:40) was slightly faster than the release from coevaporates. This could be due to the fact that dissolution mechanism of solid dispersion with PEG 6000 might be predominantly diffusion-controlled, and presumably the high viscosity of this carrier in stagnate layer is the main factor to control the dissolution rate.
Powder X-Ray Diffraction (PXRD)
The extent of crystallinity affects dissolution of drugs. An amorphous or metastable form will dissolve promptly because of its higher internal energy and greater molecular motion, which increase thermodynamic properties compared to crystalline materials.23,24 Crystallinity was determined by comparing some representative peak heights in diffraction patterns of the solid dispersions with those of physical mixtures. Our findings revealed that prednisolone was in its crystalline forms in the physical mixtures and solid dispersions prepared with lactose without any change compared to the corresponding treated physical mixtures and physical mixtures of the drug. On the contrary solid dispersions prepared with dextrin were characterized by the complete absence of any diffraction peaks, suggesting a complete amorphization in the amorphous carrier. In the case of solid dispersions prepared with PEG 6000, prednisolone was still detectable in crystalline form. Therefore no complete amorphization occurred.
Fourier-transform infrared spectroscopy
Fourier-transform infrared (FT-IR) spectroscopy was used to further characterize possible interactions between the drug and carrier in the solid state. From the structures of the prednisolone, lactose, dextrin and PEG 6000, it can be assumed that possible interaction could occur between thehydroxyl or carbonyl group of prednisolone and the hydroxyl group of these carriers. Any sign of interaction would be reflected by a change in the position of C=O vibration and disappearance of O-H stretching depending on the extent of interaction.
The results from FT-IR spectroscopy showed that incorporation of prednisolone into PEG 6000, lactose, did not modify their peak positions and trends. These results further indicate the absence of well-defined interaction between prednisolone and PEG 6000, lactose as already confirmed from the X-ray diffraction study. The spectrum of prednisolone showed characteristic bands of the OH group which were found at 3200-3500 cm-1 (OH involved in intermolecular association). Two carbonyl stretching peaks appears as a very strong band at 1708 cm-1 and 1654 cm-1. The spectrum of dextrin showed, amongst others, important bands at 2953 cm-1 (C–H stretching) and 1109 cm-1 (C–O stretching). A very broad band was also visible at 3446 cm-1, which was attributed to the presence of water. Important vibrations detected in the spectrum of PEG 6000 are the C–H stretching at 2890 cm-1 and the C–O stretching at 1110 cm-1 and OH stretching at 3350 cm-1. Comparing the spectra of physical mixtures and solid dispersions of prednisolone with lactose, PEG 6000, and dextrin no difference was shown in the position of the absorption bands. The spectra can be simply regarded as the superposition of those of prednisolone and carrier. Although it could be expected to have hydrogen bonding between the hydrogen atom of the OH of the drug and one of the ione pairs of the oxygen atom in carrier, this could not be demonstrated.
Conclusion
An increased dissolution rate of prednisolone solid dispersions was observed with increasing the ratio of dextrin to the drug. Controversially, the dissolution rate of the drug was not varied upon increasing the ratio of the carrier to the drug when the drug was dispersed in lactose. However decreased dissolution rate of the solid dispersions was achieved by increasing the ratio of PEG 6000.
In conclusion improving the dissolution rate of prednisolone solid dispersions using polysaccharides like lactose is more appropriate than using dextrin and PEG 6000.
Conflict of interests
The authors declare no conflict of interests.
References
- 1.Kim EJ, Chun MK, Jang JS, Lee IH, Lee KR, Choi HK. Preparation of a solid dispersion of felodipine using a solvent wetting method. Eur JPharm Biopharm. 2006;64:200–5. doi: 10.1016/j.ejpb.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 2.Chiou WL, Riegelman S. Pharmaceutical applications of solid dispersion systems. J Pharm Sci. 1971;60:1281–302. doi: 10.1002/jps.2600600902. [DOI] [PubMed] [Google Scholar]
- 3.Ford JL. The current status of solid dispersions. Pharm Acta Helv. 1986;61:69–88. [PubMed] [Google Scholar]
- 4.Leuner C, Dressman J. Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm. 2000;50:47–60. doi: 10.1016/s0939-6411(00)00076-x. [DOI] [PubMed] [Google Scholar]
- 5.Habib MJ. Pharmaceutical Solid Dispersion Technology. Pennysylvania: Technomic Publishing; 2001. 7–36 [Google Scholar]
- 6.Serajuddin ATM. Solid dispersion of poorly water soluble drugs: early promises, subsequent problems, and recent breakthroughs. J Pharm Sci. 1999;88:1058–1066. doi: 10.1021/js980403l. [DOI] [PubMed] [Google Scholar]
- 7.Charman WN. Lipids, lipophilic drugs, and oral drug delivery: some emerging concepts. J Pharm Sci. 2000;89:967–978. doi: 10.1002/1520-6017(200008)89:8<967::aid-jps1>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 8.Craig DQM. The mechanism of drug release from solid dispersion in water-soluble polymers. Int J Pharm. 2002;231:131–144. doi: 10.1016/s0378-5173(01)00891-2. [DOI] [PubMed] [Google Scholar]
- 9.Spireas S, Sadu S. Enhancement of prednisolone dissolution properties using liquisolid compacts. Int J Pharm. 1998;166:177–188. [Google Scholar]
- 10.Kwan LC, Feldman S. The role of surfactants in the release of very slightly soluble drugs from tablets. J Pharm Sci. 1982;71:1038–45. doi: 10.1002/jps.2600710921. [DOI] [PubMed] [Google Scholar]
- 11.Sahin NO, Arslan H. Inclusion complex of prednisolone with skimmed milk. part I: physicochemical characterization. Yakugaku Zasshi. 2007;127:1255–61. doi: 10.1248/yakushi.127.1255. [DOI] [PubMed] [Google Scholar]
- 12.Dastmalchi S, Garjani A, Maleki N. et al. Enhancing dissolution, serum concentration and hypoglycemic effect of glibenclamide using solvent deposition technique. J Pharm Pharm Sci. 2005;8:175–81. [PubMed] [Google Scholar]
- 13.Barzegar-jalali M, Nayebi AM, Valizadeh H. et al. Evaluation of in vitro-in vivo correlation and anticonvulsive effect of carbamazepine after cogrinding with microcrystalline cellulose. J Pharm Pharm Sci. 2006;9:307–316. [PubMed] [Google Scholar]
- 14.Nokhodchi A, Talari R, Valizadeh H, Barzegar Jalali M. An investigation on the solid dispersions of chlordiazepoxide. Int J biomed sci. 2007;3:210–216. [PMC free article] [PubMed] [Google Scholar]
- 15.Carelli V, Di colo G, Nannipieri E. Effect of water-soluble additives on drug release from silicone rubber matrices. III. A study of release mechanism by differential scanning calorimetry. Int J Pharm. 1986;30:9–16. [Google Scholar]
- 16.Bisrat M, Nyström C, Edman P. Influence of dissolution rate of sparingly soluble drugs on corneal permeability in vitro. Int J Pharm. 1990;63:49–56. [Google Scholar]
- 17.Valizadeh H, Zakeri-milani P, Barzegar-jalali M, Mohammadi G, Danesh-bahreini MA, Adibkia K, Nokhodchi A. Preparation and characterization of solid dispersions of piroxicam with hydrophilic carriers. Drug Dev Ind Pharm. 2007 Jan;33:45–56. doi: 10.1080/03639040600814965. [DOI] [PubMed] [Google Scholar]
- 18.Valizadeh H, Nokhodchi A, Qarakhani N. et al. Physicochemical characterization of solid dispersions of indomethacin with PEG 6000, Myrj 52, lactose, sorbitol, dextrin, and eudragit e100. Drug Dev Ind Pharm. 2004;30:303–17. doi: 10.1081/ddc-120030426. [DOI] [PubMed] [Google Scholar]
- 19.Khan KA, Rhodes CT. Further studies of the effect of compaction pressure on the dissolution efficiency of direct compression systems. Pharm Acta Helv. 1974;49:258–261. [PubMed] [Google Scholar]
- 20.Craig DQM. The mechanisms of drug release from solid dispersions in water-soluble polymers. Int J Pharm. 2002;231:131–144. doi: 10.1016/s0378-5173(01)00891-2. [DOI] [PubMed] [Google Scholar]
- 21.Ford JL. The current status of solid dispersions. Pharm Acta Helv. 1986;61:69–88. [PubMed] [Google Scholar]
- 22.El-zein H, Riad L, El-bary AA. Enhancement of carbamazepine dissolution: in vitro and in vivo evaluation. Int J Pharm. 1998;168:209–220. [Google Scholar]
- 23.Hancock BC, Zografi G. Characteristics and Significance of the Amorphous State in Pharmaceutical Systems. J Pharm Sci. 1997;86:1–12. doi: 10.1021/js9601896. [DOI] [PubMed] [Google Scholar]
- 24.Matsumoto T, Zografi G. Physical properties of solid molecular dispersions of indomethacin with poly(vinylpyrrolidone) and poly(vinylpyrrolidone-co-vinyl-acetate) in relation to indomethacin crystallization. Pharm Res. 1999;16:1722–1728. doi: 10.1023/a:1018906132279. [DOI] [PubMed] [Google Scholar]