

Abstract
Coding a wide range of light intensities in natural scenes poses a challenge for the retina: adaptation to bright light should not compromise sensitivity to dim light. Here we report a novel form of activity-dependent synaptic plasticity, specifically, a “weighted potentiation” that selectively increases output of Mb-type bipolar cells in the goldfish retina in response to weak inputs but leaves the input–output ratio for strong stimuli unaffected. In retinal slice preparation, strong depolarization of bipolar terminals significantly lowered the threshold for calcium spike initiation, which originated from a shift in activation of voltage-gated calcium currents (ICa) to more negative potentials. The process depended upon glutamate-evoked retrograde nitric oxide (NO) signaling as it was eliminated by pretreatment with an NO synthase blocker, TRIM. The NO-dependent ICa modulation was cGMP independent but could be blocked by N-ethylmaleimide (NEM), indicating that NO acted via an S-nitrosylation mechanism. Importantly, the NO action resulted in a weighted potentiation of Mb output in response to small (≤−30 mV) depolarizations. Coincidentally, light flashes with intensity ≥2.4 × 108 photons/cm2/s lowered the latency of scotopic (≤2.4 × 108 photons/cm2/s) light-evoked calcium spikes in Mb axon terminals in an NEM-sensitive manner, but light responses above cone threshold (≥3.5 × 109 photons/cm2/s) were unaltered. Under bright scotopic/mesopic conditions, this novel form of Mb output potentiation selectively amplifies dim retinal inputs at Mb → ganglion cell synapses. We propose that this process might counteract decreases in retinal sensitivity during light adaptation by preventing the loss of visual information carried by dim scotopic signals.
Introduction
The retina codes for contrast, color, and motion information of an image projected to the photoreceptor mosaic (Wässle, 2004) under light intensities changing across 10 orders of magnitude (Dowling, 1987; Sterling, 2003). To fit this wide range of inputs to the output limited by the maximal spiking rate of ganglion cells (GCs) (∼102), the retina adapts to the average light intensity (“light adaptation”) as well as to the range of intensities around the mean (“contrast adaptation”) by shifting the dynamic range of coding (Demb, 2002; Rieke and Rudd, 2009). Both light and contrast adaptation are accomplished by implementing various strategies at different stages of retinal processing and on multiple time scales. However, the fundamental challenge for all retinal adaptation processes is similar: prevent saturation of retinal output at high stimulus intensity/contrast while maintaining coding capability for weak inputs.
Contrast adaptation of GC responses is resolved using two opposing forms of plasticity in parallel: after high-contrast stimulation, some GC responses were depressed in response to consecutive low-contrast stimuli, whereas others were sensitized (Smirnakis et al., 1997; Kim and Rieke, 2001; Baccus and Meister, 2002; Kastner and Baccus, 2011). In addition to adaptation processes intrinsic to GCs (Kim and Rieke, 2001; Baccus and Meister, 2002), short-term synaptic plasticity mechanisms that affect bipolar cell (BC) output onto GCs also play a critical role in both adaptation and sensitization during contrast adaptation of GCs (Rieke, 2001; Kastner and Baccus, 2011; Nikolaev et al., 2013). To adapt to changing light intensity and sustain vision across all light conditions, the retina uses a highly sensitive rod pathway for night vision and a less sensitive cone pathway for daylight vision (Walraven et al., 1990). The light sensitivities of rods and cones overlap, forming a substantial mesopic range that covers behaviorally relevant environmental light intensities for many vertebrates (Wu, 1994; Krizaj, 2000). The dynamic interaction between the rod and cone pathways is influenced by neuromodulators. For example, during light adaptation, dopamine increases signal flow through cone circuits, diminishes that through rod circuits (Witkovsky, 2004), and reduces GC responses to prevent saturation (Hayashida et al., 2009), which ultimately reduces overall retinal sensitivity.
Here we report a novel form of activity-dependent synaptic plasticity in the retina, mediated by retrograde nitric oxide (NO) signaling. Mbs in the goldfish retina receive and process both rod and cone input (Wong et al., 2005; Joselevitch and Kamermans, 2009). The synaptic plasticity described here results in selective, weighted potentiation of Mb output in response to weak rod- but not cone-mediated inputs. We propose that this new form of NO-mediated, activity-dependent synaptic plasticity plays an important role under bright scotopic and mesopic conditions. Selective enhancement of weak, rod-mediated inputs at the Mb → GC synapses may counteract sensitivity loss during light adaptation, ultimately preventing the loss of visual information carried by low-intensity signals. Our results suggest that, similar to contrast adaptation (Kastner and Baccus, 2011), adaptation to increasing luminance also uses opposing forms of plasticity in parallel to extend the dynamic range of retinal signaling.
Materials and Methods
Retinal preparations
Retinal slices and isolated bipolar cells were prepared from retinal tissue dissected from adult 4- to 5-inch-long Goldfish (Carassius auratus) of either sex that had been dark-adapted for at least 1 h. Goldfish were kept on a 12 h light/dark schedule. All experimental procedures conformed to the guidelines of the Colorado State University Institutional Animal Care and Use Committee.
Retinal slice preparation.
Retinal slices (200–250 μm) were prepared as described previously (Palmer et al., 2003). In experiments requiring light stimulation, slice preparation and dissection procedures were performed under infrared illumination as previously described (Vigh et al., 2011). After the slicing procedure, slices in the recording chamber were moved to an upright microscope (Axioskop 2, Zeiss) and superfused at 2–5 ml/min with Ringer's solution containing the following (in mm): 100 NaCl, 2.5 KCl, 1.0 MgCl2, 2.5 CaCl2, 25 NaHCO3, 0–0.2 ascorbic acid, and 12 glucose or with bicarbonate buffered Ames' medium (US Biological) supplemented with 1.15 mm CaCl2. The pH was set to 7.45 with NaOH. The osmolarity was 260 ± 2 mOsmol for both Ringer's solution and Ames' medium, and both were gassed continuously with 95% O2/5% CO2. To block inhibitory feedback to Mb terminals, picrotoxin (PTX, 100 μm) was present (Vigh et al., 2005, 2011) in all retinal slice experiments unless otherwise noted. Slices were viewed using a 40× water-immersion objective and infrared differential contrast and through an infrared CCD camera with 2.5 premagnification (XC-75, Sony). The camera was connected to a Camera Controller C2741–62 (Hamamatsu), which directed the output to a 19 inch monitor (Westinghouse). Identification of Mb terminals within the inner plexiform layer was based on the large, bulbous terminal morphology and Cm increase after depolarization (Palmer et al., 2003).
Dissociated Mb preparation.
Solitary bipolar cells were isolated by conventional methods (Tachibana and Kaneko, 1987). Briefly, retinal pieces were dissociated by mechanical trituration with a Pasteur pipette after papain digestion (10 U/ml, Worthington Biochemical). Dissociated cells were plated on clean glass bottom dishes (MatTek) coated with poly-d-lysine, and stored at room temperature in low Ca2+ (0.2 mm CaCl2) Ringer's solution containing the following (in mm): 117 NaCl, 2.6 KCl, 1.0 MgCl2, 0.2 CaCl2, 10 glucose, 5 HEPES, and supplemented with 0.5 mg/ml BSA. The pH 7.45 was set with NaOH; osmolarity was 260 ± 2 mOsmol. Plated cells were viewed using DIC optics through a 40× air objective coupled to a 2.5× Optovar cube (Axio Observer Z1; Zeiss) and AxioCam HRm CCD camera (Zeiss) and were superfused at 2–5 ml/min with Ringer's solution (see above).
Electrophysiology and light stimulation
Whole-cell voltage- and current-clamp recordings were made directly from Mb terminals in both slice and dissociated preparations using a EPC-10 USB patch-clamp amplifier and Patchmaster software (version 2.3; both HEKA) at room temperature during daytime (10 A.M. to 6 P.M.). Membrane current and voltage data were filtered at 3 kHz, sampling rates were set at 5 kHz for voltage ramp protocols, 20 kHz for voltage step protocols, and 25 kHz for recording spiking membrane potential responses in either current-clamp ramp protocol or using light stimulation. The current-clamp ramp protocol used in these studies stretched from −20 pA to 65 pA over 2 s and was applied relative to the holding current that kept the resting membrane potential at −60 mV. The holding current was determined in voltage-clamp mode when establishing whole-cell mode (“patch break”) and was maintained when switching to current-clamp mode using the “Gentle CC-switch” option of Patchmaster. The threshold of current ramp-evoked spikes was determined as follows: The leak was estimated based on the slope of the membrane voltage trace between −100 mV and −60 mV. The leak-subtracted data were normalized to the first (initial) spike amplitude. A current ramp-evoked membrane potential change was considered a spike if its peak voltage exceeded 2 SD of the baseline noise between −100 mV and −60 mV. The threshold was defined as 5% of the peak membrane voltage for the first spike. Current–voltage relationships were determined by whole-cell voltage-clamp ramps and conventional stepwise reconstructions. The whole-cell voltage-clamp ramp protocol used in these studies was a linear 2 s ramp from −100 to 50 mV. The stepwise I-V protocol consisted of 3 ms steps to potentials (from −80 mV to 60 mV, 10 mV increments) with 5 s between each step. For real-time measurement of membrane capacitance, the Sine+DC technique was used (Gillis, 2000). The capacitive current was measured by the lock-in amplifier after a 1 kHz sinusoidal voltage command (20 mV peak to peak) applied to the holding potential of −60 mV. Patch pipettes of 8–12 mΩ were pulled using a horizontal puller (model p-97, Sutter) from 1.5-mm-diameter, thick-walled borosilicate glass (World Precision Instruments) and were coated with dental wax (Cavex) to reduce stray pipette capacitance. For isolation of ICa in whole-cell voltage-clamp experiments, a Cs-gluconate-based internal solution was used (in mm) as follows: 85 Cs-gluconate, 10 phosphocreatine-di(tris) salt, 10 l-ascorbic acid, 10 TEA-Cl, 10 methylamine HCl, 2 EGTA, 3 Mg-ATP, 0.5 Na-GTP, pH 7.2 (adjusted with CsOH), and osmolarity of 260 ± 2 mOsmol. For current-clamp (including light-evoked response) recordings, a K-gluconate based internal was used (in mm) as follows: 106 K-gluconate, 10 phosphocreatine-di(tris), 3 l-ascorbic acid, 10 KCl, 3 Mg-ATP, 2 EGTA, 0.5 Na-GTP, pH 7.2 (adjusted with NaOH), and osmolarity of 260 ± 2 mOsmol. During experiments using BAPTA, Exo 1, KT5823, or N-ethylmaleimide (NEM) in the pipette solution, at least 1 min was allowed for intracellular perfusion with the pipette solution before any recording and at least 45 s with standard pipette solution. To keep the osmolarity constant, when 10 mm BAPTA was included in the pipette solution, the amount of Cs-gluconate was reduced accordingly.
In voltage-clamp experiments voltage commands were not corrected for liquid junction potential. Series resistance (Rs) was tightly monitored during recording and was not compensated electronically. Data obtained from Mb terminals were excluded if the uncompensated Rs exceeded 30 mΩ, as in such recordings we found the slope of ICa activation curve to exceed 1/Rs indicating the lack of voltage-clamp (Marty and Neher, 1995). Rs change by 20% any time during the recording also resulted in exclusion of the recording from further evaluation. Recordings with greater than ±1 mV drift of electrode potential, measured at the beginning and conclusion of the recording, were also excluded. Light stimulation was performed as previously described (Vigh et al., 2011). Briefly, 500-ms-long green (λ = 505 nm) or red (λ = 660 nm) LEDs (American Bright Optoelectronics) were used for full-field illumination of retinal slices. The LED voltage was controlled by the EPC-10 through D/A output. The light intensity was calibrated with an optical meter (model 1918-C, sensor 918D-SL-OD3; Newport).
Drugs and chemicals
Kainic acid (KA), NBQX, d-(−)-2-amino-5-phosphonopentanoic acid (d-AP5), 1-(2-trifluromethylphenyl)imidazole (TRIM), 8-bromoguanosine cyclic 3′,5′-monophosphate sodium salt (8-Br-cGMP), Exo 1, KT5823, and picrotoxin were obtained from Tocris Bioscience. All other chemicals and salts were obtained from Sigma.
Diethyltriamine-NO/NO (DETA/NO) was synthesized and validated as an NO donor as previously described (Maragos et al., 1991; Hrabie et al., 1993; Damodaran et al., 2012). Briefly, 500 mg of diethyltriamine was added to a thick-walled glass reaction vessel equipped with a stir bar. The vessel contained 40 ml of dehydrated acetonitrile dried over 40 nm molecular sieves. The reaction vessel was capped with a gas tight lid and was attached to a custom-made NO reactor. Oxygen was flushed from the reaction vessel using argon purges. NO was introduced to the vessel and kept at a pressure of 80 psi for 24 h under stirring conditions. The resulting white DETA/NO precipitate was collected by filtration, washed with acetonitrile, and dried under vacuum. The DETA/NO was then validated by a characteristic UV absorbance feature at 252 nm in 0.01 m NaOH with a molar extinction coefficient of 7600 m−1 cm−1. Direct and real-time NO measurements were performed using GE Analytical Nitric Oxide Analyzer equipped with a custom sample cell. A 3 ml aliquot of the media containing DETA/NO was added to the sample cell. The temperature of the sample cell was maintained at 21°C. Measurements were collected at 5 s intervals at the following instrument operating parameters: cell pressure (1.37 kPa), supply pressure (38.6 kPa), and cooler temperature (−12°C). The DETA solution was prepared 20 min before evaluation. Upon injection into the NOA sample cell, NO release with a steady rate was observed for the duration of the measurement. An average NO release rate of 2.00 ± 0.19 nm s−1 was measured (n = 4). This value corresponds to an average instantaneous concentration of NO of 9.99 ± 0.93 nm, which matches retinal NO concentrations measured extracellularly 10–20 μm away from relatively strong sources of NO production (Eldred and Blute, 2005). In all of our experiments, the DETA/NO solution (1 mm) was made fresh every day at least 1 h before the experiment began and used for a maximum of 12 h.
Data analysis
All data were analyzed off-line using IgorPro software (version 5.03; Wavemetrics). Voltage-clamp ramp and stepwise I-V curves were leak-subtracted and normalized to the peak to analyze the Ca2+ or K+ currents. Estimation of the leak current was accomplished using a procedure similar to that reported by Hirasawa and Kaneko (2003). The leak subtraction procedure consisted of extrapolating the slope of the line between −100 mV and −60 mV. The estimated leak current was subtracted from the raw value providing the “pure” ICa or IK. Normalized, leak-subtracted ramp-evoked IK I-V curves were fit using the following Boltzmann equation:
where V1/2 is the half-activation potential and S is the slope of the voltage dependency (Griguer and Fuchs, 1996). For the stepwise ICa I-V curve, the first 3 points (−80, −70, −60 mV) were used to estimate the leak current for extrapolation. Leak-subtracted stepwise and ramp-evoked ICa I-V curves were fit using the following modified Boltzmann equation:
where Vrev is the reversal potential, Gmax is the maximum Ca2+ conductance, V1/2 is the half-activation potential, and kG is the slope factor (Leuranguer et al., 2003). Curve fits were done using SigmaPlot (version 11; Systat Software). For ramp evoked ICa I-V data, analysis of I-V kinetics was also determined directly by using MiniAnalysis software (version 6.0.3; Synaptosoft). “Activation” (V(5%)) was defined as the voltage at which the resulting current was 5% of the peak current, “half-activation” (V(50%)) was defined as the voltage at which the resulting current was halfway between “activation” and the peak current, and “maximum” (V(Max)) was defined as the voltage that resulted in the peak current.
Threshold of calcium spikes, in current-clamp ramp protocols, was determined using baseline-subtracted and normalized recordings. A baseline subtraction procedure similar to that used for voltage clamp was implemented for the current-clamp recordings. Then, the data were normalized to first (initial) spike amplitude. For light-evoked spikes, the latency to first spike was determined by the time point corresponding to the peak of the first evoked spike relative to the onset of the light stimulus. Where appropriate, depolarization triggered transmitter release was calculated based on the increase in membrane capacitance (Cm jump or ΔCm) by the equation ΔCm= Cm Response − Cm Baseline (Vigh and von Gersdorff, 2005). Statistics were calculated using SigmaPlot (version 11; Systat Software), GraphPad Prism (version 6.01), IgorPro software (version 5.03; Wavemetrics), and Excel (Microsoft). Paired or unpaired Student's t tests were used for comparisons between groups; data are presented as mean ± SEM, with p < 0.05 considered a significant difference. For I-V curves and current-clamp ramps, paired Student's t tests were performed on parameters obtained from normalized, leak-subtracted pairs of traces unless otherwise noted. For light-evoked spiking responses, paired Student's t tests were performed on latencies determined from original recordings of membrane potentials. One-way ANOVA with Tukey's multiple-comparison test was used for analyzing light-evoked spiking response latencies obtained after sensitizing light stimulations with different intensities.
Results
Strong depolarization shifted calcium spike threshold in Mb-type bipolar cells
The classical view by which visual signals are coded by slow, graded potentials in BCs (Werblin and Dowling, 1969) has been challenged by observations made in many species: some BCs signal with light-evoked sodium spike in ground squirrel (Saszik and DeVries, 2012) and with calcium (Ca2+) spikes in goldfish (Saito et al., 1979), zebrafish (Dreosti et al., 2011; Baden et al., 2011), and mouse (Baden et al., 2013a). However, the role of Ca2+spike-coded, digital signaling by BCs in visual information processing is not fully understood (for review, see Baden et al., 2013b). Voltage-gated Ca2+ channels, expressed at the axon terminal region, play a critical role in the spiking light responses of Mb-type BCs in the goldfish retina (Zenisek and Matthews, 1998; Protti et al., 2000); therefore, Ca2+ spikes have been proposed to play a role in transmitter release (Zenisek and Matthews, 1998). We made whole-cell current-clamp recordings directly from the large axon terminals of intact Mbs in goldfish retinal slice preparation. With a K+-based internal solution in the recording pipette, a current-ramp protocol from −20 pA to 65 pA over 2 s induced membrane potential spikes in Mbs (Fig. 1Ai,Aii). The depolarizing current threshold of calcium spike initiation was determined using baseline-subtracted and normalized recordings (for details, see Materials and Methods) from which the current value, in turn, was used to determine the membrane potential threshold on the original traces. We found that, in intact Mbs, the depolarizing current needed to reach the spike threshold was between 25.5 pA and 59.3 pA, averaging 46.76 ± 5.76 pA, giving the average membrane potential threshold for Ca2+ spikes a value of −38.66 ± 5.13 mV (n = 5) at the axon terminal. These values fell into the range of Ca2+ spike threshold for BCs in the zebrafish retina (Baden et al., 2011). Interestingly, when we applied a second current-clamp ramp at least 2.5 min after the completion of the first, we found that less depolarizing current (between 13.1 pA and 51.7 pA, averaging 40.24 ± 6.91 pA) was necessary to induce a Ca2+ spike (Fig. 1Aii). In other words, the membrane potential threshold for Ca2+ spike initiation was lower during the second current-ramp (−42.95 ± 5.92 mV) than that during the first (control). Concurrently, the decrease in membrane potential threshold (−4.30 ± 1.46 mV) was significant (p = 0.02, paired Student's t test). Importantly, the holding current (Ihold) needed to keep Mbs at −60 mV did not change during the recordings (Ihold first ramp: −99.25 ± 31.16 pA vs Ihold second ramp: −99.75 ± 32.98 pA, ΔIhold: −0.5 ± 1.81 pA), indicating that the input resistance was unaltered between consecutive ramps.
Figure 1.
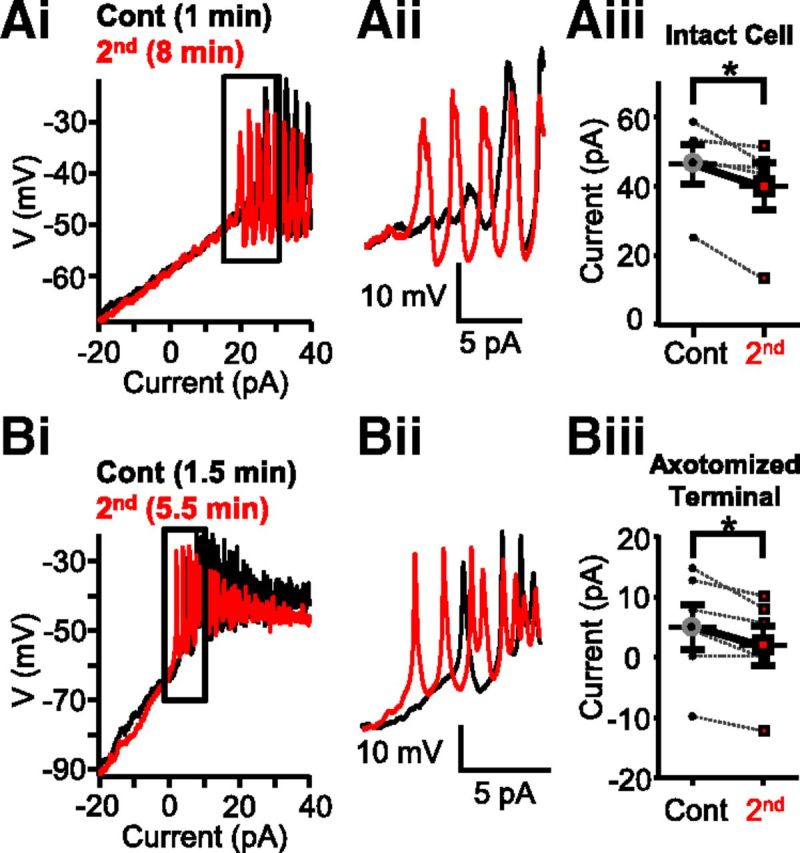
Strong depolarization shifted calcium spike threshold in Mb-type bipolar cells. Ai, Consecutive current ramp recordings made directly from the large axon terminals of intact Mbs in goldfish retinal slice preparation. Less depolarizing current was needed to evoke spikes when a second current ramp was applied 8 min (red) after the first (black). Aii, Enlargement of Ai focusing on the region of spike initiation. The initiation of spikes is shifted leftward for the 8 min trace. Aiii, Summary showing less current required for the initiation of Ca2+ spikes in terminals of whole Mbs in response to current ramp when preceded by strong depolarization. Data are taken from leak-subtracted and normalized traces. *p = 0.03 (paired Student's t test). n = 5. Bi, Current ramp evoked Ca2+ spikes recorded from an axotomized Mb terminal in slice preparation. The second current ramp (red represents 5.5 min after break-in) triggered spikes with less depolarizing current than the first current ramp (black represents 1.5 min after break-in). Bii, Enlargement of the regions of spike initiation from Bi. There is a leftward shift in the red trace. Biii, Summary showing that less current is required for the initiation of Ca2+ spikes in axotomized Mb terminals in response to current ramp when preceded by strong depolarization. Data are taken from leak-subtracted and normalized traces. *p = 0.02 (paired Student's t test). n = 5. Data are mean ± SEM.
Next, we tested whether the shift in Ca2+ spike threshold was related to the perfusion, over time, of intact cells with complex morphology. In the next set of experiments, we targeted terminals of Mbs whose axon was severed. In concert with their simple morphology, axotomized Mb terminals exhibit a single-exponential membrane time constant and have high input resistance (i.e., “less leaky”) in the absence of dendritic and somatic inputs (Palmer et al., 2003). The data included for processing were obtained from axotomized Mb terminals with Ihold recorded at patch-break between −3 pA and −38.5 pA, averaging −24.35 ± 6.16 pA (n = 5). Consistent with the high input resistance, the depolarizing current threshold during the first ramp for Ca2+ spike initiation was much lower (8.06 ± 2.65 pA) than that for intact Mbs. Importantly, during a second ramp, which was delivered with at least 2.5 min delay, Ca2+ spikes were triggered earlier in axotomized Mb terminals (Fig. 1Bi,Bii) in a similar fashion to that observed in intact Mbs. In other words, a significant reduction in depolarizing current threshold for initiation of Ca2+ spikes was seen (4.84 ± 2.07 pA, p = 0.02, paired Student's t test) (Fig. 1Biii) without a change in Ihold (23.7 ± 6.03 pA) compared with that recorded at patch break (ΔIhold: 0.60 ± 0.15 pA). Converting the depolarizing current threshold values into membrane potentials, the data showed that, in axotomized Mb terminals, the initial Ca2+ spike threshold of −44.81 ± 2.37 mV measured during the first ramp dropped to −50.84 ± 3.33 mV during the second ramp, and this reduction was statistically significant (p = 0.004, paired Student's t test). The spike threshold values obtained in axotomized Mb terminals were lower than those obtained from intact Mbs for both the first and second ramps (−44.81 ± 2.37 mV vs −38.66 ± 5.13 mV, and −50.84 ± 3.33 mV vs −42.95 ± 5.92 mV, respectively) demonstrating that depolarizing current injections triggered Ca2+ spikes more efficiently in axotomized Mb terminals with more compact structure than in intact cells possessing complex morphology. However, when the corresponding spike thresholds were compared (i.e., spike threshold during first ramp in intact cells vs spike threshold during first ramp in axotomized terminals and thresholds during the second ramps in a similar manner), we found no statistical difference between them (p = 0.3 and p = 0.2 for first and second ramp thresholds, respectively, unpaired Student's t test). These results also indicated that the observed reduction of Ca2+ spike threshold was independent of any process in the somatodendritic compartment of Mbs and/or the outer retinal circuitry.
Strong depolarization induced modulation of voltage-gated calcium (ICa) but not voltage-gated potassium (IK) currents in Mb terminals
In the next set of experiments, we tested whether the Ca2+ spike threshold shift we described above might have been caused by changes in the membrane currents mediating Ca2+ spikes. The ionic currents responsible for the spiking response of Mb terminals consist of rapidly activating (Mennerick and Matthews, 1998) voltage-gated influx of Ca2+ (ICa) through l-type voltage-gated Ca2+ channels (Heidelberger and Matthews, 1992; von Gersdorff and Matthews, 1996; Tachibana, 1999) that appear to be CaV1.3 type (Logiudice et al., 2006) and subsequent efflux of K+, made up by voltage-gated K+ (IK) and big conductance (BK) Ca2+-dependent K+ (IK(Ca)) currents (Kaneko and Tachibana, 1985; Sakaba et al., 1997; Palmer, 2006).
To study which one of these ionic currents might be responsible for the shift in spike initiation, we performed whole-cell voltage-clamp recordings on axotomized Mb terminals using pairs of consecutive voltage-clamp ramp protocols (from −100 mV to +50 mV, in 2 s) at least 3 min apart. First, we isolated IK using a K+-based internal solution (see Materials and Methods) and by complete blockade of ICa with 100 μm CdCl2 in the external solution (Fig. 2Ai). Blockers of ICa have been shown to eliminate IK(Ca) in Mbs as well (Kaneko and Tachibana, 1985; Palmer, 2006). Figure 2Ai shows a representative recording of IK traces evoked by consecutive voltage ramps, applied 3 min apart. Holding potentials <−60 mV were excluded from the figure for expansion of the physiologically relevant membrane potential region, although the entire current trace was included in the analysis. Analysis was performed on leak-subtracted IK recordings fit with a Boltzmann function (see Materials and Methods). Once fit, kinetics of the control ramp I-V and second ramp I-V were reflected by the slope factor (S) of the IK activation and the membrane potential resulting in 50% activation of IK (V(1/2)) (Griguer and Fuchs, 1996). No significant difference was found in the slope factors of control ramp I-Vs compared with those of the second ramp I-Vs (0.66 ± 0.08 mV vs 0.63 ± 0.09 mV, respectively, Fig. 2Aii; paired Student's t test, p = 0.4, n = 5) or between V(1/2) values (control: −11.42 ± 1.13 mV vs second ramp I-V: −11.01 ± 1.66 mV; Fig. 2Aiii; paired Student's t test, p = 0.7, n = 5).
Figure 2.
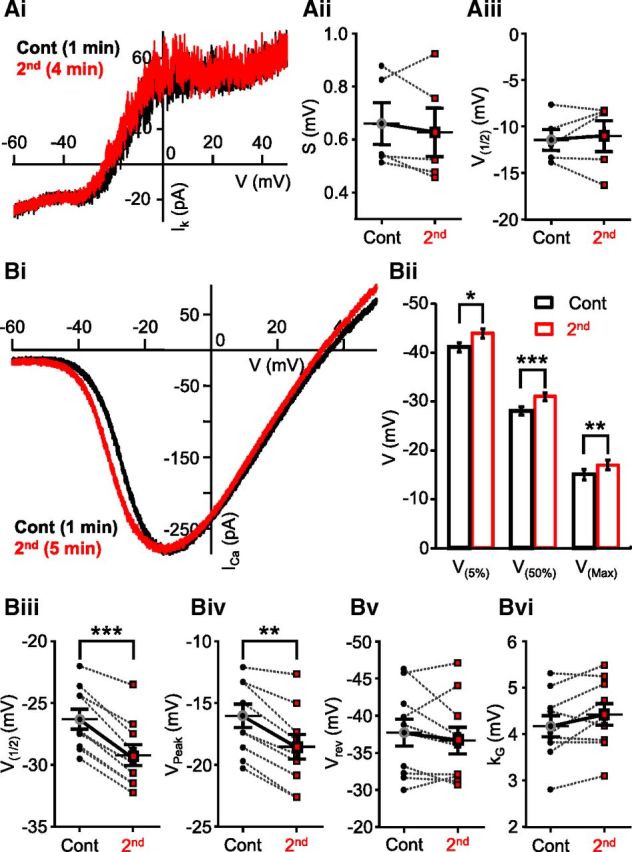
Strong depolarization induced modulation of ICa activation but not IK kinetics. Ai, Potassium current (IK) in axotomized Mb terminal in response evoked by a voltage ramp from −100 mV to 50 mV for 2 s. Points below −60 mV have been removed for illustration purposes. Black trace represents 1 min after break-in; red trace represents 4 min after break-in. IK was isolated by blocking ICa with 100 μm CdCl2. The second trace (red) coincides with the control (black). Aii, No significant differences were observed between the slope factor (S) of control and second traces. Black circles represent control; red squares represent second. n = 5. Aiii, The membrane potential at 50% of the maximal current (V(50%)) did not differ in the first and second recordings. Black circles represent control; red squares represent second. n = 6. Bi, Consecutive ICa I-Vs generated from an axotomized terminal in slice preparation, in response to a 2 s voltage ramp. There is a shift in the trace recorded at 5 min after break-in (red) compared with the control trace recorded 1.5 min after break-in (black). Bii, Summary graph showing ICa activation (V(5%)), half-activation (V(50%)), and peak (V(Max)). Black represents control; red represents second. *p = 0.00003 (paired Student's t test). ***p = 0.000001 (paired Student's t test). **p = 0.00001 (paired Student's t test). n = 10. Biii, Summary graph showing half-activation (V1/2) parameter from Boltzmann fits is significantly less for the second voltage-ramps compared with control. Black circles represent control; red squares represent second. **p = 0.000002 (paired Student's t test). n = 10. Biv, Summary graph showing significant shift in peak parameter obtained from Boltzmann fits. Black circles represent control; red squares represent second. **p = 0.00004 (paired Student's t test). n = 10. Bv, Summary graph of pairwise comparisons displaying no difference in the reversal potential (Vrev) parameter obtained from Boltzmann fits. Black circles represent control; red squares represent second. p = 0.31 (paired Student's t test). n = 10. Bvi, Summary graph showing no significant difference in the slope factor (kG) parameter obtained from Boltzmann fits for control and second voltage ramps. Black circles represent control; red squares represent second. p = 0.052 (paired Student's t test). n = 10. Data are mean ± SEM.
ICa, in axotomized Mb terminals, was evoked by applying the same linear ramp protocol used for triggering IK in voltage-clamp mode, but in the presence of Cs+- based internal solution containing TEA-Cl to block IK and IK(Ca) (Kaneko and Tachibana, 1985). When kinetics of ICa I-V traces recorded under these conditions (Fig. 2Bi) were evaluated by obtaining measurements of the membrane potential relating to 5%, 50%, and peak ICa (V(5%), V(50%), and V(Max), respectively; see Materials and methods), we found that, on average, V(5%) was −40.5 ± 0.86 mV, V(50%) was −27.6 ± 0.80 mV, and V(Max) was −14.7 ± 1.00 mV for the ICa I-V curves evoked by the first (control) voltage ramp. These values, obtained in slice preparation, were in perfect agreement with earlier reports studying parameters of ICa in dissociated, solitary Mbs (Kaneko and Tachibana, 1985; Heidelberger and Matthews, 1992). However, when we applied an identical second voltage-clamp ramp stimulus with at least 2.5 min delay, a portion of the resulted ICa did not overlap with that evoked by the first ramp protocol (Fig. 2Bi): ICa triggered by the second ramp activated at more negative membrane potentials. On average, for the second ramp, evoked ICa I-V V(5%) was −43.5 ± 0.79 mV, V(50%) was −30.35 ± 0.75 mV and V(Max) was −17.2 ± 0.95 mV (Fig. 2Bii). The difference between the first and second ramp I-V parameters was −3.0 ± 0.39 mV for V(5%), −2.75 ± 0.25 mV for V(50%), and −2.5 ± 0.31 mV for V(Max), all proven to be statistically significant (V(5%): p = 0.00003; V(50%): p = 0.000001; V(Max): p = 0.00001; paired Student's t test, n = 10) (Fig. 2Bii). It is important to note that, once shifted, we found that (1) subsequent ramp I-Vs were insufficient to shift ICa activation kinetics further to the left and (2) shifted ICa I-Vs did not return to their initial control values within the timeframe of a recording (up to 20 min), set by >10% rundown of peak ICa (data not shown).
ICa I-V traces evoked by consecutive depolarizing voltage ramps were also analyzed by comparing the parameters of their modified Boltzmann function fits (Leuranguer et al., 2003). We found a difference in half-activation (V(1/2)first: −26.34 ± 0.80 mV vs V(1/2)second: −29.24 ± 0.84 mV; Figure 2Biii) and in the peak ICa (VPeak1st: −16.03 ± 0.95 mV vs VPeak2nd: −18.55 ± 1.0 mV; Fig. 2Biv); the differences in both parameters were statistically significant (V(1/2): p = 0.000002; VPeak: p = 0.00004; paired Student's t test, n = 10). These results were entirely consistent with the results of analysis based on I-V characteristics obtained directly from the I-V curves. Although the half-activation (V(50%) vs V(1/2)) and peak ICa membrane potential (V(Max) vs VPeak) values for the same ICa I-V curves obtained by the two methods were not identical, the differences between the corresponding values were not statistically significant (V(50%) vs V(1/2) first: p = 0.14, second: p = 0.16; V(Max) vs VPeak first: p = 0.35, second: p = 0.34; paired Student's t test, n = 10). There was no significant difference in the reversal potential (Vrev) of ICa between the first (control) and second ramp I-Vs (Vrev1st: 37.70 ± 1.84 mV vs Vrev2nd: 36.65 ± 1.78 mV; p = 0.31; paired Student's t test, n = 10; Fig. 2Bv), which supported the view that the leftward shift in ICa activation did not originate from an artifact caused by a drift in electrode potential over the course of our experiments. Although the average slope factor (kG) slightly increased between the corresponding first (control) and second ICa I-Vs (kG1st: 4.18 ± 0.23 mV vs kG2nd: 4.44 ± 0.23 mV), this increase was not statistically significant (p = 0.052, paired Student's t test, n = 10). This was consistent with the fact that the slope increased in only 6 of 10 recordings, whereas in the rest of the cases it decreased (4 of 10) or remained the same (1 of 10; Fig. 2Bvi). These results suggested that a change in voltage dependence of Ca2+ channel gating charges (Hille, 2001) was not a critical mediator of the leftward shift observed in ICa activation.
The threshold values for Ca2+ spikes in axotomized Mb terminals measured with current ramps and ICa activation (V(5%)) measured with voltage-clamp ramps were in great agreement, suggesting that the leftward shift in ICa activation threshold was responsible for the reduction in Ca2+ spike threshold.
Although it was not tested, we considered direct contribution of (BK) IK(Ca) to the shift of Ca2+ spike threshold to be highly unlikely: to open (BK) IK(Ca) channels require large membrane depolarization (Vergara et al., 1998; Sah and Davies, 2000) and 10–20 μm [Ca2+]i in Mb terminals (Sakaba et al., 1997). Thus, (BK) IK(Ca) channels are not active at membrane potentials below ICa activation.
The depolarization induced shift in ICa activation at Mb terminals was calcium-dependent and required glutamate-mediated synaptic signaling
Subjecting Mb terminals to 2-s-long depolarizing ramp protocols in retinal slice preparation changed the activation kinetics of ICa. To explore whether this change was related to the massive influx of Ca2+ triggered by these protocols, we generated consecutive I-V curves for ICa at least 2.5 min apart in a stepwise manner using 3-ms-long square pulse voltage steps between −80 and 60 mV (see Materials and Methods). This stepwise protocol dramatically reduced the total Ca2+ influx at the Mb terminal over the course of experiment: between −80 and 40 mV, the cumulative step induced Ca2+ charge transfer (Q) was only ∼0.02% of that triggered by a 2 s ramp (data not shown). In addition, the delay between consecutive short depolarizing steps presumably allowed mobile and fixed Ca2+ buffering systems of the Mb terminals (Burrone et al., 2002) to control the spatial and temporal parameters of intracellular Ca2+ microdomains (Neher, 1998), formed during each short depolarizing step, at higher efficacy than during a long ramp depolarization. It is important to emphasize that, in our whole-cell recordings, the standard internal solutions contained 2 mm EGTA (see Materials and Methods), which slightly overestimated the Ca2+ buffering capacity of mobile endogenous buffers of Mb terminals (Burrone et al., 2002).
Analysis of parameters obtained from a modified Boltzmann function fit of the data points was used to determine potential differences between the characteristics of consecutive stepwise I-V curves (Fig. 3A). On average, the slope factor (kG) was 5.27 ± 0.29 mV for first (control) and 5.27 ± 0.31 mV for the second stepwise I-V, V(1/2) was −27.80 ± 1.00 mV for first (control) and −28.36 ± 0.95 mV for the second stepwise I-V, and VPeak was −12.27 ± 0.93 mV for control and −12.87 ± 1.11 mV for the second stepwise I-V. No significant difference between the parameters of control and second stepwise I-V was found (kG: p = 0.96; V(1/2): p = 0.27 and VPeak: p = 0.11, paired Student's t test, n = 13). The lack of change in ICa activation kinetics under these conditions indicated that the negative shift in ICa activation observed in Mbs in experiments using consecutive depolarizing linear ramp protocols was dependent upon (large) Ca2+ influx-mediated intracellular processes, specifically, those processes that could be triggered by ramp protocols but not by short depolarizing steps.
Figure 3.
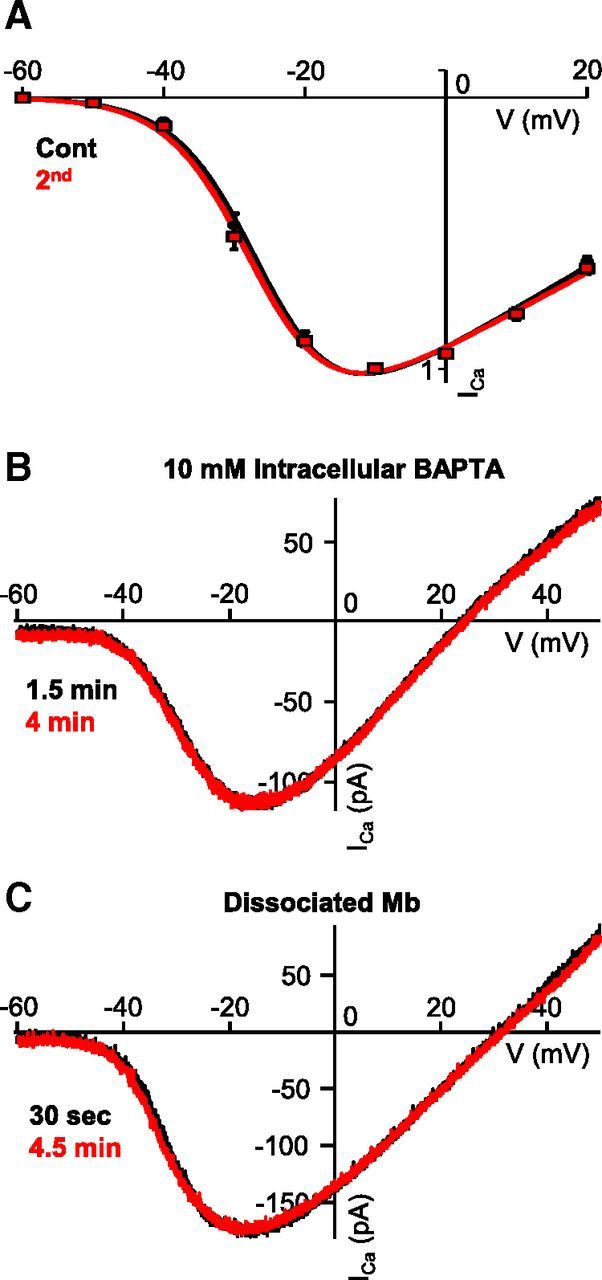
Modulation of ICa at the Mb terminal is Ca2+-dependent and required synaptic signaling. A, Average (leak-subtracted and normalized) stepwise I-V curve from axotomized terminals in slice preparation (n = 13 for steps to ≤−10 mV, n = 9 for steps to 0 mV and 20 mV). Consecutive I-V curves were generated 3 min apart by using 3 ms voltage steps. Data were fitted with a modified Boltzmann equation, smooth line (black represents first, control I-V; red represents second I-V). Data are mean ± SEM. B, Chelation of ramp-evoked Ca2+ influx using BAPTA (10 mm) inhibited strong depolarization-induced modulation of ICa kinetics of axotomized terminals in retinal slice preparation. C, Consecutive ICa I-Vs recorded from the axon terminal of an enzymatically dissociated, solitary Mb. The voltage ramp protocol was the same used in slice preparation. No shift was observed between the control (black) and the second (red) ICa I-Vs.
Supporting this notion, when the pipette solution was supplemented with 10 mm BAPTA in place of the regularly used 2 mm EGTA, ICa I-Vs generated by consecutive voltage-clamp ramp protocols (−100 mV to 50 mV, over 2 s, at least 2.5 min apart) overlapped (Fig. 3B). On average, V(5%) was −39.0 ± 1.75 mV for control and −39.30 ± 1.95 mV for the second ramp I-V, V(50%) was −23.95 ± 1.09 mV for control and −23.71 ± 2.11 mV for the second ramp I-V, and V(Max) was −12.59 ± 1.17 mV for control and −12.18 ± 2.22 mV for the second ramp I-V. Statistical analysis revealed that there was no difference between control and second ramp I-V for V(5%), V(50%), and V(Max) (p = 0.8, p = 0.9, and p = 0.8, respectively, paired Student's t test; n = 5). Together, these results suggested that modulation of ICa kinetics at the Mb axon terminal required a strong depolarization, resulting in a large influx of Ca2+, which initiated a Ca2+-dependent presynaptic process that ultimately led to modulation of ICa activation kinetics.
To test whether the ICa activation shift was indeed solely mediated by intracellular process triggered by the enormous Ca2+ influx during long ramp depolarizations, ICa I-Vs were generated by applying the previously used voltage-clamp ramp protocols (−100 mV to 50 mV, over 2 s, at least 2.5 min apart) to the axon terminals of enzymatically dissociated, solitary Mbs using our standard pipette solution containing 2 mm EGTA. We observed no shift in the I-V curves resulted by consecutive ramps (Fig. 3C): we found V(5%) of −37.7 ± 2.04 mV for control and −37.14 ± 2.26 mV for the second ramp I-V, V(50%) of −21.93 ± 1.84 mV for control and −22.71 ± 1.95 mV for the second ramp I-V, and V(Max) of −6.14 ± 1.75 mV for control and −8.29 ± 1.78 mV for the second ramp I-V, with no significant difference between control and second ramp I-Vs for V(5%), V(50%), and V(Max) (p = 0.6, p = 0.4, and p = 0.4, respectively, paired Student's t test; n = 7). This was in concert with the fact that previous studies of ICa of cultured solitary Mbs using voltage ramps did not report apparent changes in the ICa activation (Kaneko and Tachibana, 1985; Heidelberger and Matthews, 1992). Together, these results suggested that the pathway that mediated large Ca2+ influx-evoked modulation of ICa kinetics at Mb terminals is likely to have synaptic components driven by the glutamate released from Mb terminals.
To investigate the role of glutamate release from the Mb terminal in ICa modulation further, we inhibited exocytosis pharmacologically by adding 50 μm Exo1 (Feng et al., 2003) to the internal solution of the recording pipette. In the presence of Exo1 (50 μm), the control and second whole-cell voltage-clamp ramp generated ICa I-Vs overlapped (Fig. 4A). With depolarizing voltage ramps in the presence 50 μm Exo1, for ICa we recorded an average V(5%) of −41.3 ± 1.56 mV for control and −40.94 ± 1.70 mV for the second ramp I-V, V(50%) of −28.73 ± 1.30 mV for control and −28.10 ± 1.36 mV for the second ramp I-V, and V(Max) of −16.68 ± 1.40 mV for control and −16.57 ± 1.33 mV for the second ramp I-V. Statistical analysis revealed no difference between control and second ramp I-Vs (V(5%): p = 0.4; V(50%): p = 0.4; and V(Max): p = 0.9, paired Student's t test; n = 10). The lack of leftward shift of ICa activation was consistent with the notion that inhibiting the ramp-evoked glutamate release from Mb terminals prevented the activation of processes responsible for the ICa modulation. We could not obtain direct evidence for the reduction of glutamate release in these experiments as membrane capacitance measurements triggered by our standard 2 s voltage-clamp ramp were unreliable because of the slowly decaying tail currents (Gillis, 2000) that often lasted for >10 s after the ramps. Nonetheless, in our hands, intracellular perfusion of axotomized Mb terminals with Exo1 (50 μm) for at least 4 min in slice preparation reduced the control membrane capacitance increase in response to 250 ms depolarizing steps from −60 to −30 mV by 57.89 ± 10.09% (n = 11) without altering the depolarization-evoked ICa charge transfer (103.10 ± 12.80%, data not shown).
Figure 4.
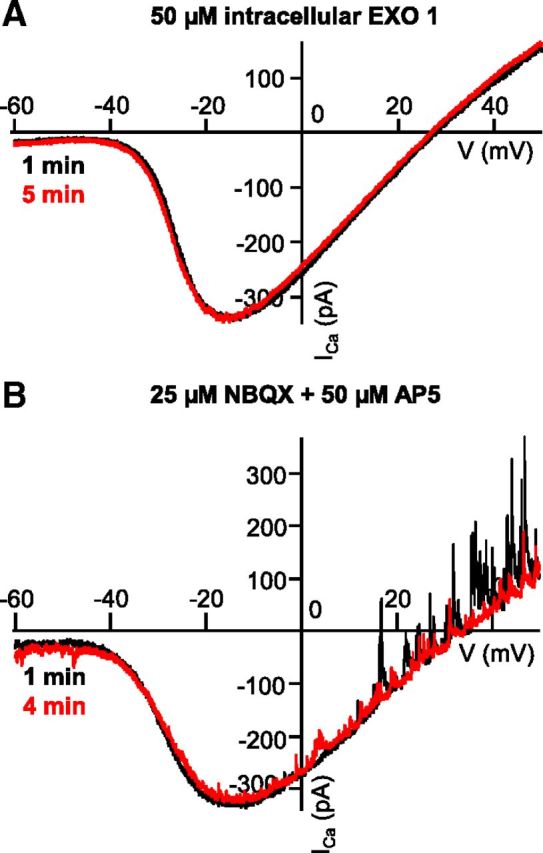
Exocytosis of glutamate and activation of neurons postsynaptic to the Mb terminal are required for modulation of ICa. A, Consecutive ICa traces recorded from an axotomized Mb terminal in slice preparation in response to voltage ramps 4 min apart in the presence of intracellular Exo1 (50 μm). The exocytosis inhibitor Exo1 prevented the leftward shift of the second ramp I-V (red) compared with the control ramp I-V (black). B, Consecutive ICa traces recorded from an axotomized Mb terminal in slice preparation, in response to voltage ramps 3 min apart in the presence of iGluR antagonists NBQX (25 μm) and d-AP5 (50 μm). The antagonists prevented the leftward shift of the second trace (red) from control (black). There are spontaneous outward GABAergic IPSCs at depolarized potentials in the absence of PTX.
These results collectively suggested that inhibition of glutamate release from Mb terminals by Exo 1 prevented the large depolarization-evoked modulation of their own ICa. This notion was also supported by two sets of results presented earlier showing lack of ICa modulation (1) when ICa I-Vs were generated by a series of short, stepwise depolarizations in an attempt to reduce Ca2+ influx into Mb terminals, but that protocol coincidentally also reduced overall glutamate release; and (2) in the presence of 10 mm intracellular BAPTA that was used to prevent Ca2+-triggered intracellular processes; BAPTA at this concentration was shown to effectively eliminate depolarization evoked exocytosis of glutamate from BC terminals (Singer and Diamond, 2003) including Mbs (Vigh and von Gersdorff, 2005).
Addition of the selective AMPA/kainate receptor antagonist NBQX (25 μm) and NMDA receptor antagonist d-AP5 (50 μm) to the external solution also prevented the characteristic leftward shift in ICa kinetics seen in response to the second voltage-clamp ramp (Fig. 4B). In these experiments, PTX was not present in the recording medium. Thus, spontaneous IPSCs, triggered by GABA release from amacrine cells (ACs), are superimposed on the ICa I-V although the reciprocal GABAergic feedback IPSCs, triggered by the depolarization-evoked glutamate release from Mb terminals, was blocked by ionotropic glutamate receptor (iGluR) antagonists NBQX and d-AP5 (Vigh et al., 2005). In the presence of NBQX (25 μm), an d-AP5 (50 μm) we found V(5%) of −40.64 ± 0.67 mV for control and −40.53 ± 0.96 mV for the second ramp I-V, V(50%) of −27.05 ± 0.49 mV for control and −27.09 ± 0.75 mV for the second ramp I-V, and V(Max) of −13.46 ± 0.76 mV for control and −13.66 ± 0.65 mV for the second ramp I-V. Statistical analysis indicated no significant difference between control and second ramp I-V (V(5%): p = 0.5; V(50%): p = 0.9; V(Max): p = 0.4; paired Student's t test; n = 9).
As these recordings were obtained exclusively from axotomized Mb terminals that do not express either presynaptic AMPA/kainate- or NMDA receptors, the site of action of iGluR antagonists by which they prevented a shift in ICa I-V can be attributed to neurons postsynaptic to the Mb terminals. Together, these data suggested that modulation of ICa kinetics at the Mb terminal required large depolarization of Mb terminals, followed by exocytosis of a large amount of glutamate to activate postsynaptic neurons, thereby initiating a feedback signal that positively modulated ICa in Mb terminals.
We tested further whether iGluR activation can trigger modulation of ICa in axotomized Mb terminals. Specifically, we recorded ICa from axotomized Mb terminals in response to a 10 ms depolarizing step from the holding potential of −60 mV to −40 mV, the membrane potential associated with threshold of L-type ICa in Mbs (Tachibana, 1999) (see also control V(5%); Fig. 2Bi,Bii). As expected, upon depolarization, a tiny inward ICa was seen (Fig. 5A, black ICa trace), which was associated with a barely detectable (if any) increase in the membrane capacitance (4.09 ± 1.72 fF, n = 8) of the Mb terminals (Fig. 5A, black Cm trace), indicating negligible glutamate release at this membrane potential (Tachibana, 1999). We then mimicked a large glutamate release from the Mb terminal by superfusing the retinal slices with the iGluR agonist KA (10 μm) for 3 min. The repeated 10 ms depolarizing step to −40 mV after KA application triggered a substantially larger ICa (Fig. 5A, red ICa trace): the peak ICa increased from an average value of −20.66 ± 5.38 pA under control conditions to −46.95 ± 12.19 pA after KA application (Fig. 5B), which was statistically significant (p = 0.04, paired Student's t test; n = 8). Importantly, the elevated ICa triggered a clear increase in the membrane capacitance (Fig. 5A, red Cm trace) indicating glutamate release. For the tested Mb terminals, ΔCm after KA increased to 12.49 ± 4.08 fF (p = 0.02, paired Student's t test; n = 8; Fig. 5A,C).
Figure 5.

Activation of postsynaptic neurons potentiated ICa and exocytosis from Mb terminals. A, Depolarization of an axotomized Mb terminal from the holding potential (HP) of −60 to −40 mV for 10 ms activated tiny calcium influx through voltage-gated calcium channels (ICa), which triggered no glutamate release, as evidenced by the absence of increase in Cm. The protocol used is shown in the bottom trace. The fast voltage sine wave used to measure Cm was not delivered during the depolarization. Bath application of 10 μm KA facilitated ICa and enhanced exocytosis (Cm) from the axotomized Mb terminals in response to a depolarizing step from −60 mV to −40 mV. Black represents control; red represents KA treatment. B, Summary figure displaying KA (10 μm) effect on peak ICa in response to 10 ms step from −60 mV to −40 mV. Black circles represent control; red squares represent KA. *p = 0.04 (paired Student's t test). n = 8. C, Summary figure displaying KA (10 μm) effect on ΔCm in response to 10 ms step from −60 mV to −40 mV. Black circles represent control; red squares represent KA. *p = 0.02 (paired Student's t test). n = 8. Data are mean ± SEM.
KA did not trigger any membrane currents in axotomized Mb terminals in the presence of PTX; therefore, these results suggested that KA depolarized neurons postsynaptic to the Mb terminal and initiated a feedback pathway. The increase in the depolarizing step evoked ICa, associated with a significant increase in the glutamate release from Mb terminals at a physiologically relevant membrane potential in the presence of KA was consistent with the notion that KA might have triggered some endogenous feedback signal generation in the inner retina, which then shifted ICa activation to more negative potentials.
NO as a retrograde messenger-modulated ICa at Mb terminals
Our data presented so far implicated the presence of a retrograde signal that plays a critical role in (strong) stimulation evoked modulation of ICa at Mb axon terminals. A prime candidate for such a retrograde signaling molecule is NO as it has been shown to shift ICa activation to more negative potentials in both rod photoreceptors (Kurenny et al., 1994) and cultured GCs (Hirooka et al., 2000) and is known to be released from inner retinal neurons by light (Eldred and Blute, 2005; Giove et al., 2009) or by glutamate (Tsumamoto et al., 2002).
First we tested whether the NO donor DETA/NO could alter ICa activation in dissociated Mbs. We found that bath application of DETA/NO (1 mm) resulted in a leftward shift in ramp-evoked ICa I-V (Fig. 6A) similar to that seen under control conditions for the second ramp-evoked ICa I-V of axotomized Mb terminals in slice preparation (Fig. 2Bi,Bii). On average, V(5%) was −41.2 ± 1.01 mV for control and −47.5 ± 2.16 mV for the second ramp I-V, V(50%) was −23.6 ± 0.74 mV for control and −28.9 ± 1.36 mV for the second ramp I-V, and V(Max) was −6.1 ± 0.99 mV for control and −10.4 ± 1.55 mV for the second ramp I-V. On average, the second ramp I-V exhibited an average difference of −6.3 ± 1.49 mV for V(5%), −5.3 ± 0.84 mV for V(50%), and −4.3 ± 0.73 mV for V(Max). Statistical analysis determined a significant difference in V(5%), V(50%), and V(Max) for the ramp I-V obtained in DETA/NO compared with the control (p = 0.002, p = 0.0001, and p = 0.0002, respectively, paired Student's t test; n = 10).
Figure 6.
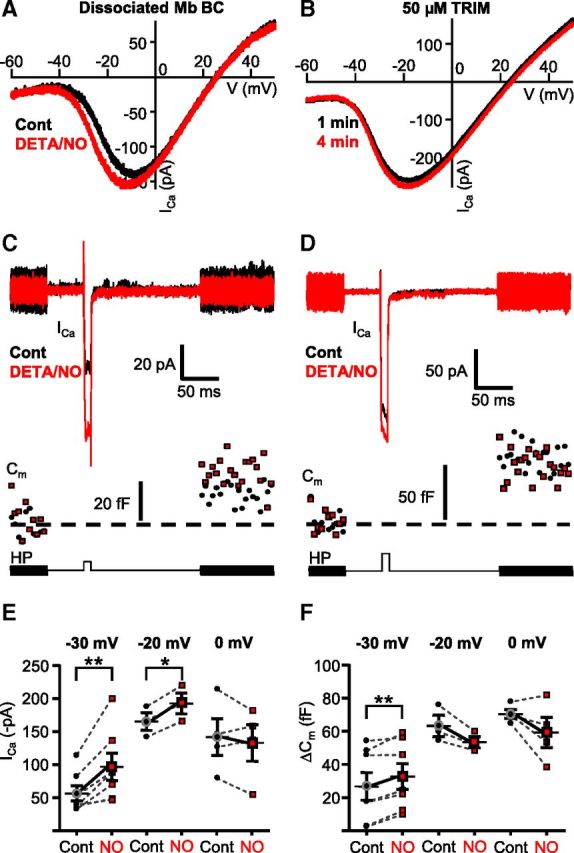
The NO donor-mediated shift of ICa caused weighted potentiation of Mb output selectively in response to weak stimuli. A, Application of NO donor DETA/NO (1 mm) for 1.5 min shifted the ramp-evoked ICa activation to more negative potentials in enzymatically dissociated Mbs (black represents control; red represents DETA/NO). B, Inhibition of endogenous NO synthases by TRIM (50 μm) prevented the leftward shift of ICa activation in axotomized Mb terminals in slice preparation during consecutive ramp stimulations (black represents control; red represents second ramp I-V). C, Bath application of DETA/NO (1 mm) facilitated ICa and enhanced exocytosis (Cm) from the axotomized Mb terminals in response to a depolarizing step from −60 mV to −30 mV. HP, Holding potential. Black represents control; red represents DETA/NO treatment. D, Bath application of DETA/NO (1 mm) slightly increased ICa, but this increase was not associated with increased exocytosis from axotomized Mb terminals in response to a depolarizing step from −60 mV to −20 mV (bottom trace). Black represents control, red represents DETA/NO treatment. E, Summary figure displaying DETA/NO (1 mm) effect on peak ICa in response to 10 ms step from −60 mV to −30, −20, or 0 mV. Black circles represent control; red squares represent DETA/NO. **p = 0.004 (paired Student's t test). n = 7. *p = 0.008 (paired Student's t test). n = 3. F, Summary figure displaying DETA/NO (1 mm) effect on ΔCm evoked by 10 ms step from −60 mV to −30, −20, or 0 mV. Black circles represent control; red squares represent DETA/NO. **p = 0.003 (paired Student's t test). n = 7. n = 3 for −20 mV; n = 4 for 0 mV. Every terminal was tested at one depolarization level in control then in the presence of DETA/NO; thus, each pair of measurements shown originated from different cells. Data are mean ± SEM.
Next, we examined whether endogenous NO generation and release, triggered by synaptic activity, contribute to the modulation of ICa at Mb terminals in retinal slice preparation. Specifically, we tested whether pharmacological inhibition of NO production by a nonselective inhibitor of both inducible and neuronal NO synthase (iNOS and nNOS, respectively), TRIM, can reduce the negative shift in ICa activation after depolarizing ramps applied to the Mb terminals. We found that continuous bath application of TRIM (50 μm) eliminated the leftward shift in ICa activation kinetics following ramp depolarization of axotomized Mb terminals (Fig. 6B). On average, V(5%) was −42.1 ± 1.63 mV for control and −42.5 ± 2.01 mV for the second ramp I-V, V(50%) was −27.9 ± 1.57 mV for control and −28.63 ± 1.92 mV for the second ramp I-V, and V(Max) was −13.75 ± 1.58 mV for control and −14.8 ± 1.90 mV for the second ramp I-V. In the presence of TRIM, there was no significant difference in V(5%), V(50%), and V(Max) between control and second ramp evoked ICa I-Vs (p = 0.6, p = 0.3, and p = 0.07, respectively; paired Student's t test; n = 8).
Together, these data suggested that the endogenous retinal retrograde messenger responsible for modulation of ICa at Mb terminals could be NO. The NO donor-evoked shift of ICa activation predicted an increase of ICa amplitude in the membrane potential range between −50 mV and −20 mV, which is physiologically relevant for Mb function (Saito et al., 1979; Wong et al., 2005). To test this notion directly, we measured depolarization evoked ICa in response to a 10 ms depolarizing step from −60 mV to −30 mV, −20, or 0 mV along with the depolarization-evoked increase in the membrane capacitance (ΔCm) of axotomized Mb terminals under control conditions and after at least 3 min of continuous bath application of DETA/NO (1 mm). We found that bath application of DETA/NO increased the peak ICa in response to a depolarizing step to −30 mV (Fig. 6C) from −56.61 ± 11.47 pA to −96.81 ± 20.62 pA (Fig. 6E; p = 0.004; paired Student's t test; n = 7). The NO-mediated increase of ICa at −30 mV was associated with enhanced exocytosis (Fig. 6C; Cm traces) from an average ΔCm of 26.80 ± 8.31 fF under control conditions to 32.73 ± 7.79 fF after application of DETA/NO (Fig. 6F; p = 0.003, paired Student's t test; n = 7). Treatment with the NO donor also increased the peak ICa in response to steps to −20 mV (Fig. 6D) from −165.33 ± 13.27 pA to −192.59 ± 15.62 pA (Fig. 5E; p = 0.008; paired Student's t test, n = 3). However in these terminals, the ICa increase was not associated with an increase in exocytosis (ΔCm: 63.27 ± 6.45 fF vs 53.46 ± 3.37 fF in control and in DETA/NO, respectively; Fig. 6F; p = 0.09; paired Student's t test; n = 3). Neither the control ICa amplitude (−117.69 ± 17.15 pA) nor the corresponding membrane capacitance increase (ΔCm: 67.99 ± 1.67 fF) was increased by DETA/NO (ICa: −116.69 ± 31.56 pA; ΔCm: 59.31 ± 10.54 fF) significantly (ICa: p = 0.5; ΔCm: p = 0.2, paired Student's t test; n = 4) in response to depolarizing steps from −60 to 0 mV (Fig. 6E,F). These results were in perfect agreement with the NO donor evoked leftward shift in ICa activation (Fig. 6A) in that the weaker depolarization evoked voltage-gated ICa amplitude was enhanced more. Importantly, these results demonstrated that the NO-induced ICa modulation was reflected in weighted potentiation of glutamate output from Mb terminals.
NO modulated ICa activation threshold through S-nitrosylation in Mb terminals
Within recent years, NO's role as a retinal signaling molecule has been expanding rapidly (Vielma et al., 2012). The classical intracellular signaling cascade that underlies the effects of NO involves NO detection by soluble guanylate cyclase, which stimulates the production of cGMP. The increased levels of cGMP, in turn, stimulate protein kinase G (PKG), resulting in increased transmitter release in cones (Savchenko et al., 1997) and increased sensitivity of cone BCs (Snellman and Nawy, 2004).
To parse out the intracellular pathway associated with modulation of ICa in Mb terminals by NO, first we attempted to simulate the NO effect in enzymatically dissociated Mbs using the membrane permeable form of cGMP, 8-Br-cGMP. Application of an external solution containing 8-Br-cGMP (100–500 μm) induced a large (“leak”) current in 2 of 5 intact Mbs at −60 mV holding potential, making voltage-ramp evoked ICa I-Vs unreliable and inconsistent in the presence of 8-Br-cGMP (data not shown). These results were consistent with the expression of cyclic nucleotide-gated channels (CNGCs) in Mbs (Henry et al., 2003) that mediate a cation current when activated by focal application of 8-Br-cGMP onto the dendrites of Mbs in retinal slice (Ping et al., 2008). In the rest of the dissociated Mbs (3 of 5), the leak was not increased during 8-Br-cGMP application, which we interpreted as a consequence of potentially damaged dendritic CNGCs. Nonetheless, in those Mbs, 8-Br-cGMP did not alter ICa activation (V(5%): p = 0.9; V(50%): p = 0.6; V(Max): p = 0.7; paired Student's t test; n = 3). In our hands, 8-Br-cGMP (500 μm) did not induce a current in axotomized Mb terminals in slice preparation either (Ihold control: −26.45 ± 9.67 pA vs cGMP: −36.92 ± 16.11 pA, p = 0.4; paired Student's t test; n = 4), further suggesting that functional CNGCs in Mbs might be restricted to the somatodendritic compartment. More importantly, in the presence of NBQX (25 μm) and d-AP5 (50 μm) or, in other words, under conditions when the reciprocal endogenous retinal pathway mediating the shift in ICa activation was blocked (Fig. 4B), 8-Br-cGMP (500 μm) did not alter the kinetics of voltage ramp-evoked ICa I-Vs (Fig. 7A). Statistical analysis revealed no significant difference between the control and subsequent ramp I-Vs recorded in the presence of 8-Br-cGMP (V(5%): p = 0.9, V(50%): p = 0.7 and V(Max): p = 0.4; paired Student's t test; n = 4).
Figure 7.
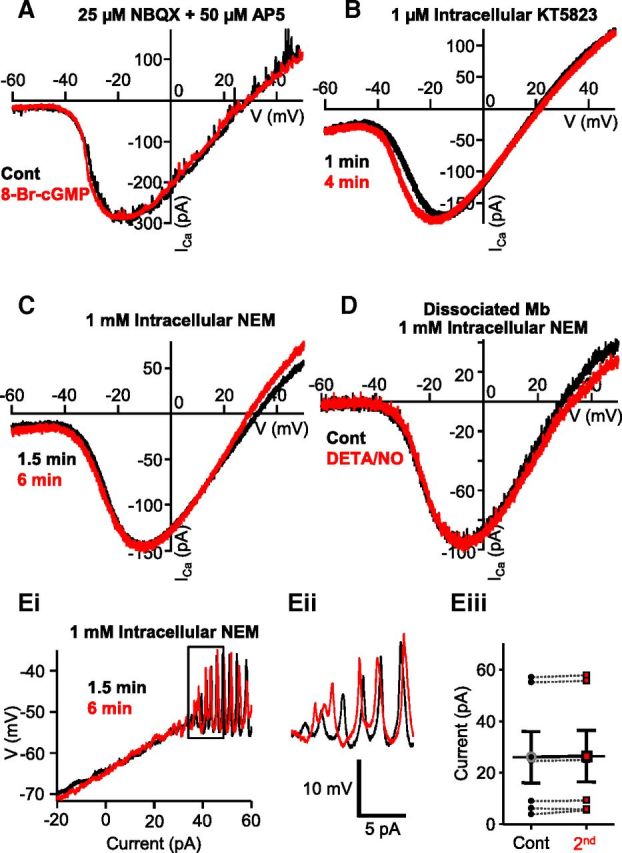
NO modulated ICa in Mb terminals through a cGMP-independent pathway that involved S-nitrosylation reactions. A, In the presence of NBQX and d-AP5, bath application of the membrane-permeable cGMP analog 8-Br-cGMP (500 μm) for 3 min failed to shift ICa activation of axotomized Mb terminals in slice preparation. B, KT5823 (1 μm), a selective inhibitor of PKG, was unable to prevent the shift in ICa activation seen during consecutive ramp stimulations of Mb terminals in slice preparation. C, The leftward shift of the Ica I-V seen during consecutive ramp stimulations of Mb terminals in slice preparation was occluded by intracellular application of NEM (1 mm). D, In the presence of NEM (1 mm) in the intracellular solution, the NO donor, DETA/NO (1 mm), did not induce a leftward shift in the ICa activation threshold of dissociated, solitary Mbs. Ei, In the slice preparation, with NEM (1 mm) in the intracellular solution, no reduction in Ca2+ spike threshold was noticed during consecutive current ramp protocols applied to the axon terminal of Mbs. Eii, enlargement of boxed area in Ei illustrating that initiation threshold for Ca2+ spikes remained similar to that of control when S-nitrosylation reactions were prevented by NEM. Eiii, Summary diagram showing that, in the presence of NEM, no significant change was observed in the depolarizing current threshold of Ca2+ spikes between control and second current ramp-evoked responses. Black circles represent control; red squares represent second; n = 6; 4 intact cells, 2 axotomized pooled. Data are mean ± SEM.
KT5823 was shown to block the NO signaling cascade in retinal neurons by inhibiting PKG (Hirooka et al., 2000; Snellman and Nawy, 2004). Nonetheless, in the presence of 1 μm KT5823 in the pipette solution, consecutive depolarizing ramps shifted the ICa activation to the left in axotomized Mb terminals in slice preparation (Fig. 7B) similar to what was found under control conditions (Fig. 2Bi,Bii). On average, V(5%) was −44.0 ± 1.58 mV for control and −47.2 ± 1.77 mV for the second ramp I-V, V(50%) was −29.5 ± 1.71 mV for control and −32.6 ± 1.73 mV for the second ramp I-V, and V(Max) was −15.0 ± 1.95 mV for control and −18.0 ± 1.84 mV for the second ramp I-V. The difference in ICa I-V curve parameters (V(5%): −3.2 ± 0.37 mV; V(50%): −3.1 ± 0.19 mV; and V(Max): −3.0 ± 0.32 mV) was statistically significant (V(5%): p = 0.001; V(50%): p = 0.00007; V(Max): p = 0.0006; paired Student's t test; n = 5).
These findings discounted the role of an NO-stimulated cGMP-dependent pathway in altering ICa activation at Mb terminals and are in concert with the report showing that NO donor application failed to trigger cGMP elevation in Mbs in the goldfish retina (Baldridge and Fischer, 2001). An alternative mechanism of action by which NO can influence cellular function is through direct nitrosylation, in which NO covalently binds to the thiol side-chains of cysteine residues of various proteins to form S-nitrosothiols (S-nitrosylation) (for review, see Ahern et al., 2002). To assess whether NO-mediated S-nitrosylation played a role in the observed leftward shift of ICa activation in response to ramp I-Vs of axotomized Mb terminals, we introduced NEM (1 mm) into the intracellular solution. NEM is a potent inhibitor of S-nitrosylation as it irreversibly reacts with and binds to sulfhydryl groups thereby preventing NO from engaging in an S-nitrosylation reaction. NEM prevented the leftward shift of ICa during consecutive voltage ramps in axotomized Mb terminals in retinal slice (Fig. 7C). On average, V(5%) was −43.2 ± 0.92 mV for control and −44.0 ± 1.14 mV for the second ramp I-V, V(50%) was −28.5 ± 0.71 mV for control and −27.6 ± 0.54 mV for the second ramp I-V, and V(Max) was −12.6 ± 0.68 mV for control and −11.6 ± 1.12 mV for the second ramp I-V. Statistical analysis determined no significant difference in V(5%), V(50%), and V(Max) between the control and second ramp evoked ICa I-Vs (p = 0.7, p = 0.9, and p = 0.4, respectively; paired Student's t test; n = 5). Similarly, in the presence of NEM (1 mm) in the pipette solution, bath application of 1 mm DETA/NO (Fig. 7D) failed to alter the parameters of voltage ramp evoked ICa I-V in enzymatically dissociated Mbs. On average, V(5%) was −41.5 ± 1.26 mV for control and −41.8 ± 0.87 mV for the second ramp I-V, V(50%) was −25.3 ± 1.45 mV for control and −25.7 ± 1.41 mV for the second ramp I-V, and V(Max) was −9.2 ± 2.32 mV for control and −9.7 ± 2.54 mV for the second ramp I-V. Statistical analysis revealed no difference between control and second ramp I-V parameters (V(5%): p = 0.8; V(50%): p = 0.7 and V(Max): p = 0.6, paired Student's t test; n = 6).
Last, we assessed whether preventing S-nitrosylation in Mb terminals also prevents the shift in Ca2+ spike threshold observed during stimulation of Mb terminals with consecutive depolarizing current ramps (Fig. 1). We found that blocking S-nitrosylation of proteins within the Mb terminal with NEM prevented a change in the threshold for Ca2+ spike initiation (Fig. 7Ei). An enlargement of the region of the trace where the spikes begin to originate shows no obvious difference in the threshold for initiation (Fig. 7Eii). On average, initiation of spikes during the first (control) ramp required 25.92 ± 9.98 pA, almost identical to that required during the second ramp applied 3 min later (26.47 ± 10.03 pA), and no significant difference between the thresholds of calcium spikes were detected (p = 0.1, paired Student's t test, n = 6; 4 intact cells, 2 axotomized terminals; Fig. 7Eiii). Further, there was no shift in the membrane potential associated with the spike threshold. With 1 mm NEM in the internal solution, on average, spikes initiated at −45.35 ± 2.46 mV for control and at −44.82 ± 2.73 mV for the second trace (p = 0.4, paired Student's t test, n = 6; 4 intact cells, 2 axotomized terminals).
Light induced NEM-sensitive modulation of Mbs response to scotopic, rod-mediated inputs in an intensity-dependent manner
Although direct nitrosylation of thiol side-chains of cysteine residues by NO plays an important role in numerous physiological processes (Ahern et al., 2002), it is a particularly important process in neurodegenerative diseases (Nakamura et al., 2013). Our results presented until this point show that this pathway can be triggered by endogenous, synaptically released glutamate, which initiates retrograde modulation of ICa at the same presynaptic BC axon terminals. Nonetheless, the depolarizing ramp protocols that were shown to trigger this pathway consistently in our experiments were far from physiologically relevant depolarizations for Mbs. To study whether our findings have functional consequences for normal visual processing or whether this NO-mediated process can only exist under experimental conditions in retina, we designed a set of experiments where depolarizations of Mbs were evoked by light stimulations, based on the observation that: (1) NO release in the retina is triggered by increasing light intensity (Eldred and Blute, 2005; Giove et al., 2009); (2) our preceding experiments suggesting that small depolarizations of Mb terminals might be preferentially potentiated by endogenous NO, which appeared to be released in our prep by large synaptic depolarizations of inner retinal neurons (Figs. 2Bi and 5); and (3) magnitude of depolarization in ON BCs is related to strength and wavelength of illumination (Joselevitch and Kamermans, 2007; Jarsky et al., 2011). Although we did not intend to simulate Mb responses to natural underwater visual scenes, the wavelength and intensity of light stimulations were selected to match those behaviorally relevant in shallow water (Munz and McFarland, 1973; McFarland and Munz, 1975; Loew and McFarland, 1990). Additionally, the light stimulations were chosen in accordance with the observations that Mbs in the fish retina receive direct inputs from both rods and cones (Wong et al., 2005; Joselevitch and Kamermans, 2007), similar to many BCs in cold-blooded vertebrate retinas (Wu, 1994) and certain mammalian BCs (Protti et al., 2005; Haverkamp et al., 2008; Pang et al., 2010).
In these experiments, light-evoked responses of intact Mbs were recorded directly from their axon terminals in current-clamp mode from dark-adapted retinal slices. Mbs were slightly hyperpolarized to keep their membrane potential at ∼−51 mV, which prevented spontaneous Ca2+ spike firing in the dark. Figure 8Ai shows representative control responses recorded form the axon terminal of an intact Mb, evoked by two sets of 3 consecutive dim (1.6 × 107 photons/cm2/s) green flashes (λ = 505 nm, 500 ms) applied 5 s apart and with 4 min between the two sets of light stimuli. The responses to the same light stimulation showed great variability in the number and amplitude of the Ca2+ spikes (Fig. 8Ai), but they were rather consistent in terms of the latency within sets as well as across consecutive sets (Fig. 8Aii): for the Mb shown, the latency of first light-evoked spikes varied between 245 and 258 ms, averaging at 251.33 ± 3.76 ms for the first set and varied between 249 and 250 ms averaging at 249.67 ± 0.3 ms for the second set of stimulations (Fig. 8Aiii). When latencies of these dim green light (1.6 × 107 photons/cm2/s, λ = 505 nm, 500 ms) evoked spikes between the first and second sets (248.6 ± 14.6 ms vs 247.02 ± 13.8 ms, respectively) were compared across multiple cells (n = 6), we found no significant statistical difference (p = 0.6, paired Student's t test).
Figure 8.
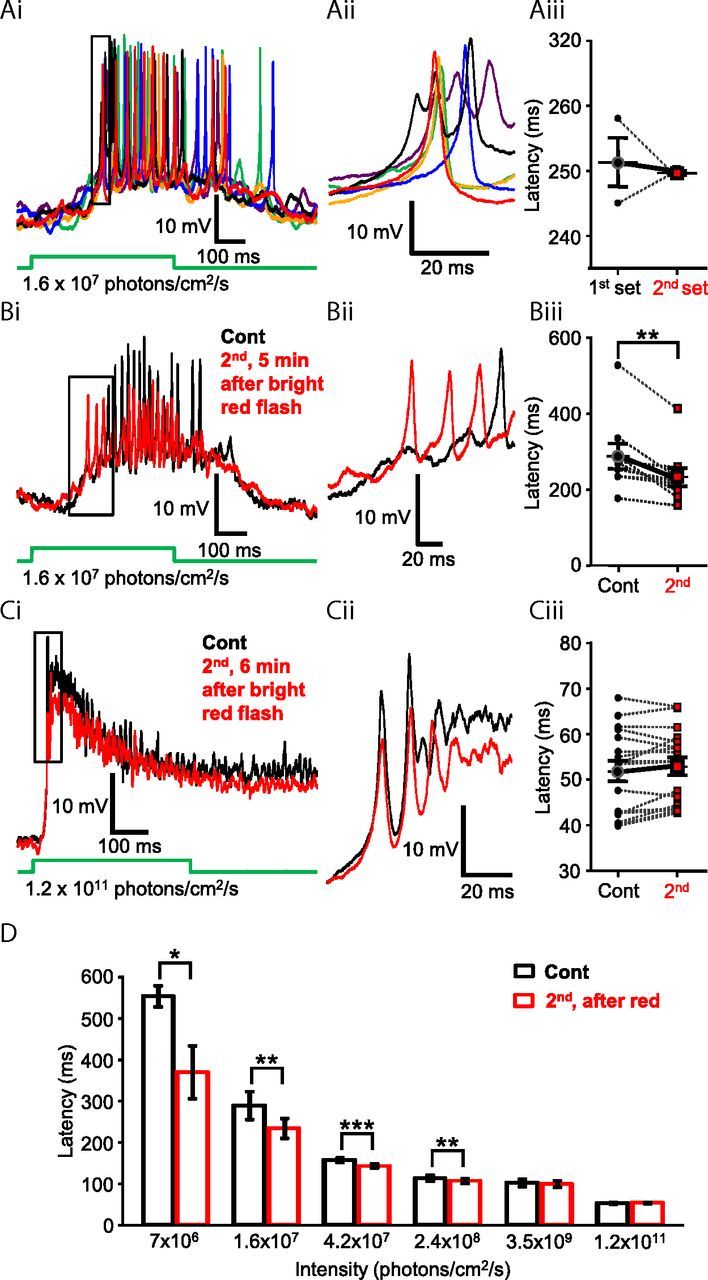
Photopic light stimulation modulated scotopic light responses of Mb axon terminals. Ai, Repeated stimulations of rod-specific scotopic green light flashes (500 ms, λ = 505 nm, 1.6 × 107 photons/cm2/s) did not significantly alter the latency of evoked Ca2+ spikes from the axon terminal of an intact Mb. The first set of responses (black, blue, and green traces) were evoked by three distinct flashes of dim green light with a 5 s delay between flashes. After a 4 min delay, a second set of responses (purple, red, and orange traces) was evoked by three distinct flashes of dim green light. Membrane potentials at the beginning of the traces are −51.8 mV (black), −48.9 mV (blue), −52.4 mV (green), −47.9 mV (purple), −50.8 mV (red), and −52.1 mV (orange). Aii, Enlargement of boxed area in Ai illustrating consistency in the latency of scotopic green light-induced Ca2+ spikes. Membrane potentials at the beginning of the traces are −44.6 mV (black), −44.7 mV (blue), −43.3 mV (green), −41.9 mV (purple), −44.6 mV (red), and −46.6 mV (orange). Aiii, Summary graph showing paired comparison of latency to first spikes of corresponding responses triggered by the first and second set of dim green (505 nm, 1.6 × 107 photons/cm2/s) light flashes. The latency to first spike was not significantly different between sets. Black circles represent first set; red squares represent second set. Bi, Rod-mediated scotopic green (505 nm, 1.6 × 107 photons/cm2/s) full-field light flash (500 ms) evoked responses recorded from the axon terminal of an intact Mb, in dark-adapted retina slice preparation (black), were modulated by bright red light stimulation (500 ms, 660 nm, 5 × 1013 photons/cm2/s, Mb response not shown here; but see Fig. 9Ci). Five minutes after the bright red flash, the repeated dim green stimulus evoked spiking response with decreased latency (red trace) compared with control. Membrane potential at the beginning of both traces was −51 mV. Bii, Enlargement of boxed area in Bi illustrating the decreased latency of scotopic green light-induced Ca2+ spikes, after the bright red stimulation. Membrane potentials at the beginning of traces are −50.5 mV (red) and −52.8 mV (black). Biii, Summary graph showing paired comparison of latency to first spike triggered by dim green (505 nm, 1.6 × 107 photons/cm2/s) flash before and after presentation of bright red light recorded from different Mb terminals. The latency to first spike was significantly reduced for the second stimulus, on average by 55.18 ± 14.98 ms. Black circles represent control; red squares represent second scotopic response latency. **p = 0.006 (paired Student's t test). n = 9. Ci, Responses of Mb terminal to mesopic green (500 ms, 505 nm, 1.2 × 1011 photons/cm2/s) light flashes, before (black) and 5 min after (red) the bright red flash (660 nm, 5 × 1013 photons/cm2/s, 500 ms, Mb response not shown). No difference in the delay of mesopic light responses was noticeable. Membrane potential at the beginning of the traces was −52 mV. Cii, Enlargement of boxed area in Ci illustrating no apparent difference in the latency of scotopic green light-induced Ca2+ spikes, after the bright red stimulation. Membrane potentials at the beginning of traces are −50.25 mV (red) and −50.1 mV (black). Ciii, Summary graph of paired comparison of mesopic green light (500 ms, 505 nm, 1.2 × 1011 photons/cm2/s) response latency before and after presentation of bright red light. No significant difference was detected. Black circles represent control; red squares represent second scotopic response latency. n = 17. D, Summary graph showing the average latency to first spike from green light responses triggered by six intensities before (black) and 5 min after (red) bright red stimulation. Of the six different intensities of green light presented, a significant decrease in latency was observed in response to dim, scotopic intensities known to stimulate rods, but not cones (≤3.5 × 109 photons/cm2/s): 7 × 106 photons/cm2/s; n = 3. *p = 0.04. 1.6 × 107 photons/cm2/s; n = 9. **p = 0.006. 4.2 × 107 photons/cm2/s; n = 17. ***p = 0.0001. 2.4 × 108 photons/cm2/s; n = 17. **p = 0.02. 3.5 × 109 photons/cm2/s; n = 17. 1.2 × 1011 photons/cm2/s: n = 17 (paired Student's t test). Data are mean ± SEM.
In the next set of experiments, we devised a protocol with the intent to determine whether the Mb spiking response could be sensitized by light. The general framework of the light stimulation consisted of an initial light flash, followed by a brighter, “sensitizing” light flash, and then a repeat presentation of the initial flash. This paradigm allowed us to compare the light-evoked spiking response of the Mb terminal before and after a sensitizing light stimulus and use the observed differences as a metric for whether or not the terminal had been “sensitized.” Specifically, the retina was exposed to a series of full field green (λ = 505 nm) light flashes (500 ms) with increasing intensities ranging from scotopic (7 × 106 photons/cm2/s) to dim photopic (1.2 × 1011 photons/cm2/s) intensities (Busskamp et al., 2010). Then a 500 ms bright red light (sensitizing) stimulus (λ = 660 nm, 5 × 1013 photons/cm2/s) was delivered. With a minimum delay of 5 min after the red light flash, the series of (initial) green light flashes were repeated. Figure 8Bi shows a representative recording where we found that the second dim green flash (1.6 × 107 photons/cm2/s), applied after sensitizing red stimulation, triggered a spiking response from the Mb axon terminal with shorter delay compared with that evoked by the same intensity before the red flash (Fig. 8Bi,Bii) as if the terminal was “sensitized” by the bright red light stimulation. Similar shortening of first spike delay was seen in every cell tested (9 of 9; Fig. 8Biii) at this green light intensity and was found to be statistically significant (p = 0.006, paired Student's t test). Interestingly, the delay of light responses to bright (1.2 × 1011 photons/cm2/s) green light of the same Mb was not different before and after the bright red simulation (Fig. 8Ci,Cii). Across cells, the latency to first spike increased in 7 of 17 Mbs, decreased in 6 of 17, and remained virtually unaltered in 4 of 17 cells (Fig. 8Ciii); and when summed, no statistically significant difference was detected (p = 0.1, paired Student's t test, n = 17). Of the six different intensities of green light presented in this set of experiments, a significant decrease in latency was observed in those under intensities known to stimulate only rods (≤108 photons/cm2/s) and was not observed in intensities above cone threshold (∼109 photons/cm2/s in the goldfish retina) (Joselevitch and Kamermans, 2009). The dimmest green light stimulus used in our experiments (7 × 106 photons/cm2/s) produced a spiking response with an average latency of 553.50 ± 25.74 ms in control (first) and with 369.37 ± 63.80 ms after the red flash (second); in response to a 1.6 × 107 photons/cm2/s flash, the average latency was 288.71 ± 33.36 ms in control (first) and 233.54 ± 24.44 after the red flash (second); in response to a 4.2 × 107 photons/cm2/s flash, the average latency was 156.92 ± 5.12 ms in control (first) and 142.55 ± 4.69 after the red flash (second); in response to a 2.4 × 108 photons/cm2/s flash, the average latency was 112.99 ± 7.19 ms in control (first) and 105.93 ± 5.97 after the red flash (second); in response to a 3.5 × 109 photons/cm2/s flash, the average latency was 101.57 ± 8.59 ms in control (first) and 98.85 ± 7.99 after the red flash (second); in response to a 1.2 × 1011 photons/cm2/s flash, the latency was 51.89 ± 2.24 ms in control (first) and 52.93 ± 1.97 after the red flash (second) (Fig. 8D). The latency, as well as the variability in latency across cells, decays as light intensity increases. These results are consistent with the observation that temporal precision of spikes produced by BCs, evoked by high-contrast light stimulation, is in the millisecond range, which correlates with that of AC and GC spikes in the fish retina (Baden et al., 2011).
The intensity of sensitizing light stimulus used in the previous experiment was sufficient to activate both rod and cone inputs to Mbs (Joselevitch and Kamermans, 2007). In the next set of experiments, we sought to parse out the contribution of rod- and cone-mediated signaling in triggering the light-induced sensitization of dim scotopic light (λ = 505 nm, 500 ms, 1.6 × 107 photons/cm2/s) responses. This particular scotopic intensity was selected as it reliably evoked spiking responses in Mbs with relatively high temporal precision (Fig. 8Ai,Aii) and exhibited a latency that did not change over the time course (∼5 min) of these experiments (Fig. 8Aiii). However, the latency of responses to this scotopic intensity showed significant reduction, after bright red sensitizing light stimulation, consistently across cells (Fig. 8Biii). We found that a full field green flash (λ = 505 nm, 500 ms) with an intensity of 2.4 × 108 photons/cm2/s, which is just below cone threshold in the fish retina (Joselevitch and Kamermans, 2009), could also reduce the latency of consecutive light responses evoked by the dim scotopic green flash. On average, the latencies of the spiking response were reduced from 281.12 ± 11.28 ms to 249.86 ± 10.75 ms (Fig. 9Ai,Aii) reliably (in 11 of 11 experiments, Fig. 9Aiii), and the reduction was proven statistically significant (p = 0.0001, paired Student's t test, n = 11). In the next experiments, we increased the sensitizing green flash (λ = 505 nm, 500 ms) stimulus intensity to 1010 photons/cm2/s. This mesopic intensity saturates rods (Sterling, 2003; Busskamp et al., 2010) and starts to activate cones (Joselevitch and Kamermans, 2009). As expected, flashes at this intensity also reduced the latency of consecutive dim green, flash-evoked responses in every cell tested (Fig. 9Bi,Bii) (control: 275.29 ± 9.41 ms vs second: 230.84 ± 10.17 ms) in a statistically significant manner (p = 0.00008, paired Student's t test, n = 7) (Fig. 9Biii). Compared across experiments, we found that the reduction of dim scotopic green (λ = 505 nm, 500 ms, 1.6 × 107 photons/cm2/s) response latencies triggered by the sensitizing light was intensity-dependent: it was the largest (55.18 ± 14.98 ms, n = 9) for bright photopic red (λ = 660 nm, 500 ms, 5 × 1013 photons/cm2/s), followed by the reduction of (44.44 ± 4.71 ms, n = 7) caused by rod-saturating mesopic green (λ = 505 nm, 500 ms, 1010 photons/cm2/s) flashes and was the smallest (31.25 ± 3.12 ms, n = 11) for bright scotopic green (λ = 505 nm, 500 ms, 2.4 × 108 photons/cm2/s) stimuli. However, when the latency changes caused by these three sensitizing light stimulations were statistically analyzed, no significant difference was detected among them (one-way ANOVA, with Tukey's multiple-comparison test). These results indicated that bright scotopic signals, which are processed and conveyed to the inner retina by Mbs, were sufficient to potentiate processing of consecutive weak rod inputs by Mbs. Light stimuli bright enough to activate cones might trigger further activity-dependent weighted potentiation of Mb signaling. Together, the data demonstrated that the strength of Mb depolarization drove the retrograde, NO-mediated modulation of Mb terminal signaling.
Figure 9.
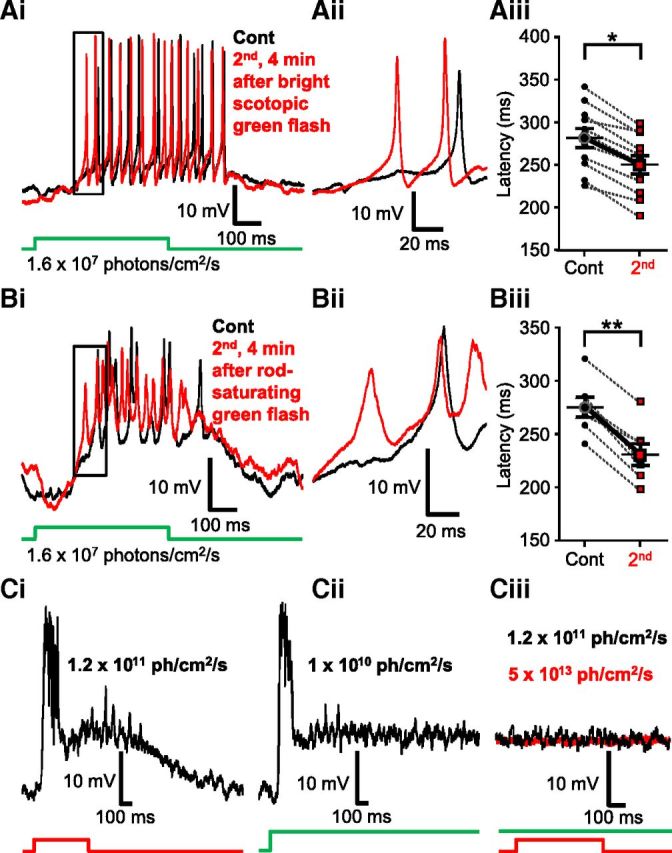
Scotopic light responses of Mb axon terminals can be modulated by light through rod-specific pathways. Ai, Rod-mediated responses recorded from the axon terminal of an intact Mb to a full-field light flash (500 ms) of scotopic green (λ = 505 nm, 1.6 × 107 photons/cm2/s) before (black) and 4 min after (red) a bright scotopic green (λ = 505 nm, 2.4 × 108 photons/cm2/s) full-field light flash (500 ms). After stimulation with the bright scotopic green light, the repeated dim green stimulus evoked a spiking response with decreased latency compared with control. Membrane potentials at the beginning of the traces are −51.9 mV (red) and −49.2 mV (black). Aii, Enlargement of boxed area in Ai illustrating the decreased latency of rod-mediated, light-induced Ca2+ spikes when the green scotopic stimulus is increased by one order of magnitude. Membrane potentials at the beginning of the traces are −49.5 mV (red) and −48.9 mV (black). Aiii, Summary graph showing paired comparison of latency to first spike triggered by dim green (505 nm, 1.6 × 107 photons/cm2/s) flash before and after presentation of sensitizing green light (505 nm, 2.4 × 108 photons/cm2/s) recorded from different Mb terminals. The latency to first spike was significantly reduced for the second stimulus, on average by 31.25 ± 3.12 ms. Black circles represent control; red squares represent second scotopic response latency. **p = 0.0001 (paired Student's t test). n = 11. Data are mean ± SEM. Bi, Responses of an Mb terminal evoked by a scotopic green (505 nm, 1.6 × 107 photons/cm2/s) light flash (500 ms) of a dark-adapted retina (black) were modulated by a rod-saturating bright green (500 ms, 505 nm, 1.0 × 1010 photons/cm2/s) light flash. Four minutes after the bright green flash, a repeated dim green stimulus evoked a spiking response with decreased latency (red) compared with control. Membrane potential at the beginning of the trace was −47.14 mV (red) and −49.47 mV (black). Bii, Enlargement of boxed area in Bi illustrating the decrease in latency observed in scotopic green light-evoked Ca2+ spikes after rod saturating green light stimulation. Membrane potentials at the beginning of the traces are −48.79 mV (red) and −48.92 mV (black). Biii, Summary graph showing paired comparison of latency to first spike triggered by dim green (505 nm, 1.6 × 107 photons/cm2/s) flash before and after presentation of bright green light (505 nm, 1.0 × 1010 photons/cm2/s). The latency to first spike was significantly reduced for the second stimulus, on average by 44.44 ± 4.71 ms. Black circles represent control; red squares represent second scotopic response latency. **p = 0.00008 (paired Student's t test). n = 7. Data are mean ± SEM. Ci, Responses recorded from an Mb terminal evoked by a weak photopic red flash (500 ms, λ = 660 nm, 1.2 × 1011 photons/cm2/s). Membrane potential at the beginning of the trace is −50.5 mV. Cii, Response from the same Mb axon terminal evoked by a sustained presentation of rod saturating green light (λ = 505 nm, 1.0 × 1010 photons/cm2/s). Membrane potential at the beginning of the trace is −50.9 mV. Ciii, Neither weak photopic (1.2 × 1011 photons/cm2/s) nor brighter (5 × 1013 photons/cm2/s, applied 9 min later) red flash (500 ms, λ = 660 nm) evoked measurable responses in Mb terminals (black and red traces, respectively) when applied on top of sustained rod-saturating green background illumination (λ = 505 nm, 1.0 × 1010 photons/cm2/s). Membrane potentials at the beginning of the traces are −40.3 mV (black) and −43.9 mV (red).
Next, we tested whether similar modulatory mechanism affects photopic signals processed by Mbs; specifically, we tested whether weak photopic responses of Mb terminals evoked by red flashes (λ = 660 nm, 500 ms, 1.2 × 1011 photons/cm2/s) can be also sensitized by bright photopic stimulation (λ = 660 nm, 500 ms, 5 × 1013 photons/cm2/s) in a similar manner. When delivered on a dark background, the weak photopic red (λ = 660 nm, 500 ms, 1.2 × 1011 photons/cm2/s) and the rod-saturating green (λ = 505 nm, 1010 photons/cm2/s) stimulation evoked identical responses in Mb terminals (Fig. 9Ci and 9Cii, respectively): the spikes merged into a transient initial depolarization that was followed by a depolarized plateau with superimposed small spikes/membrane potential oscillations, which exceeded the length of the illumination. The membrane potential of the plateau varied between −33.25 mV and −40.05 mV and averaged −37.22 ± 2.41 mV (n = 7). To isolate cone responses, red flashes were delivered on a rod-saturating green background (λ = 505 nm, 1010 photons/cm2/s), which was turned on 4 s before the first red stimulations. Neither weak (1.2 × 1011 photons/cm2/s) nor strong (5 × 1013 photons/cm2/s) photopic red stimulation (λ = 660 nm, 500 ms), when superimposed on the rod-saturating green background, evoked detectable responses (Fig. 9Ciii). These results remain consistent with the observation that Mbs process rod-dominant visual signals and nearly saturate at intensities of 1010 photons/cm2/s (Joselevitch and Kamermans, 2009).
Collectively, these data suggested that the latency of light-evoked Ca2+ spikes in Mb terminals is subject to plasticity; in our experimental protocol, a flash of light with intensity ≥2.4 × 108 photons/cm2/s shortened the latency, especially for Ca2+ spikes evoked by scotopic light intensities. This pattern closely resembled what was reported for NO-mediated changes in cone BC sensitivity in mammalian retinas: NO increased BC sensitivity to weak inputs but did not alter strong ones (Snellman and Nawy, 2004). However, our results could also be explained by an NO-mediated effect localized to the Mb terminals, in which NO via S-nitrosylation reaction caused a shift in ICa activation that resulted in reduction of Ca2+ spike threshold, leading to shortened first spike delay in response to weak inputs.
To test this notion directly, we repeated previous experiments where we observed light-induced changes in the Mb terminal's scotopic responses under conditions that prevented S-nitrosylation by including NEM (1 mm) in the recording pipette. We found that, in the presence of NEM, the latency of dim scotopic green responses (1.6 × 107 photons/cm2/s) was slightly increased in 3 of 4 experiments (Fig. 10Ai,Aii) and was reduced in 1 of 4 experiments after a bright scotopic (2.4 × 108 photons/cm2/s) green flash (Fig. 10Aiii). On average, no significant change was detected (control: 243.04 ± 19.09 ms vs second: 246.06 ± 16.05 ms, n = 4; Fig. 10Aiii; p = 0.6, paired Student's t test). To test whether a photopic-sensitizing stimulus also alters the scotopic response latency in Mb terminals in an NEM-sensitive manner, we performed a similar experiment using 500-ms-long flashes of bright red (λ = 660 nm) light with intensity of 5 × 1013 photons/cm2/s to sensitize scotopic green flash (4.2 × 107 photons/cm2/s) responses. In the presence of intracellular NEM (1 mm), no reduction of spike latency was observed in response to scotopic green stimulations (Fig. 10Bi,Bii) in any tested cell (n = 3) (Fig. 10Biii; p = 0.1, paired Student's t test). However, there is reduction of light-evoked Ca2+ spikes in the presence of intracellular NEM in both sets of experiment (Fig. 10Ai,Bi), indicating a possible inhibitory NEM effect on dendritic signaling in addition to preventing S-nitrosylation in intact Mbs (Shapiro et al., 1994). It is important to note that light-induced modulation of rod-mediated dim light responses recorded at the Mb terminals could be influenced by multiple upstream NO-mediated processes acting on (1) rods (Kurenny et al., 1994), (2) cones (Savchenko et al., 1997), and/or (3) horizontal cells (Baldridge and Fischer, 2001), making the use of NOS inhibitor TRIM in these experiments inappropriate. Nonetheless, it is unlikely that the NO-mediated, cGMP-dependent increase in Mb sensitivity for weak inputs (Snellman and Nawy, 2004) played a role in modulation of dim light responses because, unlike in horizontal cells, NO donor failed to visibly increase cGMP levels in Mb terminals in the goldfish retina, even in the presence of PDE blocker (Baldridge and Fischer, 2001).
Figure 10.

Both bright scotopic and photopic light induced modulation of scotopic light responses in Mb terminals in an NEM-sensitive manner. Ai, Including NEM (1 mm) in the pipette solution prevented bright scotopic light stimulation (500 ms, λ = 505 nm, 2.4 × 108 photons/cm2/s) from reducing the latency of dim scotopic green light (500 ms, λ = 505 nm, 1.6 × 107 photons/cm2/s) responses of Mb terminals. Membrane potentials at the beginning of traces shown in Ai are −48.7 mV (black) and −48.4 mV (red). Aii, Enlargement of boxed area in Ai illustrating that the delay of dim scotopic green light-induced Ca2+ spikes, after the bright scotopic green stimulation, remained similar to that of control in the presence of NEM. Membrane potentials at the beginning of traces are −49.5 mV (black) and −47.6 mV (red). Aiii, Pairwise comparison showing no significant difference in latency of scotopic light responses (500 ms, λ = 505 nm, 1.6 × 107 photons/cm2/s) compared before and 5 min after bright scotopic green flash (500 ms, λ = 505 nm, 2.4 × 108 photons/cm2/s) in the presence of NEM. Black circles represent control; red squares represent second scotopic response latency; n = 4. Bi, When NEM (1 mm) was included in the pipette solution the bright red flash (500 ms, 660 nm, 5 × 1013 photons/cm2/s) did not induce a leftward shift in the latency to first spike triggered by scotopic green stimulation (505 nm, 4.2 × 107 photons/cm2/s). Membrane potential at the beginning of both traces shown in Bi is −52 mV. Bii, Enlargement of boxed area in Bi illustrating that the delay of scotopic green light-induced Ca2+ spikes, after the bright red stimulation, remained similar to that of control in the presence of NEM. Membrane potentials at the beginning of traces are −48 mV (red) and −50 mV (black). Biii, Pairwise comparison showing no significant difference in latency of scotopic light responses (505 nm, 4.2 × 107 photons/cm2/s) compared before and 5 min after bright red flash in the presence of NEM. Black circles represent control; red squares represent second scotopic response latency; n = 3. Data are mean ± SEM.
Discussion
The present study describes a novel NO-dependent modulatory mechanism that alters the output of Mb-type BCs. The major findings of this investigation were as follows: (1) Ca2+ spike threshold in Mb terminals is subject to activity-dependent plasticity; (2) glutamate released by strong depolarization of a single Mb terminal is sufficient to trigger generation and delivery of NO; (3) NO can act in a retrograde manner to lower ICa threshold in strongly stimulated Mb terminals via an NEM-sensitive S-nitrosylation mechanism; (4) Mb terminals subjected to endogenous or exogenous NO show weighted potentiation: they respond to weak depolarizing inputs with enhanced glutamate output while maintaining input–output ratio (gain) for strong stimuli; and (5) exposure to light with intensity ≥2.4 × 108 photons/cm2/s induced weighted potentiation of scotopic responses in Mb terminals (expressed as reduced Ca2+ spike delay) in an NEM-sensitive manner.
NO modulation of retinal signaling
In vertebrate retinas, NO is synthetized mainly by the neuronal-type NO synthase (Dawson et al., 1991; Tsumamoto et al., 2002), although endothelial (Haverkamp et al., 1999) as well as the inducible (Palamalai et al., 2006) types are also present. Extensive investigations using immunohistochemistry and in situ hybridization detected these enzymes in subtypes of all retinal cell classes with species-dependent patterns (Vielma et al., 2012). The best known retinal effect of NO is the uncoupling of gap junctions between horizontal cells in a variety of species (DeVries and Schwartz, 1989; Lu and McMahon, 1997; Daniels and Baldridge, 2011) that is mediated by increased intracellular cGMP via soluble guanylate cyclase. Similarly, gap junctions between AII ACs and BCs are regulated by NO in a cGMP-dependent manner (Mills and Massey, 1995). Indeed, a number of reports demonstrated increased retinal cGMP synthesis in response to NO donor application (Blute et al., 1998; Gotzes et al., 1998; Baldridge and Fischer, 2001; Sáenz et al., 2002). In addition, NO-triggered increases in cGMP can open CNGCs and in turn trigger transmitter release from cones in salamander (Rieke and Schwartz, 1994) and lizard (Savchenko et al., 1997) retinas.
Not all actions of NO are mediated through cGMP-dependent pathways in the retina: NO was shown to modulate ICa of rods in salamander independent of cGMP (Kurenny et al., 1994). Most interestingly, the activation threshold of ICa in salamander rods was shifted to the left, resulting in a decrease of V1/2 by −4.3 ± 0.6 mV (Kurenny et al., 1994). Those results are strikingly similar to our data (Fig. 6A, V50%: ∼−5.5 mV); thus, our findings and conclusions support the speculation of Kurenny et al. (1994), suggesting S-nitrosylation as a possible mechanism by which NO could alter ICa threshold in salamander rods. The similarities between the results of the two studies suggest that ICa modulation through NO-triggered S-nitrosylation reactions could be a ubiquitous regulatory process in the vertebrate retina.
Can NO-mediated S-nitrosylation in Mbs occur under physiological conditions?
Direct nitrosylation of thiol side-chains of cysteine residues by NO plays an important role in numerous physiological processes by triggering dynamic conformational changes, which affect protein–protein interactions or influence protein location (Ahern et al., 2002). Yet, S-nitrosylation in the nervous system is particularly characteristic during periods of oxidative stress and in neurodegenerative diseases in which NO levels are elevated (Nakamura et al., 2013). We were able to trigger this NO pathway by endogenous synaptic glutamate, released from a single presynaptic terminal, which resulted in the retrograde modulation of ICa through S-nitrosylation reactions at the same presynaptic Mb terminals. However, the depolarizing ramp protocols that were proven to trigger this pathway, and in turn consistently shifted ICa threshold in an NEM-sensitive manner (Figs. 1 and 2Bi,Bii), were robust, and Mb terminals were depolarized far above their physiologically relevant maximal membrane potential (∼−20 mV) (Saito and Kujiraoka, 1982; Protti et al., 2000). Importantly, we were able to trigger changes in dim (∼107 photons/cm2/s) light-evoked Ca2+ spike responses of Mb terminals by brighter (≥2.4 × 108 photons/cm2/s) light: the sensitizing bright light decreased the latency of the first light-evoked Ca2+ spike in an NEM-sensitive manner (Fig. 10), consistent with a light-evoked decrease in ICa activation threshold through S-nitrosylation reactions.
Precise measurement of increases in membrane capacitance associated with our standard voltage ramp protocol was problematic because of the long tail currents (Gillis, 2000); but based on our best measurements (n = 3), it was ∼350 fF. Direct measurement of light-evoked membrane capacitance increase of intact Mbs is also complicated because of their complex morphology (Mennerick et al., 1997) and the slowly decaying light-evoked synaptic conductances (Saito and Kujiraoka, 1982). However, when membrane potential waveforms triggered by 500 ms bright red light flash (similar to the ones used to “sensitize” the Mb terminals; Fig. 8Bi,Bii,D) recorded from the axon terminal of intact Mbs were used as command potentials of voltage-clamp protocols in separate experiments, they triggered membrane capacitance increase of 275 ± 48 fF (n = 10) in axotomized Mb terminals (Lipin and Vigh, 2013). Therefore, the magnitude of depolarizing ramp-evoked glutamate release appears to be comparable with that triggered by bright light stimuli from Mbs.
Consequences of NO-mediated weighted potentiation for glutamate release from Mb terminals
Numerous neurotransmitters inhibit presynaptic ICa via G-protein coupled receptors by inducing changes in the voltage dependence of ICa, resulting in channel activation at more depolarized potentials (Bean, 1989). Shifting the activation threshold of ICa to more depolarized potentials is an extremely potent way of reducing transmitter release (Catterall and Few, 2008) because of the power law of synaptic transmission (Katz and Miledi, 1970). Less is known about positive modulation of ICa by transmitters; to this end, only the neurosteroid pregnenolone sulfate was shown to potentiate presynaptic release at the calyx of Held by a mechanism that involved shifting ICa activation to the left (Hige et al., 2006). Despite the extensive literature published on activity-dependent short- and long-term synaptic plasticity in the CNS, to date only one reference described activity-dependent positive modulation of presynaptic ICa activation: at the calyx of Held synapse, tetanic stimulation shifted the half-activation voltage (V1/2) of the presynaptic ICa by −4.1 mV (Cuttle et al., 1998), analogous to our results. Although the underlying mechanism was not identified beyond establishing that it was independent of G-proteins, Cuttle et al. (1998) showed that a small shift in ICa activation to more negative potentials can significantly increase transmitter output from the presynaptic calyx, which is entirely consistent with our data (Fig. 6A,C,F).
Although Mb terminals release glutamate at ribbon synapses, which are specialized to release transmitters continuously over long periods and capable to code changes in illumination rather quickly (Sterling and Matthews, 2005; Thoreson, 2007), they are subject to short-term synaptic plasticity known as paired pulse depression of glutamate release (von Gersdorff and Matthews, 1997). Activity-dependent long-term potentiation of the BC → GC synapse has also been shown in the developing zebrafish retina (Wei et al., 2012), although this phenomenon disappears with maturation. Activity-dependent short-term potentiation of release seen at central synapses (Habets and Borst, 2006) from Mb or other BCs has not been reported so far. Importantly, the effect of the left-shifted ICa activation on the input–output ratio of Mbs is very unique: this weighted potentiation not only enhanced ICa triggered by weak inputs, but the enhancement gradually decreased as the input strength increased, limiting the potentiation to the physiologically relevant membrane potentiation range. The power law of synaptic transmission (Katz and Miledi, 1970) and the particularly high level of Ca2+ cooperativity in mediating release from Mb terminals (Heidelberger et al., 1994) further magnify this phenomenon at the level of glutamate output from Mb terminals: NO-mediated increase of ICa resulted in increased exocytosis at −30 mV; however, the small (yet significant) increase in ICa amplitude at −20 mV did not cause consequent ΔCm increase (Fig. 6D,F).
Intrinsic adaptation to luminance is mediated through synaptic depression of glutamate release from rod BCs (Oesch and Diamond, 2011), resulting in Ca2+ channel inactivation and vesicle depletion (Jarsky et al., 2011). It is unclear whether the rod-driven circuitry in the mammalian retina is subject to NO-mediated or other types of synaptic potentiation. Nonetheless, mammalian ON cone BCs show NO-dependent, cGMP-mediated weighted sensitization: upon NO donor exposure, their response to weak stimuli increased, whereas responses to strong inputs remained unaltered (Snellman and Nawy, 2004). We discovered a novel form of activity-dependent, NO-mediated synaptic plasticity expressed at the axon terminal of the rod-dominated Mb-type bipolar cell: it is triggered by large depolarization and, through synaptic communication, it causes positive modulation of voltage-gated Ca2+ channels, particularly in response to consecutive weak inputs. Because Mbs in the fish retina make direct synaptic contacts with GCs (Witkovsky and Dowling, 1969; Marc and Liu, 2000; Palmer, 2010), we propose that this novel mechanism enhances the representation of weak rod signals at Mb → GC synapses under bright scotopic and mesopic light conditions. The selective potentiation of weak signals may counter use-dependent depression of glutamate release from BCs during light adaptation and prevent the loss of critical visual information carried by dim scotopic signals.
Footnotes
This work was supported by the National Institutes of Health-National Eye Institute Grant EY019051 to J.V. We thank Shannon K. Gallagher and Vinod Damodaran for their assistance in the completion of this study.
The authors declare no competing financial interests.
References
- Ahern GP, Klyachko VA, Jackson MB. cGMP and S-nitrosylation: two routes for modulation of neuronal excitability by NO. Trends Neurosci. 2002;25:510–517. doi: 10.1016/S0166-2236(02)02254-3. [DOI] [PubMed] [Google Scholar]
- Baccus SA, Meister M. Fast and slow contrast adaptation in retinal circuitry. Neuron. 2002;36:909–919. doi: 10.1016/S0896-6273(02)01050-4. [DOI] [PubMed] [Google Scholar]
- Baden T, Esposti F, Nikolaev A, Lagnado L. Spikes in retinal bipolar cells phase-lock to visual stimuli with millisecond precision. Curr Biol. 2011;21:1859–1869. doi: 10.1016/j.cub.2011.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baden T, Berens P, Bethge M, Euler T. Spikes in mammalian bipolar cells support temporal layering of the inner retina. Curr Biol. 2013a;23:48–52. doi: 10.1016/j.cub.2012.11.006. [DOI] [PubMed] [Google Scholar]
- Baden T, Euler T, Weckström M, Lagnado L. Spikes and ribbon synapses in early vision. Trends Neurosci. 2013b;36:480–488. doi: 10.1016/j.tins.2013.04.006. [DOI] [PubMed] [Google Scholar]
- Baldridge WH, Fischer AJ. Nitric oxide donor stimulated increase of cyclic GMP in the goldfish retina. Vis Neurosci. 2001;18:849–856. [PubMed] [Google Scholar]
- Bean BP. Neurotransmitter inhibition of neuronal calcium currents by changes in channel voltage dependence. Nature. 1989;340:153–156. doi: 10.1038/340153a0. [DOI] [PubMed] [Google Scholar]
- Blute TA, Velasco P, Eldred WD. Functional localization of soluble guanylate cyclase in turtle retina: modulation of cGMP by nitric oxide donors. Vis Neurosci. 1998;15:485–498. doi: 10.1017/s0952523898153075. [DOI] [PubMed] [Google Scholar]
- Burrone J, Neves G, Gomis A, Cooke A, Lagnado L. Endogenous calcium buffers regulate fast exocytosis in the synaptic terminal of retinal bipolar cells. Neuron. 2002;33:101–112. doi: 10.1016/S0896-6273(01)00565-7. [DOI] [PubMed] [Google Scholar]
- Busskamp V, Duebel J, Balya D, Fradot M, Viney TJ, Siegert S, Groner AC, Cabuy E, Forster V, Seeliger M, Biel M, Humphries P, Paques M, Mohand-Said S, Trono D, Deisseroth K, Sahel JA, Picaud S, Roska B. Genetic reactivation of cone photoreceptors restores visual responses in retinitis pigmentosa. Science. 2010;329:413–417. doi: 10.1126/science.1190897. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Few AP. Calcium channel regulation and presynaptic plasticity. Neuron. 2008;59:882–901. doi: 10.1016/j.neuron.2008.09.005. [DOI] [PubMed] [Google Scholar]
- Cuttle MF, Tsujimoto T, Forsythe ID, Takahashi T. Facilitation of the presynaptic calcium current at an auditory synapse in rat brainstem. J Physiol. 1998;512:723–729. doi: 10.1111/j.1469-7793.1998.723bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damodaran VB, Place LW, Kipper MJ, Reynolds MM. Enzymatically degradable nitric oxide releasing S-nitrosated dextran thiomers for biomedical applications. J Mater Chem. 2012;22:23038–23048. doi: 10.1039/c2jm34834a. [DOI] [Google Scholar]
- Daniels BA, Baldridge WH. The light-induced reduction of horizontal cell receptive field size in the goldfish retina involves nitric oxide. Vis Neurosci. 2011;28:137–144. doi: 10.1017/S0952523810000490. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Bredt DS, Fotuhi M, Hwang PM, Snyder SH. Nitric-oxide synthase and neuronal NADPH diaphorase are identical in brain and peripheral-tissues. Proc Natl Acad Sci U S A. 1991;88:7797–7801. doi: 10.1073/pnas.88.17.7797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demb JB. Multiple mechanisms for contrast adaptation in the retina. Neuron. 2002;36:781–783. doi: 10.1016/S0896-6273(02)01100-5. [DOI] [PubMed] [Google Scholar]
- DeVries SH, Schwartz EA. Modulation of an electrical synapse between solitary pairs of catfish horizontal cells by dopamine and 2nd messengers. J Physiol. 1989;414:351–375. doi: 10.1113/jphysiol.1989.sp017692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling JE. The retina: an approachable part of the brain. Cambridge, MA: Belknap; 1987. [Google Scholar]
- Dreosti E, Esposti F, Baden T, Lagnado L. In vivo evidence that retinal bipolar cells generate spikes modulated by light. Nat Neurosci. 2011;14:951–952. doi: 10.1038/nn.2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldred WD, Blute TA. Imaging of nitric oxide in the retina. Vision Res. 2005;45:3469–3486. doi: 10.1016/j.visres.2005.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Yu S, Lasell TK, Jadhav AP, Macia E, Chardin P, Melancon P, Roth M, Mitchison T, Kirchhausen T. Exo1: a new chemical inhibitor of the exocytic pathway. Proc Natl Acad Sci U S A. 2003;100:6469–6474. doi: 10.1073/pnas.0631766100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillis KD. Admittance-based measurement of membrane capacitance using the EPC-9 patch-clamp amplifier. Pflugers Arch. 2000;439:655–664. doi: 10.1007/s004240050990. [DOI] [PubMed] [Google Scholar]
- Giove TJ, Deshpande MM, Eldred WD. Identification of alternate transcripts of neuronal nitric oxide synthase in the mouse retina. J Neurosci Res. 2009;87:3134–3142. doi: 10.1002/jnr.22133. [DOI] [PubMed] [Google Scholar]
- Gotzes S, de Vente J, Müller F. Nitric oxide modulates cGMP levels in neurons of the inner and outer retina in opposite ways. Vis Neurosci. 1998;15:945–955. doi: 10.1017/s0952523898155141. [DOI] [PubMed] [Google Scholar]
- Griguer C, Fuchs PA. Voltage-dependent potassium currents in cochlear hair cells of the embryonic chick. J Neurophysiol. 1996;75:508–513. doi: 10.1152/jn.1996.75.1.508. [DOI] [PubMed] [Google Scholar]
- Habets RL, Borst JG. An increase in calcium influx contributes to post-tetanic potentiation at the rat calyx of held synapse. J Neurophysiol. 2006;96:2868–2876. doi: 10.1152/jn.00427.2006. [DOI] [PubMed] [Google Scholar]
- Haverkamp S, Kolb H, Cuenca N. Endothelial nitric oxide synthase (eNOS) is localized to Müller cells in all vertebrate retinas. Vision Res. 1999;39:2299–2303. doi: 10.1016/S0042-6989(99)00007-3. [DOI] [PubMed] [Google Scholar]
- Haverkamp S, Specht D, Majumdar S, Zaidi NF, Brandstätter JH, Wasco W, Wässle H, Tom Dieck S. Type 4 OFF cone bipolar cells of the mouse retina express calsenilin and contact cones as well as rods. J Comp Neurol. 2008;507:1087–1101. doi: 10.1002/cne.21612. [DOI] [PubMed] [Google Scholar]
- Hayashida Y, Rodríguez CV, Ogata G, Partida GJ, Oi H, Stradleigh TW, Lee SC, Colado AF, Ishida AT. Inhibition of adult rat retinal ganglion cells by D1-type dopamine receptor activation. J Neurosci. 2009;29:15001–15016. doi: 10.1523/JNEUROSCI.3827-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidelberger R, Matthews G. Calcium influx and calcium current in single synaptic terminals of goldfish retinal bipolar neurons. J Physiol. 1992;447:235–256. doi: 10.1113/jphysiol.1992.sp019000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidelberger R, Heinemann C, Neher E, Matthews G. Calcium dependence of the rate of exocytosis in a synaptic terminal. Nature. 1994;371:513–515. doi: 10.1038/371513a0. [DOI] [PubMed] [Google Scholar]
- Henry D, Burke S, Shishido E, Matthews G. Retinal bipolar neurons express the cyclic nucleotide-gated channel of cone photoreceptors. J Neurophysiol. 2003;89:754–761. doi: 10.1152/jn.00771.2002. [DOI] [PubMed] [Google Scholar]
- Hige T, Fujiyoshi Y, Takahashi T. Neurosteroid pregnenolone sulfate enhances glutamatergic synaptic transmission by facilitating presynaptic calcium currents at the calyx of Held of immature rats. Eur J Neurosci. 2006;24:1955–1966. doi: 10.1111/j.1460-9568.2006.05080.x. [DOI] [PubMed] [Google Scholar]
- Hille B. Ion channels of excitable membranes. Sunderland, MA: Sinauer; 2001. Voltage-dependent gates have gating charge and gating current; pp. 56–59. [Google Scholar]
- Hirasawa H, Kaneko A. pH changes in the invaginating synaptic cleft mediate feedback from horizontal cells to cone photoreceptors by modulating Ca2+ channels. J Gen Physiol. 2003;122:657–671. doi: 10.1085/jgp.200308863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirooka K, Kourennyi DE, Barnes S. Calcium channel activation facilitated by nitric oxide in retinal ganglion cells. J Neurophysiol. 2000;83:198–206. doi: 10.1152/jn.2000.83.1.198. [DOI] [PubMed] [Google Scholar]
- Hrabie JA, Klose JR, Wink DA, Keefer LK. New nitric oxide-releasing zwitterions derived from polyamines. J Org Chem. 1993;58:1472–1476. doi: 10.1021/jo00058a030. [DOI] [Google Scholar]
- Jarsky T, Cembrowski M, Logan SM, Kath WL, Riecke H, Demb JB, Singer JH. A synaptic mechanism for retinal adaptation to luminance and contrast. J Neurosci. 2011;31:11003–11015. doi: 10.1523/JNEUROSCI.2631-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joselevitch C, Kamermans M. Interaction between rod and cone inputs in mixed-input bipolar cells in goldfish retina. J Neurosci Res. 2007;85:1579–1591. doi: 10.1002/jnr.21249. [DOI] [PubMed] [Google Scholar]
- Joselevitch C, Kamermans M. Retinal parallel pathways: seeing with our inner fish. Vision Res. 2009;49:943–959. doi: 10.1016/j.visres.2008.07.019. [DOI] [PubMed] [Google Scholar]
- Kaneko A, Tachibana M. A voltage-clamp analysis of membrane currents in solitary bipolar cells dissociated from carassius-auratus. J Physiol. 1985;358:131–152. doi: 10.1113/jphysiol.1985.sp015544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastner DB, Baccus SA. Coordinated dynamic encoding in the retina using opposing forms of plasticity. Nat Neurosci. 2011;14:1317–1322. doi: 10.1038/nn.2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz B, Miledi R. Further study of the role of calcium in synaptic transmission. J Physiol. 1970;207:789–801. doi: 10.1113/jphysiol.1970.sp009095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KJ, Rieke F. Temporal contrast adaptation in the input and output signals of salamander retinal ganglion cells. J Neurosci. 2001;21:287–299. doi: 10.1523/JNEUROSCI.21-01-00287.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krizaj D. Mesopic state: cellular mechanisms involved in pre- and post-synaptic mixing of rod and cone signals. Microsc Res Tech. 2000;50:347–359. doi: 10.1002/1097-0029(20000901)50:5<347::AID-JEMT4>3.0.CO%3B2-D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurenny DE, Moroz LL, Turner RW, Sharkey KA, Barnes S. Modulation of ion channels in rod photoreceptors by nitric-oxide. Neuron. 1994;13:315–324. doi: 10.1016/0896-6273(94)90349-2. [DOI] [PubMed] [Google Scholar]
- Leuranguer V, Dirksen RT, Beam KG. Potentiated L-type Ca2+ channels rectify. J Gen Physiol. 2003;121:541–550. doi: 10.1085/jgp.200308833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipin M, Vigh J. Quantifying the effect of light activated outer and inner retinal inhibitory pathways on exocytosis from mixed bipolar cells. ARVO meeting, Seattle WA. Invest Opthalmol Vis Sci. 2013;540 E-Abstract 6155. [Google Scholar]
- Loew ER, McFarland WN. The underwater visual environment. In: Douglas RH, Djamgoz MBA, editors. The visual system of fish. New York: Chapman and Hall; 1990. pp. 1–43. [Google Scholar]
- Logiudice L, Henry D, Matthews G. Identification of calcium channel alpha1 subunit mRNA expressed in retinal bipolar neurons. Mol Vis. 2006;12:184–189. [PMC free article] [PubMed] [Google Scholar]
- Lu C, McMahon DG. Modulation of hybrid bass retinal gap junctional channel gating by nitric oxide. J Physiol. 1997;499:689–699. doi: 10.1113/jphysiol.1997.sp021961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maragos CM, Morley D, Wink DA, Dunams TM, Saavedra JE, Hoffman A, Bove AA, Isaac L, Hrabie JA, Keefer LK. Complexes of .NO with nucleophiles as agents for the controlled biological release of nitric oxide: vasorelaxant effects. J Med Chem. 1991;34:3242–4247. doi: 10.1021/jm00115a013. [DOI] [PubMed] [Google Scholar]
- Marc RE, Liu W. Fundamental GABAergic amacrine cell circuitries in the retina: nested feedback, concatenated inhibition, and axosomatic synapses. J Comp Neurol. 2000;425:560–582. doi: 10.1002/1096-9861(20001002)425:4<560::AID-CNE7>3.0.CO%3B2-D. [DOI] [PubMed] [Google Scholar]
- Marty A, Neher E. Tight-seal whole-cell recording. In: Sakmann B, Neher E, editors. Single-channel recording. New York: Plenum; 1995. pp. 31–52. [Google Scholar]
- McFarland WN, Munz FW. Part II: the photic environment of clear tropical seas during the day. Vision Res. 1975;15:1063–1070. doi: 10.1016/0042-6989(75)90002-4. [DOI] [PubMed] [Google Scholar]
- Mennerick S, Matthews G. Rapid calcium-current kinetics in synaptic terminals of goldfish retinal bipolar neurons. Vis Neurosci. 1998;15:1051–1056. doi: 10.1017/S0952523898156043. [DOI] [PubMed] [Google Scholar]
- Mennerick S, Zenisek D, Matthews G. Static and dynamic membrane properties of large-terminal bipolar cells from goldfish retina: experimental test of a compartment model. J Neurophysiol. 1997;78:51–62. doi: 10.1152/jn.1997.78.1.51. [DOI] [PubMed] [Google Scholar]
- Mills SL, Massey SC. Differential properties of 2 gap junctional pathways made by AII amacrine cells. Nature. 1995;377:734–737. doi: 10.1038/377734a0. [DOI] [PubMed] [Google Scholar]
- Munz FW, McFarland WN. The significance of spectral position in the rhodopsins of tropical marine fishes. Vision Res. 1973;13:1829–1874. doi: 10.1016/0042-6989(73)90060-6. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Tu S, Akhtar MW, Sunico CR, Okamoto S, Lipton SA. Aberrant protein s-nitrosylation in neurodegenerative diseases. Neuron. 2013;78:596–614. doi: 10.1016/j.neuron.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E. Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron. 1998;20:389–399. doi: 10.1016/S0896-6273(00)80983-6. [DOI] [PubMed] [Google Scholar]
- Nikolaev A, Leung KM, Odermatt B, Lagnado L. Synaptic mechanisms of adaptation and sensitization in the retina. Nat Neurosci. 2013;16:934–941. doi: 10.1038/nn.3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oesch NW, Diamond JS. Ribbon synapses compute temporal contrast and encode luminance in retinal rod bipolar cells. Nat Neurosci. 2011;14:1555–1561. doi: 10.1038/nn.2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palamalai V, Darrow RM, Organisciak DT, Miyagi M. Light-induced changes in protein nitration in photoreceptor rod outer segments. Mol Vis. 2006;12:1543–1551. [PubMed] [Google Scholar]
- Palmer MJ. Modulation of Ca2+-activated K+ currents and Ca2+-dependent action potentials by exocytosis in goldfish bipolar cell terminals. J Physiol. 2006;572:747–762. doi: 10.1113/jphysiol.2006.105205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer MJ. Characterization of bipolar cell synaptic transmission in goldfish retina using paired recordings. J Physiol. 2010;588:1489–1498. doi: 10.1113/jphysiol.2009.185850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer MJ, Taschenberger H, Hull C, Tremere L, von Gersdorff H. Synaptic activation of presynaptic glutamate transporter currents in nerve terminals. J Neurosci. 2003;23:4831–4841. doi: 10.1523/JNEUROSCI.23-12-04831.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang JJ, Gao F, Lem J, Bramblett DE, Paul DL, Wu SM. Direct rod input to cone BCs and direct cone input to rod BCs challenge the traditional view of mammalian BC circuitry. Proc Natl Acad Sci U S A. 2010;107:395–400. doi: 10.1073/pnas.0907178107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping Y, Huang H, Zhang XJ, Yang XL. Melatonin potentiates rod signals to ON type bipolar cells in fish retina. J Physiol. 2008;586:2683–2694. doi: 10.1113/jphysiol.2008.152959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protti DA, Flores-Herr N, von Gersdorff H. Light evokes Ca2+ spikes in the axon terminal of a retinal bipolar cell. Neuron. 2000;25:215–227. doi: 10.1016/S0896-6273(00)80884-3. [DOI] [PubMed] [Google Scholar]
- Protti DA, Flores-Herr N, Li W, Massey SC, Wässle H. Light signaling in scotopic conditions in the rabbit, mouse and rat retina: a physiological and anatomical study. J Neurophysiol. 2005;93:3479–3488. doi: 10.1152/jn.00839.2004. [DOI] [PubMed] [Google Scholar]
- Rieke F. Temporal contrast adaptation in salamander bipolar cells. J Neurosci. 2001;21:9445–9454. doi: 10.1523/JNEUROSCI.21-23-09445.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieke F, Rudd ME. The challenges natural images pose for visual adaptation. Neuron. 2009;64:605–616. doi: 10.1016/j.neuron.2009.11.028. [DOI] [PubMed] [Google Scholar]
- Rieke F, Schwartz EA. A cGMP-gated current can control exocytosis at cone synapses. Neuron. 1994;13:863–873. doi: 10.1016/0896-6273(94)90252-6. [DOI] [PubMed] [Google Scholar]
- Sáenz DA, Turjanski AG, Sacca GB, Marti M, Doctorovich F, Sarmiento MI, Estrin DA, Rosenstein RE. Physiological concentrations of melatonin inhibit the nitridergic pathway in the Syrian hamster retina. J Pineal Res. 2002;33:31–36. doi: 10.1034/j.1600-079X.2002.01880.x. [DOI] [PubMed] [Google Scholar]
- Sah P, Davies P. Calcium-activated potassium currents in mammalian neurons. Clin Exp Pharmacol Physiol. 2000;27:657–663. doi: 10.1046/j.1440-1681.2000.03317.x. [DOI] [PubMed] [Google Scholar]
- Saito T, Kujiraoka T. Physiological and morphological identification of 2 types of on-center bipolar cells in the carp retina. J Comp Neurol. 1982;205:161–170. doi: 10.1002/cne.902050207. [DOI] [PubMed] [Google Scholar]
- Saito T, Kondo H, Toyoda JI. Ionic mechanisms of 2 types of on-center bipolar cells in the carp retina: 1. Responses to central illumination. J Gen Physiol. 1979;73:73–90. doi: 10.1085/jgp.73.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaba T, Ishikane H, Tachibana M. Ca2+-activated K+ current at presynaptic terminals of goldfish retinal bipolar cells. Neurosci Res. 1997;27:219–228. doi: 10.1016/S0168-0102(97)01155-3. [DOI] [PubMed] [Google Scholar]
- Saszik S, DeVries SH. A mammalian retinal bipolar cell uses both graded changes in membrane voltage and all-or-nothing Na+ spikes to encode light. J Neurosci. 2012;32:297–307. doi: 10.1523/JNEUROSCI.2739-08.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savchenko A, Barnes S, Kramer RH. Cyclic-nucleotide-gated channels mediate synaptic feedback by nitric oxide. Nature. 1997;390:694–698. doi: 10.1038/37803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro MS, Wollmuth LP, Hille B. Modulation of Ca2+ channels by PTX-sensitive G-proteins is blocked by N-ethylmaleimide in rat sympathetic neurons. J Neurosci. 1994;14:7109–7116. doi: 10.1523/JNEUROSCI.14-11-07109.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer JH, Diamond JS. Sustained Ca2+ entry elicits transient postsynaptic currents at a retinal ribbon synapse. J Neurosci. 2003;23:10923–10933. doi: 10.1523/JNEUROSCI.23-34-10923.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnakis SM, Berry MJ, Warland DK, Bialek W, Meister M. Adaptation of retinal processing to image contrast and spatial scale. Nature. 1997;386:69–73. doi: 10.1038/386069a0. [DOI] [PubMed] [Google Scholar]
- Snellman J, Nawy S. cGMP-dependent kinase regulates response sensitivity of the mouse on bipolar cell. J Neurosci. 2004;24:6621–6628. doi: 10.1523/JNEUROSCI.1474-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterling P. How retinal circuits optimize the transfer of visual information. In: Chalupa LM, Werner JS, editors. The visual neurosciences. Cambridge, MA: MIT; 2003. pp. 234–259. [Google Scholar]
- Sterling P, Matthews G. Structure and function of ribbon synapses. Trends Neurosci. 2005;28:20–29. doi: 10.1016/j.tins.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Tachibana M. Regulation of transmitter release from retinal bipolar cells. Prog Biophys Mol Biol. 1999;72:109–133. doi: 10.1016/S0079-6107(99)00003-6. [DOI] [PubMed] [Google Scholar]
- Tachibana M, Kaneko A. Gamma-aminobutyric-acid exerts a local inhibitory-action on the axon terminal of bipolar cells: evidence for negative feedback from amacrine cells. Proc Natl Acad Sci U S A. 1987;84:3501–3505. doi: 10.1073/pnas.84.10.3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoreson WB. Kinetics of synaptic transmission at ribbon synapses of rods and cones. Mol Neurobiol. 2007;36:205–223. doi: 10.1007/s12035-007-0019-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsumamoto Y, Yamashita K, Takumida M, Okada K, Mukai S, Shinya M, Yamashita H, Mishima HK. In situ localization of nitric oxide synthase and direct evidence of NO production in rat retinal ganglion cells. Brain Res. 2002;933:118–129. doi: 10.1016/S0006-8993(02)02289-8. [DOI] [PubMed] [Google Scholar]
- Vergara C, Latorre R, Marrion NV, Adelman JP. Calcium-activated potassium channels. Curr Opin Neurobiol. 1998;8:321–329. doi: 10.1016/S0959-4388(98)80056-1. [DOI] [PubMed] [Google Scholar]
- Vielma AH, Retamal MA, Schmachtenberg O. Nitric oxide signaling in the retina: what have we learned in two decades? Brain Res. 2012;1430:112–125. doi: 10.1016/j.brainres.2011.10.045. [DOI] [PubMed] [Google Scholar]
- Vigh J, von Gersdorff H. Prolonged reciprocal signaling via NMDA and GABA receptors at a retinal ribbon synapse. J Neurosci. 2005;25:11412–11423. doi: 10.1523/JNEUROSCI.2203-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigh J, Li GL, Hull C, von Gersdorff H. Long-term plasticity mediated by mGluR1 at a retinal reciprocal synapse. Neuron. 2005;46:469–482. doi: 10.1016/j.neuron.2005.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigh J, Vickers E, von Gersdorff H. Light-evoked lateral GABAergic inhibition at single bipolar cell synaptic terminals is driven by distinct retinal microcircuits. J Neurosci. 2011;31:15884–15893. doi: 10.1523/JNEUROSCI.2959-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Gersdorff H, Matthews G. Calcium-dependent inactivation of calcium current in synaptic terminals of retinal bipolar neurons. J Neurosci. 1996;16:115–122. doi: 10.1523/JNEUROSCI.16-01-00115.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Gersdorff H, Matthews G. Depletion and replenishment of vesicle pools at a ribbon-type synaptic terminal. J Neurosci. 1997;17:1919–1927. doi: 10.1523/JNEUROSCI.17-06-01919.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walraven J, Enroth-Cugell C, Hood DC, MacLeod DIA, Schnapf JL. In visual perception: the neurophysiological foundations. In: Spillman L, Werner JZ, editors. San Diego: Academic; 1990. pp. 53–101. [Google Scholar]
- Wässle H. Parallel processing in the mammalian retina. Nat Rev Neurosci. 2004;5:747–757. doi: 10.1038/nrn1497. [DOI] [PubMed] [Google Scholar]
- Wei HP, Yao YY, Zhang RW, Zhao XF, Du JL. Activity-induced long-term potentiation of excitatory synapses in developing zebrafish retina in vivo. Neuron. 2012;75:479–489. doi: 10.1016/j.neuron.2012.05.031. [DOI] [PubMed] [Google Scholar]
- Werblin FS, Dowling JE. Organization of retina of mudpuppy necturus maculosus: 2. Intracellular recording. J Neurophysiol. 1969;32:339–355. doi: 10.1152/jn.1969.32.3.339. [DOI] [PubMed] [Google Scholar]
- Witkovsky P. Dopamine and retinal function. Doc Ophthalmol. 2004;108:17–40. doi: 10.1023/B:DOOP.0000019487.88486.0a. [DOI] [PubMed] [Google Scholar]
- Witkovsky P, Dowling JE. Synaptic relationships in the plexiform layers of carp retina. Z Zellforsch Mikrosk Anat. 1969;100:60–82. doi: 10.1007/BF00343821. [DOI] [PubMed] [Google Scholar]
- Wong KY, Cohen ED, Dowling JE. Retinal bipolar cell input mechanisms in giant danio: II. Patch-clamp analysis of ON bipolar cells. J Neurophysiol. 2005;93:94–107. doi: 10.1152/jn.00270.2004. [DOI] [PubMed] [Google Scholar]
- Wu SM. Synaptic transmission in the outer retina. Annu Rev Physiol. 1994;56:141–168. doi: 10.1146/annurev.ph.56.030194.001041. [DOI] [PubMed] [Google Scholar]
- Zenisek D, Matthews G. Calcium action potentials in retinal bipolar neurons. Vis Neurosci. 1998;15:69–75. doi: 10.1017/s0952523898151064. [DOI] [PubMed] [Google Scholar]