

Abstract
Introduction: The purpose of this investigation was to evaluate microencapsulated controlled release preparation of theophylline using Eudragit RS 100 as the retardant material with high entrapment efficiency. Methods: Microspheres were prepared by the emulsion-solvent evaporation method. A mixed solvent system consisting of methanol and acetone and light liquid paraffin as oily phase were chosen. Sucrose stearate was used as the surfactant to stabilize the emulsification process. The prepared microspheres were characterized by drug loading, Fourier-transform infrared spectroscopy (FTIR), differential scanning colorimetry (DSC) and scanning electron microscopy (SEM). The in vitro release studies were performed at pH 1.2 and 7.4 aqueous medium. Results: Increasing the concentration of emulsifier, sucrose fatty acid ester F-70, decreased the particle size which contributed to increased drug release rate. The drug loading microparticle Eudragit RS100(1:6) showed 60-75% of entrapment and mean particle size 205.93-352.76 μm.The results showed that, an increase in the ratio of polymer: drug (F5, 6: 1) resulted in a reduction in the release rate of the drug which may be attributed to the hydrophobic nature of the polymer. Conclusion: The release of theophylline is influenced by the drug to polymer ratio and particle size. Drug release is controlled by diffusion and the best-fit release kinetic is Higuchi model.
Keywords: Microspheres, Eudragit RS 100, Theophylline anhydrous, Ester sucrose
Introduction
Theophylline anhydrous has been widely used as a bronchodilator for the treatment of chronic asthma. The bronchodilator effect of theophylline increases with serum concentration over a range 5 to 20 µ ml-1 but, at a plasma level above 20 µg ml-1 there is an increased risk of toxic effects.1 The desirability of maintaining theophylline plasma levels within this narrow range had led to the development of several controlled release formulations.
Conventional and sustained released dosage forms of theophylline with different doses have been formulated and marketed. It is clear from investigations that there are fluctuations between plasma concentrations in sustained released given once daily and conventional dosage forms given several times daily with respect to their bioavailability and bronchodilator effect.
Solvent evaporation method is the preparation technique that is widely preferred for the preparation of controlled release microspheres. The first stage of this method is to prepare emulsion by adding the dispersed phase consisting of the drug, polymer and appropriate dispersion agent in the organic solvent to dispersion medium which is immiscible with the dispersed phase. At the second stage, minimatrix forms are obtained by removing the solvent used at the dispersed phase from the droplets which are formed in the emulsion. The selection of the organic solvents is done according to the polarity of dispersion medium and their dielectric constants related too miscibility with dispersion medium.2-5
Eudragit RS 100 is a copolymer of acrylic and methacrylic acid esters containing 5% of functional quaternary ammonium groups. Since Eudragit RS 100 contains the ammonium groups its permeability is pH independent. 2
Dispersing agents can be various polymeric materials, proteins, or surfactants, which simplify the formation of microspheres by decreasing the interfacial tension between the lipophilic and hydrophilic phases.6 The dispersing agent forms a thin protective layer around the droplets and hence reduces the extent of their collision and coalescence.6 Sucrose esters have been proposed as dispersing agents because of the advantages of low toxicity and biodegradation. According to earlier studies, the droplet stabilizer, sucrose stearate, was used to produce nicardipine-Eudragit RS100 microspheres by the evaporation method.7
In the present study, emulsion-solvent evaporation technique was used to prepare a sustained-release system of theophylline. The influence of several factors on various physical characteristics, including particle size, drug loading, and dissolution properties of the resulting microspheres were investigated.
Materials and Methods
Theophylline anhydrous (Merck, Germany), Edragit RS 100 (RÖhm Pharma GMBh, Weiterstadt, Germany), Sucrose stearate (Crodesta F70) (Croda GmbH, Mettelal, Germany). All solvents and reagents were of analytical grade.
Experimental Methods
The solubility of theophylline in various organic solvents was measured by adding excess amount of drug to 10 ml of solvents. The resulting drug suspension was agitated for 24 h at room temperature in glass vials sealed with Teflon-lined closure. These samples were centrifuged and filtered (0.45 µ nylon filter millipore). An aliquot of the filtrate was weighted and diluted appropriately for UV spectrophotometer measurement.
Preparation of microspheres
Microspheres were prepared by solvent evaporation method.7,8 A mixture of acetone and methanol (3:1 ratio) used as common solvent of theophylline and polymer. Theophylline and Eudragit RS 100 were dissolved completely in common solvent consisting of acetone and methanol (3:1 ratio) by stirring at 500 rpm with magnetic stirrer. Sucrose stearate was added to the resulting mixture and sonicated for complete dispersion. This mixture was cooled to 10 ºC in an ice bath while stirring at 500 rpm. The resulting mixture was poured into the liquid paraffin previously cooled to 10 ºC. After agitation at 400 rpm by 10 ºC the resulting emulsion was heated to 35 ºC gradually (1 ºC/min) and stirred at this temperature for 4h. During this period acetone and methanol were removed by evaporation. The solidified microspheres were filtered, washed six times with 50 ml of n-hexane, dried under vacuum at a room temperature for 12 h and stored in desiccators containing CaCl2.2
The effect of the process variables such as drug: polymer ratio, stirring rate, emulsifier concentration and volume of dispersing medium (liquid paraffin) on the mean particle size of the microparticles were investigated. Theophylline microspheres were prepared using various drug: polymer ratios (1:2, 1:3 …and 1:6), while keeping the other variables (stirring rate of 400 rpm, emulsifier concentration 3% w/v, volume of dispersing medium 200 ml) constant. Similarity to determine the effect of stirring rate on the mean particle size of microparticles, microspheres were prepared at various stirring rates (200, 400 and 600 rpm), while keeping the drug: polymer ratio at 1:6, emulsifier concentrations at 3%, solvent volume at 15 ml and liquid paraffin volume at 200 ml (Table 1).
Table 1. Anhydrous theophylline microsphere formulations prepared by Emulsion Solvent Evaporation Method (ESE).
| Formulations | Drug: Polymer ratio | Emulsion(O1/O2) | |||||
| Internal organic phase (O1) | External oily phase (O2) | ||||||
|
Theophylline (g) |
Eudragit RS 100 (g) |
Sucrose stearate (%) |
acetone (ml) |
Methanol (ml) |
Liquid paraffin (ml) |
||
| F1 | 1:2 | 1.5 | 3 | 3 | 11.5 | 3.5 | 200 |
| F2 | 1:3 | 1.5 | 4.5 | 3 | 19 | 3.5 | 200 |
| F3 | 1:4 | 1.5 | 6 | 3 | 26.5 | 3.5 | 200 |
| F4 | 1:5 | 1.5 | 7.5 | 3 | 34 | 3.5 | 200 |
| F5 | 1:6 | 1.5 | 9 | 3 | 41.5 | 3.5 | 200 |
Microspheres were also prepared using different emulsifier concentration of ester sucrose (1, 3 and 5% w/v) while keeping the drug: polymer ratio at 1:6 (drug: polymer ratio. Three microspheres were prepared that the amount of dispersing medium (outer phase, O2), was varied (100, 200 and 300 ml), while keeping the other variables constant as F5 microspheres.
Determination of drug loading and loading efficiency
Drug amount in microspheres was determined by dissolving 10 mg of each sample in 10 ml of methanol. The drug concentration was determined spectrophotometrically (UV-160, Shimadzu, Japan) at 212 nm.
The loading efficiency (%) was calculated according to the following equation:
Loading efficiency (%) = (actual drug content in microparticles/theoretical drug content) × 100
The production yield of the microparticles was determined by calculating accurately the initial weight of the raw materials and the final weight of the microspheres obtained .3,9 All of the experiments were performed in triplicate.
Viscosity measurement
A Brookfield rotational digital viscometer DVLV-II was used to measure the viscosity (cP) of the internal (dispersed) and external phases (continuous phase) at 10 ˚C. Spindle numbers 1 was rotated at 100 rpm for this measurement.
X-ray Powder diffractometry (X-RPD)
X-ray diffraction analysis was performed with a (Siemens D5000, Munich, Germany) using nickel-filtered CuKα radiation (a voltage of 40 KV and a current of 20 mA). The scanning rate was 2˚/min over a 2θ range of 20-60˚ and with an interval of 0.02˚.
Differential Scanning Calorimetry (DSC)
Typically, about 5 mg of sample was weighed into an aluminum pan, the pan crimped non-hermetically, and heated in the differential scanning calorimeter (DSC 60, Shimadzu, Japan) from 30 to 300 ˚C at a rate of 10 ˚C per min.
Fourier-Transform Infrared spectroscopy (FTIR)
The infrared spectrum of the drug, microspheres containing the drug were obtained in potassium bromide discs (0.5% w/w) using a FTIR spectrophotometer (Bomen Hartmann & Brann, Canada).
Particle size analysis and morphological studies
A laser light scattering particle size analyzer (SALD-2101, Shimadzu, Japan) was used to determine the particle size of the drug and microparticulate formulations. Samples were suspended in distilled water in a 1 cm cuvette and stirred continuously during the particle size analysis. The morphology of microparticles was examined with a scanning electron microscope (LEO 440i, England) operating at 15 kV. The samples were mounted on a metal stub with double adhesive tape and coated with platinum/palladium alloy under vacuum.
In vitro release study
A drug release test was carried out using a dissolution tester (USP basket method) at 37 ˚C and 100 rpm. 900 milliliters of simulated gastric fluid (pH 1.2) and phosphate buffer (pH 7.4) were equilibrated at 37±0.5 ˚C. Polysorbate 80 (0.02%, w/v) was added to each dissolution media to improve the wetting of the microspheres. Microspheres (200 mg drug) and tablet were placed in the apparatus and 3 ml aliquots of medium were withdrawn at pre-set times over 2h and replaced by 3 ml of fresh medium. The samples were filtered through 0.45 µm filters and used for the spectroscopic determination of the drug. After 2h, 17 ml of 0.2 M phosphate buffer stock, pre-equilibrated at 37 ˚C, were added to the dissolution vessel. The pH was immediately adjusted, if necessary, with 0.2N HCl or 0.2N NaOH to pH 7.4 .2
Drug concentration in the samples was measured by UV spectrophotometric analysis at 206 and 203 nm for the acidic and enteric buffers, respectively. Each experiment was repeated three times.
Results and discussion
Solubility of drug in organic solvent
According to Mehta et al., solubility of drug and polymer in the organic solvent determines the solidification rate of the polymer during the microparticle preparation process, which in turn affects microparticle properties such as drug incorporation, matrix porosity, and solvent residues.10,11
In order to find suitable solvent, the solubility of Theophylline in various solvents was determined and the results are shown in Table 2. Theophylline anhydrous was soluble in polar solvents such as ethanol and methanol.
Table 2. Solubility of anhydrous theophylline in various solvents .
| Solvent solubility (mg/ml) | solubility (mg/ml) |
| Dichloromethane | 0.91 |
| Isopropyl alcohol | 1.04 |
| Acetonitrile | 0.784 |
| Ethanol | 15.19 |
| Methanol | 0.699 |
| Acetone | 3.142 |
Preparation of microparticles
Microspheres were formed after a series of steps like solvent extraction and solvent evaporation. Microspheres were prepared by emulsion solvent evaporation method using different polymer-drug ratios (2: 1, 3: 1 … and 6: 1) (Table 1). The drug encapsulation efficiency was high 60-75% and the production yield values were over 90%. The encapsulation efficiency of the drug depended on the solubility of the drug in the solvent and continuous phase.12 Theophylline is insoluble in liquid paraffin (continuous phase) and encapsulation efficiency of microspheres was high. Scanning electron microscopic photographs of microspheres (as F5) are shown in Figure 1.
Figure 1.
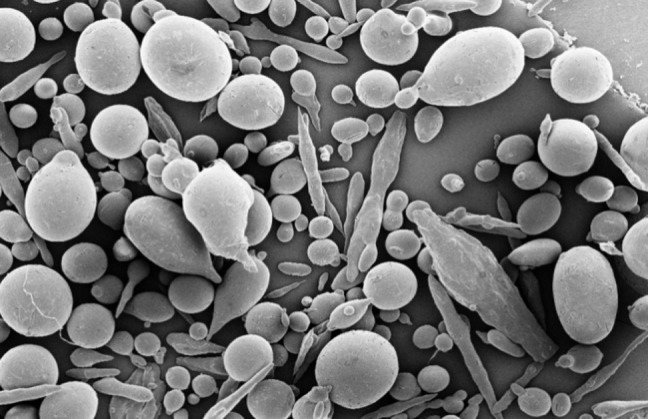
SEM of a spherical microspheres containing anhydrous theophylline F5 (drug: polymer ratio 1:6) at 50x.
The size of the prepared microspheres was in the range of 248-375 µm (Table 3). Table 3 shows that the ratio of polymer to drug plays an important role in the particle size of microspheres. As the ratio of polymer: drug is increased, particle size is decreased. Increase of polymer concentration resulted in a significant decrease (10 cPs) in the viscosity of dispersed phase, leading to smaller emulsion droplet size and finally smaller particle size. Similar observation has been reported for verapamil microspheres.2 At high polymer: drug ratio the amount of drug per microspheres was low. Volume-based size distribution of drug, polymer, and drug loaded microspheres indicated a log–probability distribution.
Table 3. Effect of drug: polymer ratio, stirring rate, Emulsifier and dispersing medium on the drug content, production yield and particle size of anhydrous theophylline microspheres.
| Parameter | Characteristics | ||||||
| Formulation code | Process variable | Production yield (%±SD) | Theoretical drug content (%) | Mean drug entrapped (%) | Mean particle Size (µm ± SD) | Drug loading efficiency (%±SD) | |
| Polymer: Drug ratio |
F1 F2 F3 F4 *F5 |
2:1 3:1 4:1 5:1 6:1 |
90.50±2.24 91.83±3.24 99.63±3.08 99.90±3.13 92.83±4.12 |
33.33 25 20 16.67 14.29 |
25.3±0.65 17.48±3.93 14.01±2.53 10.53±4.13 8.67±1.32 |
352.76±1.68 286.08±1.67 248.09±1.59 225.57±1.36 205.93±1.53 |
75.92±12.3 69.91±1.90 70.05±12.6 63.18±15.7 60.67±9.50 |
| Stirring rate (rpm) |
F5-1 *F1-2 F5-3 |
200 400 600 |
-
92.83±4.12 6.19 ±91.65 |
-
14.29 14.29 |
-
8.67±1.32 0.27±13.40 |
-
375.93±1.53 300.88±1.86 |
-
60.67±9.5 95.74±1.59 |
| Emulsifier concentration (%) |
F5-4 *F5-5 F5-6 |
1 3 5 |
1.24 ±81.01 92.83±4.12 6.59 ±92.83 |
14.29 14.29 14.29 |
0.32± 7.61 8.67±1.32 8.04±0.87 |
1.69±398.88 375.93±1.53 234.75 ±1.68 |
54.36±1.88 60.67±9.50 57.43±0.05 |
| Volume of dispersing medium (ml) |
F5-7 *F5-8 F1-9 |
100 200 300 |
1.24±94.43 92.83±4.12 77.47±3.22 |
14.29 14.29 14.29 |
8.57±1.19 8.67±1.32 10.05±0.48 |
1.71±303.76 375.93±1.53 219.36±1.76 |
59.97±0.57 60.67±9.50 70.36±2.80 |
Physical Characterization
X-ray diffraction patterns of the microspheres containing increasing polymer ratios are presented in Figure 2. DSC thermograms of microspheres are shown in Figure 3. The endothermic peaks demonstrate a reduction in the intensity. The endotherm peak of pure drug was observed at 61.68°C (Figure 3).
Figure 2.
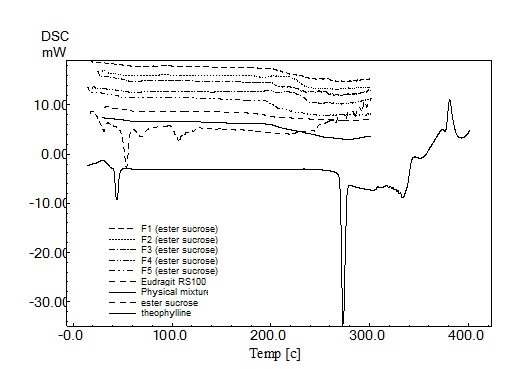
DSC thermogram of anhydrous theophylline, Eudragit RS100, physical mixture F5 (1:6 drug and polymer ratio) and different microsphere formulations.
Figure 3.

X-ray diffraction of anhydrous theophylline (a), Eudragit RS100 (b), physical mixture F5 (c), F1 (d), F2 (e), F3 (f), F4 (g) and F3 (h).
All prominent absorption bands of the drug were unchanged in the FT-IR spectra of the microspheres (Figure 4).
Figure 4.

FTIR spectrum of anhydrous theophylline (a), cellulose acetate butyrate (b), physical mixture F5 (c), F1 (d), F2 (e), F3 (f), F4 (g) and F3 (h).
Effect of formulation variables on the micromeritics properties of microspheres
The effect of drug: polymer ratio, the stirring rate, emulsifier concentration and volume of dispersing medium on the mean particle size of theophylline microspheres is shown in Table 3. In order to investigate the effect of stirring rate on the physical properties of theophylline microspheres, formulation F5(1:6 drug: polymer ratio) was chosen and the stirring rate altered in the range of 200-600 rpm. The dispersion of the internal phase of drug and polymer into the droplets in the external phase depended on the agitation speed of the systems. As stirring rate increased, the size of microparticles was reduced. For example when the rate of stirring was increased from 400 to 600 rpm, the mean particle size was decreased from 375.93±1.53 (F5-2) to 300.88±1.86 µm ((F5-3).
The effect of the ester sucrose concentration on the mean particle size of theophylline microspheres was shown in Table 3. The results showed that decreasing the amount of ester sucrose ((F5-4) increases the particle size of microspheres. The results indicated that reduction of volume of dispersing medium increased of particle size of microspheres.
In vitro release studies
The release profile of theophylline from Eudagit RS 100 microspheres is illustrated in Figure 5.
Figure 5.

Cumulative percent release of anhydrous theophylline from microspheres prepared with different polymer-to-drug ratio, physical mixture and teophylline SR® tablet
The effect of drug: polymer ratio on the drug release from microspheres depended on the polymer concentration of the prepared devices which indicates that the release rate is decreased with increasing the amount of the polymer (F5).18
The release rates of larger microspheres (F5and theophylline SR tablet) were slower than that of smaller microspheres (F1, F2, F3 and F4) and had a burst release in the initial phase (Figure 5). Drug release rates were increased with increasing amounts of theophylline in the formulation (with low polymer concentration from F5 to F1). Higher level of theophylline corresponding to lower level of the polymer in the formulation resulted in an increase in the drug release rate. Burst effect of 6:1 polymer: drug formulation was lower than that of other formulation (p<0.05). Statistical analysis of data was performed by comparing the DE (dissolution efficiency), t50% (dissolution time for 50% fractions of drug); ''similarity factor, f2 (used to compare multipoint dissolution profiles)'' (Table 4).13
Table 4. Comparision of various release characteristics of anhydrous theophylline from different microsphere formulations, physical mixture and teophylline SR® tablet.
| Formulation | aQ2 | bQ8 |
ct 50% (h) |
dDE |
Similarity factor |
| F1 | 63.3±3.6 | 100.19±0.16 | 0.75 | 89.21 | 27.29 |
| F2 | 65.3±0.3 | 99.11±0.19 | 0.5 | 95.08 | 20.07 |
| F3 | 54.9±0.2 | 92.64±1.41 | 0.5 | 89.47 | 22.95 |
| F4 | 50.6±0.5 | 94.52±0.23 | 2 | 88.39 | 24.63 |
| f | 48.5±3.2 | 92.80±1.85 | 2 | 91.28 | 27.32 |
| PM | 64.8±1.4 | 102.20±1.54 | 0.5 | 96.36 | 21.63 |
|
Theophylline SR® |
12.8±1.5 | 80.86±5.74 | 5 | 73.94 | 100 |
The change of stirring speed emulsification process also influenced the drug release profile as shown in Figure 6.
Figure 6.

Cumulative percent release of Tolmetin sodium from microspheres F5 prepared with different rates of stirring
Higher agitation rates resulted in slower drug release (as F5-3). At high concentration of ester sucrose a faster drug release was observed (Figure 7). At various volumes of dispersing medium, there is no difference on the drug released. The faster drug release and higher drug entrapment was observed from microspheres prepared using larger volume of processing medium at pH 1.2 and 7.4 (Figure 8). Mathematical models have been used extensively for the parametric representation of dissolution data.2,14 The fit parameters to Higuchi, first-order, Peppas and zero-order kinetics were determined (Table 5). High correlation to Higuchi and first order kinetics was observed for F5and theophylline SR® tablet, respectively.
Figure 7.

Cumulative percent release of Tolmetin sodium from microspheres F5 prepared with different emulsifier concentration.
Figure 8.

Cumulative percent release of Tolmetin sodium from microspheres F5 prepared with different paraffin amount.
Table 5. Fitting parameters of the in vitro release data to various release kinetic models.
| Order | F1 | F2 | F3 | F4 | F5 |
Tablet SR |
Physical Mixture |
|
|
Zero f=kt |
K | 0.0006 | 0.0003 | 0.0003 | 0.0003 | 0.0008 | 0.0007 | 0.0003 |
| RSQ | 0.9385 | 0.4461 | 0.4440 | 0.4631 | 0.9114 | 0.6905 | 0.5558 | |
| D(SS )% | 698.31 | 980.35 | 880.56 | 868.12 | 701.15 | 457.99 | 897.21 | |
|
First Ln(1-f)=kt |
k | 0.0056 | 0.0057 | 0.0018 | 0.0018 | 0.0031 | 0.0032 | 0.0061 |
| RSQ | 0.9074 | 0.9110 | 0.6101 | 0.6629 | 0.8517 | 0.9932 | 0.9772 | |
| D(SS ) % | 251.30 | 269.85 | 581.55 | 564.91 | 373.01 | 747.72 | 318.13 | |
|
Peppas Lnf=lnk+blnt |
b | 0.5267 | 0.2840 | 0.0780 | 0.0501 | 0.2076 | 1.2454 | 0.2245 |
| k | 0.1877 | 0.1905 | 0.3836 | 0.3974 | 0.1894 | 0.0004 | 0.2699 | |
| RSQ | 0.9624 | 0.9999 | 0.8360 | 0.9741 | 0.8757 | 0.9723 | 0.9476 | |
| D(SS) % | 8.4930 | 0.2167 | 6.2954 | 1.6340 | 23.9331 | 96.6275 | 6.6756 | |
| Higuchi f=kt0.5 | k | 0.0444 | 0.0171 | 0.0170 | 0.0183 | 0.0381 | 0.0339 | 0.0157 |
| RSQ | 0.9524 | 0.6556 | 0.6682 | 0.6852 | 0.9344 | 0.8740 | 0.7825 | |
| D(SS) % | 231.29 | 684.80 | 660.99 | 623.66 | 225.05 | 1399.92 | 727.68 | |
Discussion
The high solubility of the drug prevented the preparation of saturated solutions in the solvents. In Mehta’s study10, polymers having high solubility in methanol stayed longer in the semi-solid state, the dispersed phase became more concentrated before it completely solidified, resulting in denser microparticles. On the other hand, Bodmeier and co-workers found that methylene chloride result in higher encapsulation efficiency compared to other solvents.15 In order to justify this discrepancy, the authors point out that acetonitrile is more soluble in water than methylen chloride. Knowing that theophylline is slightly soluble in methylen chloride, also acetonitrile is water-miscible solvent (non-solvent for Eudragit RS 100) it can be assumed that it plays a dual function in facilitating the polymer precipitation. The high solubility allowed relatively fast mass-transfer between the dispersed and the continuous phases and leads to fast precipitation of the polymer. The importance of solubility of the organic solvent was also confirmed by the fact that the addition of water-miscible co-solvents such as acetone, methanol, ethyl acetate, or dimethyl sulfoxide (DMSO), contributes to increased encapsulation efficiency.15
Generally, the encapsulation efficiencies of the microspheres prepared with Eudragit RS100 were high; this can be attributed to the low content of the ammonium group which facilitates the diffusion of the entrapped drug to the surrounding medium during formation of the microspheres.
The surface of microspheres appeared spherical but rough and there were several cracks on the surfaces. It is observed that in spite of using the constant polymer/solvent ratio, the depth of cracks on the surface of microspheres become greater while the polymer amount of formulation was increased. This can be attributed to the increasing volume of dispersing medium, i.e. liquid paraffin (Table 1) resulting from the extended diffusion of organic solvent to the dispersing medium. Similar observation has been reported for verapamil microspheres.2
The microsphere size distribution depended on the droplet size of the emulsion and on the extent of aggregation between forming microspheres. Polymer deposition within the droplets occurred through the removal of the polymer solvent. The polymer solvent system, acetone and methanol, were soluble in liquid paraffin to a limited extent. Upon emulsification of the polymer solution some of the acetone and methanol migrate into the liquid paraffin external phase. The rate of acetone and methanol transfer depended on the size of the emulsion droplets, since partitioning is a surface area phenomenon. Therefore the rate of solvent removal was dependent upon the rate of solvent portioning and evaporation.16
The X-ray spectrum showed that addition of amorphous polymer decreased the degree of crystallinity of the drug (Figure 2). The DSC thermogram demonstrated no abrupt or drastic change in the thermal behavior of rather the drug or polymer which indicates no possible drug-polymer interaction (Figure 3). However in the thermogram of the microparticles there was no endothermic peak of the drug melting, suggesting the amorphous state of the drug in the microparticles. It was observed that sucrose stearate prevents coalescence in the emulsion system by the formation of a protective film around the droplets (Table 3). This may be due to the formation of large-size microspheres at lower emulsifier levels, which provided less surface area for drug escape to the external processing medium. These results agreed well with Kawata et al., and Yüksel et al..7,8
Ester sucrose, were used to modify solvent diffusion rates in the emulsion for the preparation of microspheres.7,18 It has been reported that ester sucrose ensured the diffusion of solvent in emulsion droplets toward outer phase, prohibiting molecular interaction between the drug and inner phase solvent. Finally, the drug and the polymer are solidifies in the form of microspheres due to their decreasing solubilities.7,19Increasing the amount of ester sucrose decreased the particle size of microspheres, because the globules of emulsion were divided to smaller particles. On the other hand the particle size of microspheres was proportional to the viscosity of the dispersing medium.
The incorporation efficiencies of theophylline in the microspheres were between 60-75%. The results showed that when the stirring rate was increased to 600 rpm, the loading efficiency increased. Increasing the amount of emulsifier from 1-5% did not affect the drug content (p>0.05). However decreasing ester sucrose concentration (from 3% to 1%) decreased production yield (p<0.05). Higher entrapment of drug was noticed when a lower concentration of ester sucrose was used (Table 3). The volume of processing medium (O2) influenced the entrapment efficiency of the microspheres (Table3). As the volume of processing medium was decreased from 200 ml to 100 ml, the entrapment efficiency did not change (p>0.05). The results also showed that when volume of dispersing medium was increased from 200 ml to 300 ml, the entrapment efficiency significantly decreased from 60% to 70% (p<0.05). As the volume of processing medium was increased, the emulsion droplets probably moved freely in the medium, thus reducing collision induced aggregation and yielding small and uniform microspheres. This could also be the reason for higher drug extraction into the processing medium resulting in lower entrapment efficiency.
This can be explained by a decreased amount of drug present close to the surface and also by the fact that the amount of uncoated drug decreases with higher polymer concentration (Figure 5).19 However, when particles are prepared by O1/O2 method, water-soluble drugs do not have tendency to migrate to the organic medium, thereby concentrating at the surface of the particles and resulting in the burst effect.21 Moreover, the burst release could also be explained by the imperfect encapsulation of the drug inside microparticles, resulting from the unstable nature of the emulsion droplets during the solvent removal step. This potential instability may cause a part of the loaded drug to relocate at the microparticle surface, thereby rapidly released.9The corresponding release rates are also reduced by increasing polymer concentrations (Figure 5). This phenomenon can be attributed to the increased thickness of the polymer matrix surrounding the drug. Similar behavior was observed in verapamil prepared with Eudragit RS100.2 In most cases, a biphasic dissolution profile was observed, the initial rapid drug leakage generally ended very; for the remaining time, nearly linear behavior was observed. It can be supposed that the first portion of the curves is due to theophylline dissolution, which starts immediately after the beginning of the test for the portion of drug on the surface of microparticles. After such a phase, two phenomena can combine in enhancing in the diffusion of the remaining dispersed drug into the bulk phase as well as the formation of pores within the matrix due to the initial drug dissolution; particle wetting and swelling which enhances the permeability of the polymer to the drug 22 (Figure 5).
Dissolution efficiency (DE) was calculated from the area under the dissolution curve at time and expressed as percentage of the area of the rectangle described by 100% dissolution in the same time. F5 microsphere and theophylline SR® tablet showed lower dissolution efficiency. Theophylline SR ® tablet and F5 had lower release in comparison with microspheres (p< 0.05), (Table 4 & Figure 5). There is no similarity between F5 and theophylline SR® tablet.
The data obtained were fitted to Korsemeyer-Peppas model in order to find out n value, which describes the drug release mechanism (Table 5). The n value of microspheres of different drug to polymer ratio was between 0.21, indicating that the mechanism of the drug release was diffusion controlled.
Conclusion
Theophylline loaded microspheres were prepared by solvent evaporation method (O1/O2). The results of this study shows that increased polymer concentration could slow down dissolution rate, decrease particle size, and influence the surface properties of microspheres. The entrapment efficiency was at range of 60-75% for all formulations. The encapsulation efficiency was more influenced with changing the stirring speed as well as dispersing medium volume. It was observed that at higher polymer concentration, the mean particle size of the microspheres is low but increasing the stirring speed and emulsifier content, resulted in smaller mean particle size of microspheres. Drug release from theophylline microspheres followed Higuchi model. It was suggested that mechanism of drug release from microspheres was diffusion controlled. Controlled release with initial burst release (loading dose) achieved with these formulations may reduce dose frequency and side effects as well as improved patient compliance.
Ethical issues
None to be declared.
Conflict of interests
The authors declare no conflict of interests.
Acknowledgement
The financial support from the research council of Tabriz University of Medical Sciences is greatly acknowledged.
References
- 1.Katzung BG, Basic and clinical pharmacology, 9th ed. 2004; pp.738-739.
- 2.Kilicarslan M, Baykara T. The effect of the drug/polymer ratio on the properties of verapamil HCL loaded microspheres. Int J Pharm. 2003;252:99–109. doi: 10.1016/s0378-5173(02)00630-0. [DOI] [PubMed] [Google Scholar]
- 3.Bogataj M, Mrhar A, Kristl A, Kozjek F. Eudragit E microspheres containgbacampicillin: preparation by solvent removal methods. J Microencapsul. 1991;8:401–406. doi: 10.3109/02652049109069567. [DOI] [PubMed] [Google Scholar]
- 4.Benoit JP, Marchais H, Rolland H, Velde VV. Biodegradable microspheres: advances in production technology. In: Benita S. (Edition), Microencapsulation Methods and Industrial Applications. New York:Marcel Dekker;1996.pp: 35-72.
- 5.Horoz BB, Kiliçarslan M, Yüksel N, Baykara T. Influenced of aluminum tristearate and sucrose stearate as the dispersing agents on physical properties and release characteristics of Eudragit RS microspheres. AAPS PharmSciTech. 2006;7:e111–e117. doi: 10.1208/pt070116. [DOI] [PubMed] [Google Scholar]
- 6.Wu PC, Huang YB, Chang JS, Tsai MJ, Hung tsai Y. Design and evaluation of sustained release microspheres of potassium chloride prepared by Eudragit®. Eur J Pharm Sci. 2003;19:115–122. doi: 10.1016/s0928-0987(03)00069-1. [DOI] [PubMed] [Google Scholar]
- 7.Yüksel N, Baykara T. Preparation and evaluation of controlled release microspheres containing nicardipine hydrochloride. Int J Pharm Adv. 1996;1:398–407. [Google Scholar]
- 8.Kawata M, Nakamura M, Goto S, Aoyama T. Preparation and dissolution pattern of Eudragit RS microcapsules containing ketoprofen. Chem Pharm Bull. 1986;34:2618–2623. doi: 10.1248/cpb.34.2618. [DOI] [PubMed] [Google Scholar]
- 9.Jelvehgari M, Barar J, Valizadeh H, Shadrou S, Nokhodchi A. Formulation, characterization and in vitro evaluation of theophylline-loaded Eudragit RS 100 microspheres prepared by an emulsion-solvent diffusion/evaporation technique. Pharm Dev Technol. 2011;16:637–44. doi: 10.3109/10837450.2010.508075. [DOI] [PubMed] [Google Scholar]
- 10.Mehta RC, Jeyanthi R, Calis S, Thanoo BC, Burton KW, Deluca PP. Biodegradable microspheres as depot system for parenteral delivery of peptide drugs. J Control Rel. 1996;29:375–384. [Google Scholar]
- 11.Yeo Y, Park K. Control of encapsulation efficiency and initial burst in polymeric microparticle systems. Arch Pharm Res. 2004;27:1–12. doi: 10.1007/BF02980037. [DOI] [PubMed] [Google Scholar]
- 12.Behera BC, Sahoo SK, Dhal S, Barik BB, Gupta BK. Characterization of glipizide-loaded polymethacrylate microsphere prepared by an emulsion solvent evaporation method. Trop J Pharm Res. 2008;7:879–885. [Google Scholar]
- 13.Pignatello R, Consoli P, Puglist G. In vitro release kinetics of tolmetin from tabletted eudragit microparticles. J Microencapsul. 2000;17:373–383. doi: 10.1080/026520400288337. [DOI] [PubMed] [Google Scholar]
- 14.Alex R, Bodmeier R. Encapsulation of water-soluble drugs by a modified solvent evaporation method. Effect of process and formulation variables on drug entrapment. J Microencapsul. 1990;7:347–355. doi: 10.3109/02652049009021845. [DOI] [PubMed] [Google Scholar]
- 15.Bodmeier R, Mcginity JW. Solvent selection in the preparation of poly(DL-Lactide) microparticles prepared by the solvent evaporation method. Int J Pharm. 1988;43:179–186. [Google Scholar]
- 16.Sunit kumar S, Abdul arif M, Barik BB, Prakash CH S. Formulation and in vitro evaluation of eudragit® microspheres of stavudine. Trop J Pharm Res. 2005;4:369–375. [Google Scholar]
- 17.Perumal D. Microencapsulation of ibuprofen and Eudragit® RS 100 by the emulsion solvent diffusion technique. Int J Pharm. 2001;218:1–11. doi: 10.1016/s0378-5173(00)00686-4. [DOI] [PubMed] [Google Scholar]
- 18.Kim BK, Hwang SJ, Park JB, Park HJ. Preparation and characterization of drug-loaded polymethacrylate microspheres by an emulsion solvent evaporation method. J Microencapsul. 2002;19:811–822. doi: 10.1080/0265204021000022770. [DOI] [PubMed] [Google Scholar]
- 19.Kawashima Y, Niwa T, Handa T, Takeuchi H, Iwamoto T, Itoh K. Preparation of contolled release microspheres ibuprofen with acrylic polymers by a novel quasi-emulsion solvent diffusion method. J Pharm Sci. 1989;78:68–72. doi: 10.1002/jps.2600780118. [DOI] [PubMed] [Google Scholar]
- 20.Moore JW, Flanner HH. Mathematical comparison of dissolution profiles. Pharm Technol. 1996;20:64–74. [Google Scholar]
- 21.Jameela SR, Suma N, Jayakrishnan A. Protein release from poly(ε-caprolactone) microspheres prepared by melt encapsulation and solvent evaporation techniques: a comparative study. J Biomater Sci Polym Ed. 1997;8:457–66. doi: 10.1163/156856297x00380. [DOI] [PubMed] [Google Scholar]
- 22.Yüksel N, Kanik AE, Baykara T. Comparison of in vitro dissolution profiles by ANOVA-based, model dependent and independent methods. Int J Pharm. 2000;209:57–67. doi: 10.1016/s0378-5173(00)00554-8. [DOI] [PubMed] [Google Scholar]