

Abstract
Abstract
Inflammatory myofibroblastic tumors (IMTs) are categorized as intermediate biologic neoplasms, whereas IMTs with genetic features of ran-binding protein 2 (RANBP2) and anaplastic lymphoma kinase (ALK) rearrangement (IMT-RAs) are possibly related to a more aggressive clinical course. However, fewer than 10 cases of IMT-RA have been reported to date. Herein, we present 2 new cases of IMT-RA in which both tumors recurred quickly after primary surgery; one patient died 3 months later from the disease, and the other patient has been living with the disease for 12 months. IMT-RAs are characterized by noncohesive epithelioid and rounded tumoral cell morphology, commonly derived from pelvic and peritoneal cavities, and frequently show larger tumor sizes. The relation between the clinicopathologic features and poor prognosis of IMT-RA is discussed.
Virtual slides
The virtual slide(s) for this article can be found here: http://www.diagnosticpathology.diagnomx.eu/vs/3314123381007714
Keywords: Inflammatory myofibroblastic tumor, RANBP2-ALK, Fluorescence in situ hybridization
Background
Inflammatory myofibroblastic tumors (IMTs) are mesenchymal neoplasms of intermediate biologic potential that are derived from myofibroblastic cells and accompanied by rich inflammatory infiltrates [1]. IMTs have a predilection for children and adolescents, and the most common anatomical locations are the abdominopelvic region, lung, and retroperitoneum. IMTs show expression of anaplastic lymphoma kinase (ALK) protein triggered by ALK gene (at 2p23) rearrangement, which has been found in 36–60% of IMTs [2]. ALK fusion oncogenes have been identified in a small proportion of IMTs, including SEC31L1 at 4q21, ATIC at 2q35, CARS at 11p15, TPM3 at 1p23, TMP4 at 19p13, CLTC at 17q23, PPFIBP1 at 12p11, and ran-binding protein 2 (RANBP2) at 2q13 [3]. Different fusion partners may lead to distinct subcellular locations of the corresponding chimeric protein, which result in distinct immunostaining patterns when detected by ALK antibody. IMTs with CARS, ATIC, and SEC31L1 fusion are generally associated with smooth cytoplasmic staining, whereas fusion with CLTC, a main structural protein of coated vesicles, displays a granular cytoplasmic pattern [4].
RANBP2 fuses with the ALK gene through balanced or unbalanced translocation [5]. The RANBP2 gene encodes a 358-kd nuclear pore protein [6], thus IMTs with RANBP2-ALK (IMT-RA) rearrangement display a unique nuclear membrane staining pattern. Most importantly, IMT-RA is possibly associated with a poor prognosis; however, this relation remains inconclusive, as fewer than 10 cases of IMTs with genetically confirmed RANBP2-ALK fusion have been reported to date [5,7-11]. Herein, we present 2 new cases of IMT-RA with follow-up information. The relations between the histopathologic features and prognosis of IMT-RAs are discussed further.
Case presentation
Case 1 was a 19-year-old female who was hospitalized due to paroxysmal abdominal pain combined with nausea and vomiting for 4 days. Ultrasound examination showed a 19 × 17 × 11 cm solid mass in the pelvic cavity and medium amounts of ascitic fluid. Intraoperatively, the tumor was situated in the mesenterium region of the terminal ileum, and had locally invaded into the adjacent ileum wall. Resections of the tumor, affected terminal ileum, and ileocecum were performed. The patient rejected further chemotherapy. Nine weeks after the operation, radiological imaging displayed recurrent intra-abdominal occupations with massive ascites. Concurrently, acute renal insufficiency was identified by laboratory tests. The patient died of the disease after 3 weeks of maintenance treatment.
Case 2 was a 39-year-old male with an initial complaint of laborious urination for more than 2 months and abdominal distention and abdominal pain for 10 days. Computer Tomography (CT) revealed a 15 × 10 × 8 cm occupation in the pelvic cavity (Figure 1). The neoplasm was found in the mesangial region at the junction of sigmoid and rectum during the operation, with invasions of the neighboring sigmoid, the upper portion of the rectum, and left ureter. The tumor and afflicted intestines and left ureter were completely excised. The patient was treated with epirubicin and iphosphamide following the operation. Four months after the first excision, CT showed multiple recurrent masses in the presacral, pararectal, and left iliac vessel regions. The patient then underwent chemoembolization therapy. Follow-up has continued to date, and the patient has lived with the tumor for 12 months.
Figure 1.

Computer tomography of Case 2 revealed a heterogeneous-density occupation in the left pelvic cavity.
Histopathological and genetic findings
The gross and histological features were similar in Case 1 and Case 2. The tumors were solid and gray-yellowish in the cut surface. Focal hemorrhage and a myxomatous appearance were observed. Microscopically, the neoplasm showed a fasciitis-like pattern and the stromal myxoid change was prominent (Figure 2A, B). The tumor cells were generally non-cohesive, and frequently demonstrated ganglion-like morphology of slightly amphophilic cytoplasm, vesicular chromatin, and large nucleoli (Figure 2C). There were also a few binucleate cells sparsely presented in the lesion (Figure 2D). In area of the neoplasm, some tumor cells were tightly packed and arranged in sheets (Figure 2E). The mitotic figures ranged from 1 to 4 per 10 high-power fields. There were abundant admixed polymorphs infiltrates, including neutrophils, eosinophils, and lymphoplasma cells, which were unevenly distributed throughout the lesion. Erythrocyte extravasation was another feature found in both cases. The tumor invaded the adjacent intestinal tissues, involving the adventitia, muscularis propria, and submucosa with a vague nodular pattern (Figure 2F).
Figure 2.

Histopathological findings of inflammatory myofibroblastic tumor with RANBP2 and ALK gene rearrangement. (A) and (B) The neoplastic cells were loosely arrayed and distributed in an abundant myxoid stroma. There were striking neutrophilic inflammatory infiltrates in the lesion, while lymphoplasma cells were only occasionally observed (inserted panel) (A and B, hematoxylin and eosin [H&E] staining, 200× original magnification; inserted panel, H&E staining, 400× original magnification). (C) The tumor cells showed ganglion-like morphology, characterized by an abundant and slightly amphophilic cytoplasm, round nuclei, vesicular chromatin, and large nucleoli (H&E staining, 400× original magnification). (D) A few binucleate cells were found in the tumor (H&E staining, 400× original magnification). (E) In focal area, the tumor cells were tightly packed around dilated vessels (H&E staining, 100× original magnification). (F) The tumor infiltrated the adjacent intestinal wall in a vague nodular pattern (H&E staining, 40× original magnification).
Immunohistochemically, the tumor cells showed distinctive nuclear membrane staining for ALK (Dako; Clone ALK1; Ready-to-use) (Figure 3A) in both cases. Similarly, positive staining for CD30 (Dako; Clone Ber-H2; 1:40 dilution) (Figure 3B), desmin (Dako; Clone D33; 1:100 dilution) (Figure 3C), and SMA (Dako; Clone 1A4; 1:100 dilution) was also observed in some tumor cells in the 2 cases. In both cases, the tumor cells were nonreactive to AE1/AE3 (Dako; Clone AE1 + AE3; 1:200 dilution), cytokeratin 8/18 (Santa Cruz Biotechnology; Clone NCL-5D3; 1:100 dilution), CD117 (Dako; polyclonal; 1:100 dilution), calponin (Santa Cruz Biotechnology; Clone CALP; 1:200 dilution), S100 (Dako; polyclonal; 1:500 dilution), CD21 (Dako; Clone 1 F8; 1:25 dilution), HMB45 (Dako; 1:50 dilution), myogenin (Dako; Clone F5D; 1:50 dilution), and Myf4 (Novocastra; Clone LO26; 1:500 dilution).
Figure 3.

Immunohistochemistry features of inflammatory myofibroblastic tumor. The tumor cells showed nuclear membranous ALK immunostaining (A), cytomembranous CD30 immunostaining (B), and cytoplasmic desmin immunostaining (C) (A and B, 400× original magnification; C, 200× original magnification). (D) Interphase fluorescence in situ hybridization (FISH) revealed 1 fused signal (arrow) in the tumor nucleus, indicating the presence of RANBP2-ALK translocation. One separate orange signal and 1 separate green signal indicated a normal ALK and a normal RANBP2 locus, respectively (1000× original magnification).
Fluorescence in situ hybridization (FISH) was performed on 4 μm-thick tissue sections with bacterial artificial chromosome probes for RANBP2 (RP11-348G16) and ALK (RP11-984I21 and RP11-62B19). In both cases, 1 fused signal was observed in the tumor cell nucleus, indicating the presence of RANBP2 and ALK gene rearrangement (Figure 3D) by unbalanced genetic rearrangement mechanism [5].
To pinpoint the fusion location of the RANBP2 and ALK genes, reverse-transcription polymerase chain reaction (RT-PCR) analysis was performed. Total RNA was extracted from 15 μm-thick paraffin sections using the RNeasy® FFPE kit (QIAGEN, Germany), and reverse-transcribed using random hexamer primers. PCR was performed using previously introduced primers [8] for 45 cycles as follows: 94°C for 30 seconds, 60°C for 30 seconds, and 72°C for 30 seconds. An expected 254-bp amplification product was detected in Case 2 (Figure 4A). No PCR product was identified in Case 1, which may be due to RNA degradation in the paraffin-embedded block since positive amplification results (285-bp) of beta-actin as a householding gene were not found either in Case 1 but present in Case 2 (Figure 4B). Direct sequencing of the chimeric cDNA product confirmed that the RANBP2-ALK fusion point was composed of exon 18 of RANBP2 to exon 20 of ALK (Figure 4C).
Figure 4.
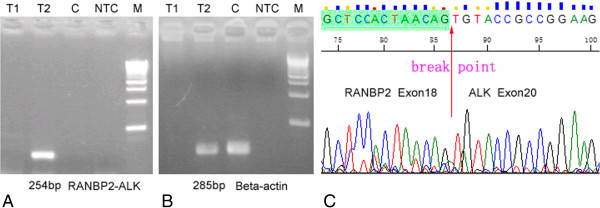
Genetic features of inflammatory myofibroblastic tumor. (A) Reverse-transcription polymerase chain reaction (RT-PCR) with RANBP2 and ALK primers. Lane T1: no amplification band was observed in Case 1. Lane T2: an expected 254-bp product was present in Case 2. Lane C: tonsil tissue was adopted as a control, and no amplification band was observed. Lane NTC: no template control. (B) Detection of house-keeping gene of beta-actin by RT-PCR. An expected 285-bp product was seen in Case 2 (Lane 2) and tonsil tissue (Lane 3). No amplification band was observed in Case 1 (Lane 1) and no-template control (Lane NTC). (C) The fusion point of the RANBP2-ALK gene as indicated by cDNA sequencing was located between exon 18 of the RANBP2 gene and exon 20 of the ALK gene.
Discussion
IMT is regarded as a neoplasm of intermediate biologic potential. Overall, the recurrence rate varies by anatomical sites, from 2% for tumors confined to the lung to 25% for extrapulmonary lesions. Distant metastasis of IMT occurs in less than 5% of cases [12]. According to the literature and the 2 cases presented herein (Table 1), the local recurrence rate of IMT-RA is 88% (7/8) with a metastasis rate of 25% (2/8), both markedly higher than those of conventional IMTs. Furthermore, 2 patients (25%, 2/8) have died from the disease within 6 months after the tumor excision. These data suggest that IMT-RAs are more invasive clinically.
Table 1.
Clinical features of 11 cases of inflammatory myofibroblastic tumor with RANBP2-ALK gene fusion
| Case | Author | Age | Sex | Anatomic site(s) | Size (cm) | Treatment | Follow-up |
|---|---|---|---|---|---|---|---|
| 1 |
Ma [5] (2003) |
7 y |
Male |
Unspecified abdominal mass |
NA |
SE + CT |
Recurred 5 weeks later after first resection. A re-excision was performed. Five months later, the tumor recurred again and was re-excised. |
| 2 |
7 m |
Male |
Mesentery and omentum |
11 |
SE |
Recurred 8 months later after first resection. A re-excision was performed. |
|
| 3 |
Patel [7] (2007) |
2 y |
Male |
Retroperitoneal abdominal mass |
10 |
SE |
No evidence of recurrence with 3 years of follow-up. |
| 4 |
Chen [8] (2008) |
34 y |
Male |
Liver |
8 |
SE |
Recurred 5 months later after resection. Died of the disease approximately 2 weeks after discovery of the recurrence. |
| 5 |
Butrynski [9] (2010) |
44 y |
Male |
Omentum |
NA |
SE + CT + ALKi |
Hepatic, peripancreatic, and perirectal masses recurred 1 year later after resection. Subsequent exploratory laparotomy with maximal debulking was performed. |
| 6 |
Marino-Enriquez [10] (2011) |
41 y |
Male |
Omentum |
26 |
SE + CT + ALKi |
Multifocal local recurrence and liver metastases. Alive with no evidence of disease for 40 months. |
| 7 |
6 y |
Male |
Omentum and Mesentery |
14 |
SE |
NA |
|
| 8 |
39 y |
Male |
Mesentery of the small bowel |
14 |
SE |
NA |
|
| 9 |
Kozu [11] (2013) |
57 y |
Male |
Pluera or chest wall |
NA |
ALKi |
NA |
| 10 |
Present cases (2013) |
19 y |
Female |
Mesentery of the small bowel |
19 |
SE |
Recurred 9 weeks later after resection. Died of the disease 3 weeks later after discovery of the recurrence. |
| 11 | 39 y | Male | Mesentery of the colon | 15 | SE + CT | Recurred 4 months later after resection. Alive with disease for 12 months. |
SE surgical excision, CT chemotherapy, ALKi ALK inhibitor, NA data not available.
The aggressive behavior of IMT-RA might be associated with the following aspects: (1) The location and size of the tumor: Coffin et al. [13] analyzed the clinicopathologic features of IMTs with an invasive course, and found that IMTs that arise in the abdominopelvic site are more likely to recur (with a recurrence rate of 85%). In addition, recurrent and metastatic IMTs were commonly larger in size, with mean diameters of 8.7 and 11 cm, respectively. Of the 11 reported cases of IMT-RAs, 10 occurred in the abdominopelvic area, and only 1 presented in the pleural cavity (no follow-up data were provided in the case). The average size on discovery was 14.6 cm. Thus, the aggressiveness of IMT-RA is possibly related at least in part to its abdominopelvic origination and bigger tumor size. It is worth noting that IMT-RA seemingly prefers to afflict male patients, as the male to female ratio is 10:1.
(2) The histological features of the tumor: The morphology of IMT-RAs is generally uniform and characterized by loosely arrayed, ganglion-like or epithelioid neoplastic cells distributed in a widespread myxoid stroma. In earlier reports, IMTs with the aforementioned features were named round cell transformation [14]. It has been well demonstrated that IMTs with round cell transformation behave aggressively with rapid recurrence and/or death, compared to conventional IMTs in which spindled tumoral cells predominate [14]. Marino-Enriquez [10] proposed to designate this type of IMT as epithelioid inflammatory myofibroblastic sarcoma based on its histological and malignant biological characteristics. Therefore, the distinctive histology of IMT-RAs dominated by epithilioid neoplastic cells also intrinsically reflects the heightened invasiveness of the tumor. However, it should be particularly noted that not all IMTs with round cell morphology carry the genetic alteration of IMT-RA [8]. Therefore, it is inappropriate to diagnosis IMT-RA based solely on morphologic features.
(3) The genetic features of the tumor: The fusion point of IMT-RAs reported to date constantly presents between exon 18 of RANPB2 and exon 20 of ALK (Table 2). Sasaki [15] found that the RANPB2-ALK fusion gene could lead to interleukin (IL)-3 independent growth of Ba/F3 cells. Similarly, another study showed that bone marrow cells transfected with the RANBP2-ALK gene acquired an enhanced colony-forming potential and a decreased dependency on cytokine [16]. These studies suggested that the chimeric RANPB2-ALK gene could promote cellular proliferation, which might be a potential mechanism for the rapid regrowth and recurrence of IMT-RA. However, there is still a lack of direct experimental evidence linking the functions of this fusion gene with the increased aggressiveness of IMT-RA.
Table 2.
Genetic and immunohistochemical characteristics of 11 cases of inflammatory myofibroblastic tumor with RANBP2-ALK gene fusion
|
Case |
Genetic features |
Immunohistochemical features |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| RANBP2toALKfusion point | Detection techniques | ALK | Desmin | SMA | Caldesmin | CD30 | CK | EMA | S100 | |
| 1 |
Exon 18 to exon 20 |
RT-PCR, FISH |
NM+ |
/ |
/ |
/ |
/ |
/ |
/ |
/ |
| 2 |
Exon 18 to exon 20 |
RT-PCR, FISH |
NM+ |
/ |
/ |
/ |
/ |
/ |
/ |
/ |
| 3 |
Exon 19 to exon 20* |
RT-PCR, FISH |
CP+ |
+ |
+ |
- |
/ |
+ |
/ |
- |
| 4 |
Exon 18 to exon 20 |
RT-PCR |
NM+ |
- |
- |
/ |
- |
- |
/ |
- |
| 5 |
Exon 18 to exon 20 |
RT-PCR |
NM+ |
+ |
- |
/ |
/ |
- |
/ |
/ |
| 6 |
Exon 18 to exon 20 |
RT-PCR |
NM+ |
+ |
- |
- |
+ |
- |
- |
- |
| 7 |
Exon 18 to exon 20 |
RT-PCR |
NM+ |
+ |
/ |
- |
+ |
/ |
/ |
- |
| 8 |
Exon 18 to exon 20 |
RT-PCR |
NM+ |
+ |
+ |
- |
+ |
- |
/ |
- |
| 9 |
Unknown |
FISH |
CP+ |
+ |
- |
/ |
- |
+ |
/ |
- |
| 10 |
Unknown |
FISH |
NM+ |
+ |
+ |
- |
+ |
- |
- |
- |
| 11 |
Exon 18 to exon 20 |
RT-PCR, FISH |
NM+ |
+ |
+ |
- |
+ |
- |
- |
- |
| Total | / | / | 100% (11/11) | 89% (8/9) | 50% (4/8) | 0% (0/6) | 71% (5/7) | 25% (2/8) | 0% (0/3) | 0% (0/8) |
NM nuclear membrane, CP cytoplasm.
*Reference 7 stated that the fusion point was exon 19 of RANBP2 to exon 20 of ALK, while Figure 2 in the paper indicated that the actual fusion location was exon 18 of RANBP2 to exon 20 of ALK.
From a diagnostic viewpoint, IMT-RA needs to be differentiated from a large group of tumors that manifest epithelioid features. Immunohistochemistry is greatly helpful in this process. Nuclear membrane staining of ALK is a unique immunophenotype of IMT-RA, which is observed in 82% (9/11) of cases. Unexplainably, a cytoplasmic pattern is also detected in 18% (2/11) of cases. In addition, the neoplasms display varied expression of desmin (89%, 8/9), CD30 (71%, 5/7), SMA (50%, 4/8), and cytokeratin (25%, 2/8), while EMA, S100, CD117, Myf4, myogenin, caldesmin, and HMB45 expression is consistently negative. Therefore, IMT-RAs could be easily distinguished from poorly differentiated carcinoma, malignant melanoma, epithelioid gastrointestinal stromal tumor, epithelioid solitary fibrous tumor [17], myofibroma [18] and alveolar rhabdomyosarcoma. It is noteworthy that a few mesenchymal mimics listed below may also show cytoplasmic ALK staining [19] and thus should particularly be discriminated to avoid misdiagnosis. (1) Epithelioid leiomyosarcoma (ELS): Compared with IMT-RA, ELS commonly displays greater cellular atypia and pleomorphism, and higher cellular density. ELS generally lacks an extensive myxoid background and inflammatory infiltrates. Furthermore, calponin and caldesmin expression could be recognized in ELS but not in IMT-RA [20]. (2) Inflammatory malignant fibrous histiocytoma (IMFH): Clinically, IMFH is more prone to occur in elderly individuals, which is different from IMT-RA that typically develops in young adults (median age, 34 years). Histologically, while the inflammatory infiltrates may be striking in both types of the tumors, the neoplastic cells in IMFH tend to be more pleomorphic, in contrast with the relative homogeneity in cellular morphology of IMT-RA. The neoplastic cells in IMFH are frequently fusiform and closely packed, and commonly arranged in a storiform pattern. Immunostaining of CD68 favors the diagnosis of IMFH. (3) Epithelioid malignant peripheral nerve sheath tumor (E-MPNST): Most E-MPNSTs develop from the setting of neurofibromatosis. E-MPNST presents with more or fewer spindled tumor cells in the lesion, and the myxoid stroma is more often focal rather than abundant [21,22]. Moreover, desmin and CD30 expression have not been discovered in E-MPNSTs. All of these features differ from those of IMT-RA. (4) Anaplastic large cell lymphoma (ALCL): The distinction between IMT-RA and ALCL has been described in a previous report [10]. Desmin and nuclear membrane ALK staining are suggestive of IMT-RA, as neither of these staining patterns has been observed in ALCL.
Most of the reported IMT-RAs are treated by surgical resection combined with chemotherapy. However, this therapeutic regimen seems to not effectively control the rapid recurrence of IMT-RAs. Recently, ALK inhibitor (ALKi) has been applied in the therapy of 2 IMT-RA cases, and a sustained partial response has been observed in at least 1 case [9]. Of note, the sensitivity of ALKi was reduced during standing treatment because of a secondary ALK gene mutation [15]. Thus, the long-term results of ALKi therapy remain to be further evaluated.
Conclusions
In comparison with conventional IMTs, IMT-RAs show enhanced aggressive behaviors, which are possibly closely associated with their abdominopelvic origination, large tumor size, epithelioid tumoral morphology, and RANPB2-ALK gene rearrangement. An in-depth understanding on this entity is urgently needed to avoid misdiagnosis and provide effective treatment schedules.
Consent
Written informed consents were obtained from the patient of Case 2 and the mother of the patient of Case 1 for publication of this report and accompanying images. The copies of the written consents are available for review by the Editor-in Chief of this Journal.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JL performed the histological, immunohistochemical and RT-PCR evaluation, literature review and drafted the manuscript. W-HY participated in histological diagnosis and immunohistochemical evaluation. KT performed FISH analysis and revised the manuscript. GH designed the study and literature review, and drafted the manuscript. Y-HH contributed to the literature review. JKCC participated in histological diagnosis and revised the manuscript. All authors read and approved the final manuscript.
Contributor Information
Jian Li, Email: lijianhouma@yahoo.cn.
Wei-hua Yin, Email: weihuayin@sina.com.
Kengo Takeuchi, Email: kentakeuchi-tky@umin.net.
Hong Guan, Email: hongguansz@sina.cn.
Yu-hua Huang, Email: yuhuahuang@yahoo.cn.
John KC Chan, Email: jkcchan@ha.org.hk.
References
- Dehner LP. Inflammatory myofibroblastic tumor: the continued definition of one type of so-called inflammatory pseudotumor. Am J Surg Pathol. 2004;28:1652–1654. doi: 10.1097/00000478-200412000-00016. [DOI] [PubMed] [Google Scholar]
- Coffin CM, Patel A, Perkins S, Elenitoba-Johnson KS, Perlman E, Griffin CA. ALK1 and p80 expression and chromosomal rearrangements involving 2p23 in inflammatory myofibroblastic tumor. Mod Pathol. 2001;14:569–576. doi: 10.1038/modpathol.3880352. [DOI] [PubMed] [Google Scholar]
- Takeuchi K, Soda M, Togashi Y, Sugawara E, Hatano S, Asaka R, Okumura S, Nakagawa K, Mano H, Ishikawa Y. Pulmonary inflammatory myofibroblastic tumor expressing a novel fusion, PPFIBP1-ALK: reappraisal of anti-ALK immunohistochemistry as a tool for novel ALK fusion identification. Clin Cancer Res. 2011;17:3341–3348. doi: 10.1158/1078-0432.CCR-11-0063. [DOI] [PubMed] [Google Scholar]
- Bridge JA, Kanamori M, Ma Z, Pickering D, Hill DA, Lydiatt W, Lui MY, Colleoni GW, Antonescu CR, Ladanyi M, Morris SW. Fusion of the ALK gene to the clathrin heavy chain gene, CLTC, in inflammatory myofibroblastic tumor. Am J Pathol. 2001;159:411–415. doi: 10.1016/S0002-9440(10)61711-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, Hill DA, Collins MH, Morris SW, Sumegi J, Zhou M, Zuppan C, Bridge JA. Fusion of ALK to the Ran-binding protein 2 (RANBP2) gene in inflammatory myofibroblastic tumor. Genes Chromosomes Cancer. 2003;37:98–105. doi: 10.1002/gcc.10177. [DOI] [PubMed] [Google Scholar]
- Yokoyama N, Hayashi N, Seki T, Panté N, Ohba T, Nishii K, Kuma K, Hayashida T, Miyata T, Aebi U. A giant nucleopore protein that binds Ran/TC4. Nature. 1995;376:184–188. doi: 10.1038/376184a0. [DOI] [PubMed] [Google Scholar]
- Patel AS, Murphy KM, Hawkins AL, Cohen JS, Long PP, Perlman EJ, Griffin CA. RANBP2 and CLTC are involved in ALK rearrangements in inflammatory myofibroblastic tumors. Cancer Genet Cytogenet. 2007;176:107–114. doi: 10.1016/j.cancergencyto.2007.04.004. [DOI] [PubMed] [Google Scholar]
- Chen ST, Lee JC. An inflammatory myofibroblastic tumor in liver with ALK and RANBP2 gene rearrangement: combination of distinct morphologic, immunohistochemical, and genetic features. Hum Pathol. 2008;39:1854–1858. doi: 10.1016/j.humpath.2008.04.016. [DOI] [PubMed] [Google Scholar]
- Butrynski JE, D’Adamo DR, Hornick JL, Dal Cin P, Antonescu CR, Jhanwar SC, Ladanyi M, Capelletti M, Rodig SJ, Ramaiya N, Kwak EL, Clark JW, Wilner KD, Christensen JG, Jänne PA, Maki RG, Demetri GD, Shapiro GI. Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N Engl J Med. 2010;363:1727–1733. doi: 10.1056/NEJMoa1007056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariño-Enríquez A, Wang WL, Roy A, Lopez-Terrada D, Lazar AJ, Fletcher CD, Coffin CM, Hornick JL. Epithelioid inflammatory myofibroblastic sarcoma: An aggressive intra-abdominal variant of inflammatory myofibroblastic tumor with nuclear membrane or perinuclear ALK. Am J Surg Pathol. 2011;35:135–144. doi: 10.1097/PAS.0b013e318200cfd5. [DOI] [PubMed] [Google Scholar]
- Kozu Y, Isaka M, Ohde Y, Takeuchi K, Nakajima T. Epithelioid inflammatory myofibroblastic sarcoma arising in the pleural cavity. Gen Thorac Cardiovasc Surg. 2013;24: . doi: 10.1007/s11748-013-0204-x. [DOI] [PubMed] [Google Scholar]
- Gleason BC, Hornick JL. Inflammatory myofibroblastic tumours: where are we now? J Clin Pathol. 2008;61:428–437. doi: 10.1136/jcp.2007.049387. [DOI] [PubMed] [Google Scholar]
- Coffin CM, Hornick JL, Fletcher CD. Inflammatory myofibroblastic tumor: comparison of clinicopathologic, histologic, and immunohistochemical features including ALK expression in atypical and aggressive cases. Am J Surg Pathol. 2007;31:509–520. doi: 10.1097/01.pas.0000213393.57322.c7. [DOI] [PubMed] [Google Scholar]
- Cook JR, Dehner LP, Collins MH, Ma Z, Morris SW, Coffin CM, Hill DA. Anaplastic lymphoma kinase (ALK) expression in the inflammatory myofibroblastic tumor: a comparative immunohistochemical study. Am J Surg Pathol. 2001;25:1364–1371. doi: 10.1097/00000478-200111000-00003. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Okuda K, Zheng W, Butrynski J, Capelletti M, Wang L, Gray NS, Wilner K, Christensen JG, Demetri G, Shapiro GI, Rodig SJ, Eck MJ, Jänne PA. The neuroblastoma-associated F1174L ALK mutation causes resistance to an ALK kinase inhibitor in ALK-translocated cancers. Cancer Res. 2010;70:10038–10043. doi: 10.1158/0008-5472.CAN-10-2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röttgers S, Gombert M, Teigler-Schlegel A, Busch K, Gamerdinger U, Slany R, Harbott J, Borkhardt A. ALK fusion genes in children with atypical myeloproliferative leukemia. Leukemia. 2010;24:1197–1200. doi: 10.1038/leu.2010.18. [DOI] [PubMed] [Google Scholar]
- Martorell M, Pérez-Vallés A, Gozalbo F, Garcia-Garcia JA, Gutierrez J, Gaona J. Solitary fibrous tumor of the thigh with epithelioid features: a case report. Diagn Pathol. 2007;2:19. doi: 10.1186/1746-1596-2-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ, Lee SH, Youk EG, Lee S, Choi JH, Cho KJ. Solitary myofibroma of the sigmoid colon: case report and review of the literature. Diagn Pathol. 2013;8:90. doi: 10.1186/1746-1596-8-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cessna MH, Zhou H, Sanger WG, Perkins SL, Tripp S, Pickering D, Daines C, Coffin CM. Expression of ALK1 and p80 in inflammatory myofibroblastic tumor and its mesenchymal mimics: a study of 135 cases. Mod Pathol. 2002;15:931–938. doi: 10.1097/01.MP.0000026615.04130.1F. [DOI] [PubMed] [Google Scholar]
- Uncu H, Tüzüner A. Epithelioid leiomyosarcoma of the gastrocolic ligament. Acta Chir Belg. 2003;103:105–107. doi: 10.1080/00015458.2003.11679376. [DOI] [PubMed] [Google Scholar]
- Carter JM, O’Hara C, Dundas G, Gilchrist D, Collins MS, Eaton K, Judkins AR, Biegel JA, Folpe AL. Epithelioid malignant peripheral nerve sheath tumor arising in a schwannoma, in a patient with “neuroblastoma-like” schwannomatosis and a novel germline SMARCB1 mutation. Am J Surg Pathol. 2012;36:154–160. doi: 10.1097/PAS.0b013e3182380802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hruban RH, Shiu MH, Senie RT, Woodruff JM. Malignant peripheral nerve sheath tumors of the buttock and lower extremity. A study of 43 cases. Cancer. 1990;66:1253–1265. doi: 10.1002/1097-0142(19900915)66:6<1253::AID-CNCR2820660627>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]