

Abstract
Alzheimer's disease (AD) is the most prevalent form of dementia in the elderly population. Accumulation, aggregation, and deposition of amyloid-β (Aβ) peptides generated through proteolytic cleavage of amyloid precursor protein (APP) are likely initiating events in the pathogenesis of AD. While Aβ production is accelerated in familial AD, increasing evidence indicates that impaired clearance of Aβ is responsible for late-onset AD. Because Aβ is mainly generated in neurons, these cells are predicted to have the highest risk of encountering Aβ among all cell types in the brain. However, it is still unclear whether they are also involved in Aβ clearance. Here we show that receptor-mediated endocytosis in neurons by the low-density lipoprotein receptor-related protein 1 (LRP1) plays a critical role in brain Aβ clearance. LRP1 is known to be an endocytic receptor for multiple ligands including Aβ. Conditional knock-out of Lrp1 in mouse forebrain neurons leads to increased brain Aβ levels and exacerbated amyloid plaque deposition selectively in the cortex of amyloid model APP/PS1 mice without affecting Aβ production. In vivo microdialysis studies demonstrated that Aβ clearance in brain interstitial fluid is impaired in neuronal Lrp1 knock-out mice. Because the neuronal LRP1-deletion did not affect the mRNA levels of major Aβ degrading enzymes, neprilysin and insulin-degrading enzyme, the disturbed Aβ clearance is likely due to the suppression of LRP1-mediated neuronal Aβ uptake and degradation. Together, our results demonstrate that LRP1 plays an important role in receptor-mediated clearance of Aβ and indicate that neurons not only produce but also clear Aβ.
Introduction
Alzheimer's disease (AD) has emerged as the most prevalent form of late-life dementia in humans. Accumulation, aggregation, and deposition of amyloid-β (Aβ) peptides generated through proteolytic cleavage of amyloid precursor protein (APP) are major features of AD pathogenesis (Hardy and Selkoe, 2002, Blennow et al., 2006, Zheng and Koo, 2011). The toxic forms of Aβ aggregates, oligomers, and fibrils, injure synapses and neurons, which leads to synaptic dysfunction, neurodegeneration, and cognitive impairments (Hardy and Selkoe, 2002, Blennow et al., 2006, Koffie et al., 2011). The majority of AD cases are sporadic and late onset, and the disease is thought to be initiated by Aβ accumulation in the brain through an overall impairment in Aβ clearance (Mawuenyega et al., 2010). A major pathway through which Aβ is cleared from the brain is cellular uptake and subsequent degradation (Bu, 2009). Several studies have shown that endocytosed Aβ can be delivered to lysosomes for degradation (Mandrekar et al., 2009, Li et al., 2012). Recently, we have demonstrated that the low-density lipoprotein receptor-related protein 1 (LRP1) mediates Aβ cellular uptake by regulating its endocytosis in neuronal cells in culture (Kanekiyo et al., 2011). LRP1 is abundantly expressed in various brain cell types including neurons and glial cells in brain parenchyma, and smooth muscle cells and pericytes in cerebral vasculature to mediate cellular uptake of an array of ligands including APP, apolipoprotein E, α2-macroglobulin, and receptor-associated protein (RAP), all of which function in either Aβ production or clearance (Herz and Strickland, 2001, Bu, 2009). Furthermore, LRP1 can couple with other cell-surface signaling receptors to regulate signal transduction (Herz and Strickland, 2001).
Previous studies have shown that externally injected radioisotope-labeled Aβ is eliminated from the brain in mice in an LRP1-dependent manner (Shibata et al., 2000). Our recent study has also shown that conditional knock-out of the Lrp1 gene in vascular smooth muscle cells exacerbated Aβ deposition in an amyloid mouse model (Kanekiyo et al., 2012). Despite increasing evidence to support important roles of LRP1 in Aβ metabolism, whether LRP1-mediated Aβ clearance plays a role in neurons in vivo is not clear. Here, using Lrp1 conditional forebrain neuronal knock-out mice, we show definitively that LRP1 plays an important role in neuronal Aβ clearance in the brain. More importantly, we demonstrate that neurons are involved in both Aβ production and clearance.
Materials and Methods
Aβ peptides.
Aβ42 and 5(6)-carboxyfluorescein (FAM)-Aβ42 were purchased from AnaSpec. Dry peptide was pretreated with neat trifluoroacetic acid, distilled under nitrogen, washed with 1,1,1,3,3,3-hexafluoro-2-propanol, distilled under nitrogen, and stored at −20°C. Aβ peptides were freshly dissolved in dimethylsulfoxide at 200 μm for each experiment.
Cell culture and LRP1-knockdown by siRNA.
Human neuroblastoma NT2 cells were cultured in DMEM with 10% fetal bovine serum under standard culture conditions. Knockdown of LRP1 was performed as described previously (Li et al., 2003). Single-stranded, LRP1-specific, sense and antisense RNA oligonucleotides were synthesized by Ambion. Cells were transfected with siRNA (100 nm) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's specifications and used for analysis 48 h after transfection.
Confocal microscopy.
Cells were cultured on eight-well slides (Lab-Tek II Chamber Slide System; Nalge Nunc International) at 37°C for at least 24 h before experiments. After incubation with FAM-Aβ42 (1 μm) at 37°C for 24 h, fluorescence associated with Aβ was observed by confocal laser-scanning fluorescence microscopy (model LSM 510 invert; Carl Zeiss). LysoTracker (50 nm; Molecular Probe) was added 30 min before confocal imaging.
Animals and tissue preparation.
Forebrain neuron-specific Lrp1-KO mice (Lrp1−/−) were generated by breeding the Lrp1 floxed mice with α-calcium-calmodulin-dependent kinase II (αCaMKII)-driven Cre recombinase mice (Liu et al., 2007). Littermates of male Lrp1−/− mice (LRP1flox/flox, αCaMKII-Cre+/−) or controls (LRP1flox/flox, αCaMKII-Cre−/−) with B6/C3H mixed background were used. Furthermore, amyloid model APP/PS1 (APPswe/PSEN1ΔE9) mice (Borchelt et al., 1997) were crossed with Lrp1−/− mice. Littermates of male APP/PS1 mice (APP/PS1, Lrp1flox/flox, αCaMKII-Cre−/−) and APP/PS1 mice lacking LRP1 in forebrain neurons, APP/PS1; Lrp1−/− (APP/PS1, Lrp1flox/flox, αCaMKII-Cre+/−) were used for analysis. After perfusion with PBS, brain tissues were dissected and kept frozen at −80°C until further analysis. Some brain tissues were fixed in 10% neutralized formalin for histological analysis. All animal procedures were approved by the Animal Study Committee at Mayo Clinic and in accordance with the regulations of the American Association for the Accreditation of Laboratory Animal Care.
Western blotting.
Samples were lysed in PBS containing 1% Triton X-100 and protease inhibitor cocktail from Roche. Protein concentration was determined in each sample using a Protein Assay kit (Bio-Rad). An equal amount of protein for each sample was used for SDS-PAGE. Immunoreactive bands were detected and quantified using Odyssey Infrared Imaging System (LI-COR Biosciences). Rabbit polyclonal anti-LRP1 antibody was produced in our laboratory. Anti-APP C-terminal, sAPPα, and sAPPβ antibodies were purchased from IBL.
Aβ ELISA.
For measurements of Aβ in APP/PS1 mouse brain, samples were sequentially homogenized in Tris-buffered saline (TBS), TBS buffer containing 1% Triton X-100 (TBS-TX), and then 5 m guanidine in 50 mm Tris-HCl, pH 8.0 (Youmans et al., 2011). Cell-associated Aβ levels were analyzed after being dissolved in guanidine in 50 mm Tris-HCl, pH 8.0. The levels of human Aβ40 and human Aβ42 were determined by ELISA as previously described (Shinohara et al., 2013) using an end-specific monoclonal antibody (13.1.1 for Aβ x-40 and 2.1.3 for Aβ x-42) and a horseradish peroxidase-conjugated detector antibody (Ab5, human Aβ 1–16 specific, all antibodies were produced in-house by the Mayo Clinic).
CTFβ ELISA.
The levels of APP-CTFβ were determined by ELISA on TBS with 1% Triton X-100 fraction of the mouse bran lysates using a rabbit anti-C terminus of APP capture antibody (a gift from Dr. Pritam Das, Mayo Clinic) and biotin-conjugated 82E1 detector antibody (IBL-America). Synthetic peptides, consisted of 15 aa of N terminus of Aβ and 20 aa of C terminus of APP, were used as standards (Shinohara et al., 2013).
Immunohistochemical imaging and image processing.
Paraffin embedded sections were immunostained using pan-Aβ antibody 33.1.1 (human Aβ1–16 specific), and visualized through the Dako Envision Plus visualization system (Chakrabarty et al., 2010). Immunohistochemically stained sections for pan-Aβ were captured using the ScanScope XT image scanner (Aperio Technologies) and analyzed using the ImageScope program (Chakrabarty et al., 2010). The final images and layouts were created using Photoshop CS2 (Adobe). Immunostained total Aβ plaque burdens in the cortex were calculated using the Positive Pixel Count program available with the ImageScope software (Aperio Technologies). All of the above analyses were performed in a blinded manner. For double immunostaining, deparaffinized sections were preincubated with citrate buffer (10 mm sodium citrate buffer with 0.05% Tween 20, pH 6.0) at 95°C for 20 min. They were incubated at 4°C overnight with a rabbit polyclonal anti-LRP1 antibody and a mouse monoclonal anti-NeuN antibody (Millipore) or a mouse monoclonal anti-GFAP antibody (Millipore), followed by Alexa488-conjugated anti-rabbit IgG (Invitrogen) and Alexa568-conjugated anti-mouse IgG (Invitrogen) for 2 h at room temperature. Rabbit polyclonal anti-LRP1 antibody was produced in our laboratory.
Reverse transcription and PCR.
Total RNA was isolated from tissues or cells using RNeasy Mini Kit (Qiagen) and subjected to DNase I digestion to remove contaminating genomic DNA. Total RNA was dissolved in nuclease-free water and stored at −80°C. Reverse transcription was performed using a SuperScript II RNase H-reverse transcriptase (Invitrogen), and the reaction mix was subjected to quantitative real-time (qRT)-PCR to detect levels of the corresponding mouse actin, neprilysin (NEP) and insulin-degrading enzyme (IDE). The set of actin primers (Qiagen) was used as an internal control for each specific gene amplification. The relative levels of expression were quantified and analyzed by using Bio-Rad iCycler iQ software. The real-time value for each sample was averaged and compared using the CT method, where the amount of target RNA (2−ΔΔCT) was normalized to the endogenous actin reference (ΔCT) and related to the amount of target gene in tissue cells, which was set as the calibrator at 1.0. The primers used to amplify target genes by reverse transcription (RT)-PCR and qPCR were as follows: mouse NEP-F (5′-GCA GCC TCA GCC GAA ACT AC-3′), mouse NEP-R (5′-CAC CGT CTC CAT GTT GCA GT-3′); and mouse IDE-F (5′-ACT AAC CTG GTG GTG AAG-3′), mouse IDE-R (5′-GGT CTG GTA TGG GAA ATG-3′).
In vivo microdialysis.
In vivo microdialysis in APP/PS1 and APP/PS1; Lrp1−/− littermates was performed essentially as described previously (Cirrito et al., 2003, 2011). After microdialysis probes were inserted into the cortex, mice recovered for at least 6 h before experiments were performed. Microdialysis perfusion buffer consisted of 0.15% bovine serum albumin (Sigma) in artificial CSF. Basal level of interstitial fluid (ISF) Aβ40 was defined as the mean concentration of Aβ over 7.5 h before drug administration using a constant flow rate of 1.0 μl/min. Compound E (200 nm), a potent γ-secretase inhibitor (by AsisChem) was administered directly to the cortex through the microdialysis probe (reverse microdialysis) into each mouse to rapidly block Aβ production. After treatment with compound E, microdialysis samples were collected every 60 min for 5 h and assayed for Aβ40 by sandwich ELISA similar to (Cirrito et al., 2011), where mHJ2 (mouse-anti-Aβ35–40) was used as the capture antibody and biotinylated mHJ5.1 (mouse-anti-Aβ13–28) as the detection antibody. The half-life of ISF Aβ40 was calculated from the slope of the semilog plot of percentage change in Aβ versus time (Cirrito et al., 2003).
Statistical analysis.
All quantified data represents an average of samples. Statistical significance was determined by two-tailed paired Student's t test, and p < 0.05 was considered significant.
Results
LRP1 mediates Aβ metabolism in neuronal cells
To determine the roles of LRP1 in Aβ metabolism in human neuroblastoma NT2 cells, we first examined if LRP1 knockdown by siRNA affects cellular Aβ uptake (Fig. 1A). We incubated control and LRP1-suppressed NT2 cells with Aβ42 for 8 h. ELISA revealed that cell-associated Aβ42 levels were significantly decreased in LRP1-suppressed cells (Fig. 1A). Consistent with our ELISA results, we observed less internalized Aβ in LRP1-suppressed NT2 cells in the lysosomal compartments by confocal microscopy when cells were incubated with FAM)-Aβ42 for 24 h (Fig. 1B). To determine whether internalized Aβ is degraded, NT2 cells were incubated with Aβ42 for 2 h at 37°C. The Aβ-containing media were then replaced with medium without Aβ, and the cells were incubated for an additional 8 h before analysis of cell-associated Aβ42 by ELISA. The internalized Aβ was cleared following 8 h of incubation in both control and LRP1-suppressed NT2 cells (Fig. 1C). When Aβ clearance was calculated under each condition, we found that it was decreased in LRP1-suppressed NT2 cells compared with control cells (Fig. 1C). These results demonstrate that LRP1 mediates Aβ uptake and subsequent degradation in neuronal cells.
Figure 1.

LRP1 mediates Aβ uptake and degradation in neuronal cells. A, NT2 cells were transfected with LRP1-siRNA and used for analysis 48 h after transfection. Western blotting showed that LRP1 expression was suppressed by LRP1-siRNA. Cellular uptake of Aβ42 (1 μm) was analyzed by ELISA in control and LRP1-suppressed NT2 cells after incubation for 24 h. B, Subcellular localization of internalized FAM-Aβ42 (1 μm) and its colocalization with lysosomal marker were observed by confocal microscopy in control and LRP1-suppressed NT2 cells after incubation for 24 h. C, Control and LRP1-suppressed NT2 cells were allowed to internalize Aβ42 (1 μm) for 2 h at 37°C (gray bars); parallel cultures were washed and incubated for an additional 8 h in medium lacking Aβ42 (black bars) and analyzed by ELISA. The decrease of internalized Aβ after 8 h of incubation is estimated as cellular clearance. Data were plotted as mean ± SD (n = 3). *p < 0.05; **p < 0.01.
Conditional knock-out of Lrp1 in forebrain neurons exacerbates Aβ deposition in the cortex of amyloid model mice
We have previously generated forebrain neuron-specific LRP1-knock-out (Lrp1−/−) mice by breeding the Lrp1 floxed mice with αCaMKII-driven Cre recombinase mice (Liu et al., 2007). To examine the effect of LRP1 deletion in neurons on Aβ metabolism in vivo, the Lrp1−/− mice were further crossed with amyloid model APP/PS1 mice (Borchelt et al., 1997). We compared amyloid plaque deposition and Aβ levels between littermates of control APP/PS1 and APP/PS1 mice lacking LRP1 in forebrain neurons. First, the brain sections of those mice were stained with an LRP1-specific antibody at 12 months of age. In cortex, LRP1 was abundantly expressed in neurons and to a lesser extent in glial cells in APP/PS1 mice (Fig. 2A). LRP1 appears to be deleted specifically in neurons in APP/PS1; Lrp1−/− mice (Fig. 2A). Western blotting also confirmed that the LRP1 level in cortex from APP/PS1; Lrp1−/− mice was significantly reduced compared with APP/PS1 mice in an age-dependent manner (Fig. 2B). The remaining LRP1 expression in APP/PS1; Lrp1−/− mice likely represents those in glial cells and in cells making up vasculature. ELISA demonstrated that the concentrations of insoluble Aβ40 and Aβ42 in the guanidine fractions were significantly higher in APP/PS1; Lrp1−/− mice than APP/PS1 mice, when they were analyzed at 12–13 and 18–20 months of age, although there was no significant difference at 6–7 months of age (Fig. 2C). Consistent with increased insoluble Aβ levels, Aβ deposition in the cortex of APP/PS1; Lrp1−/− mice was significantly higher than that of control APP/PS1 mice at 12–13 months of age (Fig. 2D,E). While the soluble Aβ40 and Aβ42 levels in the TBS fractions were increased in the cortex from APP/PS1; Lrp1−/− mice compared with APP/PS1 mice at 18–20 months of age, we did not detect any significant difference at 12–13 months of age (Fig. 2F). In addition, there was no significant difference in the detergent-soluble Aβ40 and Aβ42 levels in the TBS-TX fractions between APP/PS1 and APP/PS1; Lrp1−/− mice both at 12–13 and 18–20 months of age (Fig. 2G).
Figure 2.

LRP1 deletion in neurons exacerbates Aβ deposition in the cortex of APP/PS1 mice. A, Cortex from control APP/PS1 and APP/PS1; Lrp1−/− mice were costained with an LRP1 antibody and a neuronal marker NeuN or an astrocyte marker GFAP at 12 months of age. Scale bar, 50 μm. B, LRP1 expression in cortex from APP/PS1 and APP/PS1; Lrp1−/− mice was detected by Western blot at 6–7 (n = 4–5), 12–13 (n = 6–7), and 18–20 months (n = 5–6) of age. C, The concentrations of insoluble Aβ40 and Aβ42 levels in the cortex extracted in guanidine (GDN) from control APP/PS1 and APP/PS1; Lrp1−/− mice were analyzed by ELISA at 6–7 (n = 4–5), 12–13 (n = 6–7), and 18–20 months (n = 5–6) of age. D, Aβ plaques in brain sections from control APP/PS1 and APP/PS1; Lrp1−/− mice (12–13 months of age) were immunostained with a pan-Aβ antibody. Scale bar, 1 mm. E, Amyloid plaque burdens in the cortex from control APP/PS1 and APP/PS1; Lrp1−/− mice were quantified after scanning Aβ immunostaining by the Positive Pixel Count program (Aperio Technologies) at 12–13 months of age (n = 6–7). F, G, The concentrations of soluble Aβ40 and Aβ42 levels in the cortex extracted in TBS (F) and TBS-TX (G) from control APP/PS1 and APP/PS1; Lrp1−/− mice were analyzed by ELISA at 12–13 (n = 6–7) and 18–20 months (n = 5–6) of age. H, I, LRP1 expression in cerebellum from APP/PS1 and APP/PS1; Lrp1−/− mice was detected by Western blot at 12–13 months (n = 4), and the concentrations of insoluble Aβ40 and Aβ42 levels in the cortex extracted in GDN were analyzed (I). Data were plotted as mean ± SEM *p < 0.05; **p < 0.01. N.S., Not significant.
Insoluble Aβ levels were increased in an age-dependent manner in those mice. When the correlations of insoluble Aβ40 (Fig. 3A) and Aβ42 (Fig. 3B) levels in cortex with age (6–20 months) were analyzed, significant positive correlations were detected for both Aβ40 (R2 = 0.525, p = 0.0015) and Aβ42 (R2 = 0.510, p = 0.0019) in APP/PS1 mice (n = 16). Similarly, APP/PS1; Lrp1−/− mice (n = 16) also showed positive correlations of Aβ40 (R2 = 0.546, p = 0.0011) and Aβ42 (R2 = 0.663, p = 0.0001) with age. The age-dependent increases of Aβ levels are higher in APP/PS1; Lrp1−/− mice compared with controls in these scatter plots, suggesting a protective role of LRP1 against age-dependent increases of insoluble Aβ (Fig. 3). We next assessed whether LRP1 levels in the cortex correlate with Aβ levels in aged APP/PS1 mice (12–20 months of age, n = 12) and APP/PS1; Lrp1−/− mice (n = 11). LRP1 levels were quantified by Western blotting. When correlations of insoluble Aβ40 (Fig. 3C) and Aβ42 (Fig. 3D) levels in the cortex from both APP/PS1 mice and APP/PS1; Lrp1−/− mice with LRP1 levels were plotted, significant inverse correlations were detected for both Aβ40 (R2 = 0.344, p = 0.0033) and Aβ42 (R2 = 0.344, p = 0.0033). Since LRP1 was specifically deleted in forebrain neurons in Lrp1−/− mice (Liu et al., 2010), the expression level of LRP1 in the cerebellum of APP/PS1; Lrp1−/− mice was not changed (Fig. 2H). Accordingly, we did not detect any differences in insoluble Aβ40 and Aβ42 levels in this brain region at 12–13 months of age (Fig. 2I). To examine whether LRP1 deletion in neurons affects APP processing, PS1 levels and APP processing products in both APP/PS1 and APP/PS1; Lrp1−/− mice were analyzed. There were no significant differences in the levels of PS1 (Fig. 4A,B), full-length APP (Fig. 4C,D), and soluble forms of APP (sAPPα and sAPPβ; Fig. 4E–G) between APP/PS1 and APP/PS1; Lrp1−/− mice when analyzed by Western blotting at 12–13 months of age. Specific ELISA also did not detect any significant differences in APP C-terminal fragments β (CTFβ) levels between these mice both at 6–7 and 12–13 months of age (Fig. 4H), indicating that LRP1 expression in these aged mice does not significantly affecting APP processing.
Figure 3.

Association of Aβ levels with age and LRP1 in the cortex of APP/PS1 and APP/PS1; Lrp1−/− mice. A, B, The correlations of insoluble Aβ40 (A) and Aβ42 (B) levels in cortex with age are plotted. APP/PS1 (n = 16) and APP/PS1; Lrp1−/− mice (n = 16) were analyzed at 6–20 months of age. C, D, The correlations of insoluble Aβ40 (C) and Aβ42 (D) levels with LRP1 levels in the cortex are plotted. APP/PS1 (n = 12) and APP/PS1; Lrp1−/− mice (n = 11) were analyzed at 12–20 months of age. LRP1 levels were quantified by Western blot and normalized to β-actin. Data were plotted as the ratios to mean value of control APP/PS1 mice. The correlation coefficient (R2) and p values are shown in the graph.
Figure 4.

LRP1 deletion in neuron does not affect APP processing and mRNA levels of NEP and IDE. A–G, Levels of PS1 (A, B), full-length APP (C, D), sAPPα (E, F), and sAPPβ (E, G) were analyzed by Western blot in both APP/PS1 and APP/PS1; Lrp1−/− mice at the age of 12–13 months (n = 5). H, CTFβ levels were analyzed by ELISA at the age of 6–7 (n = 5) and 12–13 months (n = 5). I, The mRNA levels of two major Aβ degrading enzymes, NEP and IDE, in the cortex from control and Lrp1−/− mice were quantified by RT-PCR at the age of 12 months. Data are plotted as mean ± SEM (n = 6). N.S., Not significant.
LRP1 deletion in neurons suppresses Aβ clearance in the cortex of amyloid model mice
Having demonstrated that LRP1 deletion in neurons leads to increased insoluble Aβ and amyloid plaque deposition without affecting APP processing, we analyzed the mRNA levels of NEP and IDE, which are major Aβ degrading enzymes in the brain, in the cortex of control and Lrp1−/− mice (Fig. 4I). The qRT-PCR showed no significant differences in the mRNA levels of these enzymes in our experimental mice. Next, we directly assessed the roles of neuronal LRP1 in Aβ clearance. To measure the half-lives of Aβ clearance, we used in vivo microdialysis in the cortex of APP/PS1; Lrp1−/− mice and APP/PS1 littermates at 8–10 months of age. Soluble Aβ in ISF, which is exchangeable across a 38 kDa dialysis membrane, has been shown to be significantly correlated with the levels of total soluble Aβ present in extracellular pools of the brain (Cirrito et al., 2003, 2011). We infused a potent γ-secretase inhibitor directly into the cortex of APP/PS1 and APP/PS1; Lrp1−/− mice to rapidly block Aβ production, thus allowing sensitive measurement of the elimination rate of Aβ from the ISF (Fig. 5A). ISF Aβ level gradually decreased in a time-dependent manner after treatment of γ-secretase inhibitor, where APP/PS1; Lrp1−/− mice showed a slower decline compared with control APP/PS1 mice (Fig. 5C,D). The half-life of elimination from the ISF for Aβ40 was calculated as 2.4 and 4.2 h in APP/PS1 mice and APP/PS1; Lrp1−/− mice, respectively (Fig. 5C). These results indicate that deletion of LRP1 in neurons significantly suppresses the elimination of soluble Aβ from the ISF. In addition, the higher baseline concentration of ISF Aβ40 was detected in APP/PS1; Lrp1−/− mice, which is likely due to disturbed Aβ clearance in the cortical extracellular space (Fig. 5B). Together, these results indicate that LRP1 deletion in forebrain neurons exacerbates Aβ deposition through a disturbance of cellular Aβ clearance rather than affecting the levels of Aβ degrading enzymes.
Figure 5.

LRP1 deletion in neurons suppresses cortical ISF Aβ clearance in APP/PS1 mice. A, B, APP/PS1 and APP/PS1; Lrp1−/− mice were analyzed at the age of 8–10 months. The mice were treated with a γ-secretase inhibitor compound E, and the cortical ISF levels of Aβ40 were monitored for 5 h (A) after a baseline of cortical ISF Aβ40 levels was achieved through 7.5 h monitoring (B). C, D, The common logarithm of percentage baseline ISF Aβ40 concentrations versus time were plotted (C). The slope from the individual linear regressions from log [% ISFAβ40] versus time for each mouse was used to calculate the mean half-life (t1/2) of elimination for Aβ from the ISF (D). Data were plotted as mean ± SEM (n = 4–5) *p < 0.05.
LRP1 deletion in neurons does not affect insoluble Aβ levels in the hippocampus of amyloid model mice
We further assessed the effects of neuronal LRP1 deletion on insoluble Aβ levels in the hippocampus of APP/PS1 mice. As expected, LRP1 is highly expressed in CA1 neurons and to a lesser extent in glial cells in the hippocampus (Fig. 6A). Neuronal LRP1 was specifically suppressed in APP/PS1; Lrp1−/− mice at 12 months of age (Fig. 6A). Western blotting confirmed that LRP1 expression in the hippocampus of APP/PS1; Lrp1−/− mice was significantly reduced (Fig. 6B). However, different from the results of cortex, we found that the concentrations of insoluble Aβ40 and Aβ42 in the guanidine fractions were not altered in the APP/PS1; Lrp1−/− mice compared with APP/PS1 mice (Fig. 6C). In addition, no significant inverse correlations were detected in both Aβ40 (R2 = 0.0576, p = 0.2699) and Aβ42 (R2 = 0.0089, p = 0.6684) and LRP1 levels (Fig. 6D,E). These results indicate that LRP1-mediated neuronal Aβ clearance is regional specific in the brain.
Figure 6.
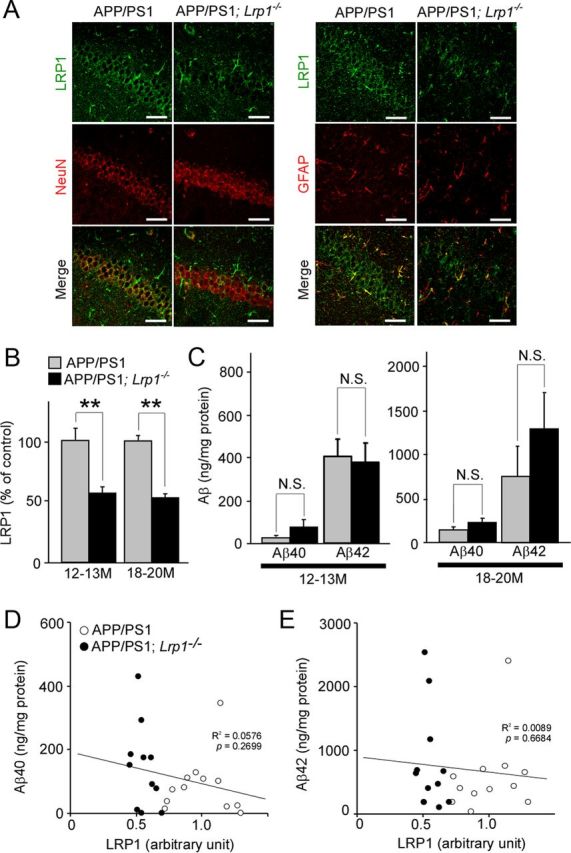
LRP1 deletion in neurons in APP/PS1 mice does not affect Aβ levels in the hippocampus. A, Hippocampus from control APP/PS1 and APP/PS1; Lrp1−/− mice were costained with an LRP1 antibody and a neuronal marker NeuN or an astrocyte marker GFAP at 12 months of age. Scale bar, 50 μm. B, LRP1 expression in the cortex from APP/PS1 and APP/PS1; Lrp1−/− mice was detected by Western blot at 12–13 (n = 6–7) and 18–20 months (n = 5–6) of age. C, The concentrations of insoluble Aβ40 and Aβ42 levels in the hippocampus from control APP/PS1 and APP/PS1; Lrp1−/− mice analyzed by ELISA at 12–13 (n = 6–7) and 18–20 months (n = 5–6) of age. Data were plotted as mean ± SEM; **p < 0.01. N.S., Not significant. D, E, The correlations of insoluble Aβ40 (D) and Aβ42 (E) levels with LRP1 levels in the hippocampus are plotted. APP/PS1 (n = 12) and APP/PS1; Lrp1−/− mice (n = 11) were analyzed at 12–20 months of age. LRP1 levels were quantified by Western blot and normalized to β-actin. Data were plotted as the ratios to mean value of control APP/PS1 mice. The correlation coefficient (R2) and p values are shown in the graph.
Discussion
In this study, we have demonstrated critical roles of neuronal LRP1 in mediating brain Aβ clearance using in vitro cellular and in vivo animal models. Previous studies have shown that astrocytes (Wyss-Coray et al., 2003, Koistinaho et al., 2004), microglia cells (Wyss-Coray et al., 2001), and vascular smooth muscle cells (Kanekiyo et al., 2012) can degrade Aβ to eliminate it from the brain. However, Aβ clearance through neurons has received little attention compared with other brain cell types despite the fact that neurons produce Aβ. Neurons are predicted to have the highest risk for encountering Aβ among all cell types in the brain, as Aβ is mainly generated locally in these cells. As Aβ is highly toxic to neurons, it is likely that neurons possess efficient mechanisms to eliminate Aβ. APP is known to be cleaved in the endosomes upon its endocytosis in both presynapses and postsynapses with Aβ secreting into extracellular space (Cirrito et al., 2005, 2008). Our findings showed that neuronal LRP1, which is predominantly expressed in the postsynaptic region (May et al., 2004) and the cell body (Bu et al., 1994), mediates Aβ uptake and subsequent degradation. Impairment of this pathway leads to acceleration of Aβ accumulation and deposition (Fig. 7). A variety of lysosomal acid hydrolases including cathepsin B and cathepsin D can efficiently degrade Aβ (Nixon et al., 2001). When the lysosomal system is modulated via Z-Phe-Ala-diazomethylketone treatment, there was a reduction in Aβ deposition by increasing neuronal cathepsin B levels in amyloid model mice (Butler et al., 2011). These findings indicate that neuronal lysosome-dependent Aβ clearance is a major pathway to eliminating Aβ and that disturbance of this pathway might be involved in AD pathogenesis.
Figure 7.

Model of neuronal Aβ clearance by LRP1. APP is cleaved and processed to Aβ in the endosomes upon endocytosis, and Aβ is secreted from the neuron into the brain ISF. LRP1 is abundantly expressed in neurons, predominantly in the postsynaptic region and cell body. As an efficient endocytic receptor, LRP1 mediates Aβ uptake and subsequent lysosomal degradation. Although Aβ is also eliminated through proteolytic enzymes, glial cell, and cerebrovascular system, disturbance of the LRP1 pathway is sufficient to induce Aβ accumulation and deposition in the cortex as shown in this work.
In previous studies, a potential role of neuronal LRP1 in brain Aβ clearance was suggested but not proven. It has been shown that clearance of exogenous Aβ from the brain is suppressed by LRP1 antagonist, RAP, or antibodies against LRP1 (Shibata et al., 2000). Furthermore, partial reductions in RAP disturbed the maturation of LRP1 and enhanced Aβ deposition in APP/PS1 mice (Xu et al., 2008). Because RAP is ubiquitously expressed in all cell types and also inhibits other members of the LDL receptor family (Bu and Schwartz, 1998), a specific role of LRP1 in neurons was not defined. In the current study, we have used Lrp1-conditional knock-out mice to demonstrate directly that deletion of LRP1 in forebrain neurons exacerbates amyloid pathology in aged APP/PS1 mice. Importantly, using in vivo microdialysis techniques, we demonstrated directly that the elimination rate of ISF Aβ was significantly slower in the cortex of APP/PS1; Lrp1−/− mice than that of control APP/PS1 mice. Because the neuronal LRP1 deletion did not affect the mRNA levels of major Aβ degrading enzymes, the disturbed Aβ clearance is likely caused by the suppression of LRP1-mediated neuronal Aβ uptake and lysosomal degradation. It is important to note that our studies are possible because Lrp1 gene deletion using the αCaMKII-Cre mice occurs only in adult mice (Liu et al., 2010), allowing us to address age-dependent effects in amyloid model mice.
While our findings showed the importance roles of neuronal LRP1 in Aβ clearance in the cortex, there was no significant difference in insoluble Aβ levels in the hippocampus between APP/PS1 mice and APP/PS1; Lrp1−/− mice. These results suggest that Aβ is differently eliminated depending on brain regions and that LRP1-mediated neuronal Aβ clearance pathway may not be a major pathway in the hippocampus. Cell populations of hippocampus are different from cortex; therefore, Aβ clearance through astrocytes or Aβ degrading enzymes may be more prominent in the hippocampus. Indeed, GFAP-positive activated astrocytes are more abundant in the hippocampus than in the cortex (Lein et al., 2007, Hewett, 2009). Further, Aβ degrading enzyme NEP is more abundant in the hippocampus compared with cortex in mouse brain (Fukami et al., 2002). Thus, Aβ clearance through proteolytic enzymes, glial cells, and/or cerebrovascular system (Fig. 7) might compensate the impaired neuronal Aβ clearance pathway upon deletion of LRP1 in hippocampus. Consistent with our results, another recent study reported that there was no significant effect of LRP1 reduction on the severity of amyloid deposition, Aβ accumulation, or the architecture of amyloid plaques in the hippocampus of APP/PS1 mice when LRP1 was deleted in neurons (Xu et al., 2012). In addition, we did not detect significant differences in insoluble Aβ levels at 6–7 months of age in our mouse models. In Lrp1−/− mice, LRP1 deletion in the cortex starts between 3 and 6 months of age and gradually reaches maximum at 12 months of age (Liu et al., 2010). Thus, the insufficient deletion of LRP1 in neurons may not induce significant effects on Aβ clearance in young APP/PS1; Lrp1−/− mice. Another possible contributing factor is that the neuronal LRP1-mediated Aβ clearance pathway might be critical after Aβ concentration reaches sufficiently high levels in aged APP/PS1 mice. Further experiments are needed to clarify these possibilities.
Previous studies have shown that LRP1 levels were significantly decreased during aging and AD (Kang et al., 2000). When LRP1 expression levels in the mid-frontal cortex of AD and normal control age-matched subjects were analyzed by Western blotting, LRP1 levels were ∼2-fold lower in AD brains compared with those of control subjects (Kang et al., 2000). Therefore, restoring LRP1 expression in neurons might be an attractive approach to prevent or treat AD by improving Aβ clearance. Enhancing LRP1 expression in neurons should also be beneficial in promoting synapse plasticity by facilitating lipid transport (Liu et al., 2010), neurite outgrowth (Holtzman et al., 1995), and neuronal survival (Fuentealba et al., 2009).
In summary, we have directly demonstrated that neuronal LRP1 critically mediates Aβ clearance in the cortex of APP/PS1 mice. We also report for the first time, to our knowledge, that neurons are capable of mediating local clearance of Aβ through receptor-mediated endocytosis in vivo. Our findings provide novel insights in to the molecular mechanisms of AD pathogenesis and further support LRP1 as a potential target for AD therapy.
Footnotes
This work was supported by National Institutes of Health (NIH) grants P01NS074969, R01AG027924, R01AG035355, and P01AG030128 (to G.B.); P01NS074969 and R01AG042513 (to J.R.C.); P01NS074969 and R37AG13956 (to D.M.H.); NIH Mayo Clinic ADRC pilot grant P50AG16574; and Mayo Clinic CRM Career Developmental Award (to T.K.). We thank Monica Castanedes Casey, Linda Rousseau, Virginia Phillips, and Dr. Dennis Dickson for histology and immunohistochemistry. We also thank Aurelie N′songo, Victoria Fasolino, Melissa C. Wren, and Caroline S. Casey for technical assistance and careful readings of this manuscript.
References
- Blennow K, de Leon MJ, Zetterberg H. Alzheimer's disease. Lancet. 2006;368:387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–945. doi: 10.1016/S0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- Bu G. Apolipoprotein E and its receptors in Alzheimer's disease: pathways, pathogenesis and therapy. Nat Rev Neurosci. 2009;10:333–344. doi: 10.1038/nrn2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu G, Schwartz AL. RAP, a novel type of ER chaperone. Trends Cell Biol. 1998;8:272–276. doi: 10.1016/S0962-8924(98)01283-5. [DOI] [PubMed] [Google Scholar]
- Bu G, Maksymovitch EA, Nerbonne JM, Schwartz AL. Expression and function of the low density lipoprotein receptor-related protein (LRP) in mammalian central neurons. J Biol Chem. 1994;269:18521–18528. [PubMed] [Google Scholar]
- Butler D, Hwang J, Estick C, Nishiyama A, Kumar SS, Baveghems C, Young-Oxendine HB, Wisniewski ML, Charalambides A, Bahr BA. Protective effects of positive lysosomal modulation in Alzheimer's disease transgenic mouse models. PLoS One. 2011;6:e20501. doi: 10.1371/journal.pone.0020501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarty P, Ceballos-Diaz C, Beccard A, Janus C, Dickson D, Golde TE, Das P. IFN-gamma promotes complement expression and attenuates amyloid plaque deposition in amyloid beta precursor protein transgenic mice. J Immunol. 2010;184:5333–5343. doi: 10.4049/jimmunol.0903382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, May PC, O'Dell MA, Taylor JW, Parsadanian M, Cramer JW, Audia JE, Nissen JS, Bales KR, Paul SM, DeMattos RB, Holtzman DM. In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-beta metabolism and half-life. J Neurosci. 2003;23:8844–8853. doi: 10.1523/JNEUROSCI.23-26-08844.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48:913–922. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Kang JE, Lee J, Stewart FR, Verges DK, Silverio LM, Bu G, Mennerick S, Holtzman DM. Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron. 2008;58:42–51. doi: 10.1016/j.neuron.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, Disabato BM, Restivo JL, Verges DK, Goebel WD, Sathyan A, Hayreh D, D'Angelo G, Benzinger T, Yoon H, Kim J, Morris JC, Mintun MA, Sheline YI. Serotonin signaling is associated with lower amyloid-beta levels and plaques in transgenic mice and humans. Proc Natl Acad Sci U S A. 2011;108:14968–14973. doi: 10.1073/pnas.1107411108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentealba RA, Liu Q, Kanekiyo T, Zhang J, Bu G. Low density lipoprotein receptor-related protein 1 promotes anti-apoptotic signaling in neurons by activating Akt survival pathway. J Biol Chem. 2009;284:34045–34053. doi: 10.1074/jbc.M109.021030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukami S, Watanabe K, Iwata N, Haraoka J, Lu B, Gerard NP, Gerard C, Fraser P, Westaway D, St George-Hyslop P, Saido TC. Abeta-degrading endopeptidase, neprilysin, in mouse brain: synaptic and axonal localization inversely correlating with Abeta pathology. Neurosci Res. 2002;43:39–56. doi: 10.1016/S0168-0102(02)00015-9. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Herz J, Strickland DK. LRP: a multifunctional scavenger and signaling receptor. J Clin Invest. 2001;108:779–784. doi: 10.1172/JCI200113992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewett JA. Determinants of regional and local diversity within the astroglial lineage of the normal central nervous system. J Neurochem. 2009;110:1717–1736. doi: 10.1111/j.1471-4159.2009.06288.x. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Pitas RE, Kilbridge J, Nathan B, Mahley RW, Bu G, Schwartz AL. Low density lipoprotein receptor-related protein mediates apolipoprotein E-dependent neurite outgrowth in a central nervous system-derived neuronal cell line. Proc Natl Acad Sci U S A. 1995;92:9480–9484. doi: 10.1073/pnas.92.21.9480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanekiyo T, Zhang J, Liu Q, Liu CC, Zhang L, Bu G. Heparan sulphate proteoglycan and the low-density lipoprotein receptor-related protein 1 constitute major pathways for neuronal amyloid-{beta} uptake. J Neurosci. 2011;31:1644–1651. doi: 10.1523/JNEUROSCI.5491-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanekiyo T, Liu CC, Shinohara M, Li J, Bu G. LRP1 in brain vascular smooth muscle cells mediates local clearance of Alzheimer's amyloid-beta. J Neurosci. 2012;32:16458–16465. doi: 10.1523/JNEUROSCI.3987-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang DE, Pietrzik CU, Baum L, Chevallier N, Merriam DE, Kounnas MZ, Wagner SL, Troncoso JC, Kawas CH, Katzman R, Koo EH. Modulation of amyloid beta-protein clearance and Alzheimer's disease susceptibility by the LDL receptor-related protein pathway. J Clin Invest. 2000;106:1159–1166. doi: 10.1172/JCI11013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koffie RM, Hyman BT, Spires-Jones TL. Alzheimer's disease: synapses gone cold. Mol Neurodegener. 2011;6:63. doi: 10.1186/1750-1326-6-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koistinaho M, Lin S, Wu X, Esterman M, Koger D, Hanson J, Higgs R, Liu F, Malkani S, Bales KR, Paul SM. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat Med. 2004;10:719–726. doi: 10.1038/nm1058. [DOI] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, Desaki AL, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Li J, Kanekiyo T, Shinohara M, Zhang Y, LaDu MJ, Xu H, Bu G. Differential regulation of amyloid-beta endocytic trafficking and lysosomal degradation by apolipoprotein E isoforms. J Biol Chem. 2012;287:44593–44601. doi: 10.1074/jbc.M112.420224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Lu W, Bu G. Essential role of the low density lipoprotein receptor-related protein in vascular smooth muscle cell migration. FEBS Lett. 2003;555:346–350. doi: 10.1016/S0014-5793(03)01272-9. [DOI] [PubMed] [Google Scholar]
- Liu Q, Zerbinatti CV, Zhang J, Hoe HS, Wang B, Cole SL, Herz J, Muglia L, Bu G. Amyloid precursor protein regulates brain apolipoprotein E and cholesterol metabolism through lipoprotein receptor LRP1. Neuron. 2007;56:66–78. doi: 10.1016/j.neuron.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Trotter J, Zhang J, Peters MM, Cheng H, Bao J, Han X, Weeber EJ, Bu G. Neuronal LRP1 knockout in adult mice leads to impaired brain lipid metabolism and progressive, age-dependent synapse loss and neurodegeneration. J Neurosci. 2010;30:17068–17078. doi: 10.1523/JNEUROSCI.4067-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandrekar S, Jiang Q, Lee CY, Koenigsknecht-Talboo J, Holtzman DM, Landreth GE. Microglia mediate the clearance of soluble Abeta through fluid phase macropinocytosis. J Neurosci. 2009;29:4252–4262. doi: 10.1523/JNEUROSCI.5572-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ. Decreased clearance of CNS beta-amyloid in Alzheimer's disease. Science. 2010;330:1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May P, Rohlmann A, Bock HH, Zurhove K, Marth JD, Schomburg ED, Noebels JL, Beffert U, Sweatt JD, Weeber EJ, Herz J. Neuronal LRP1 functionally associates with postsynaptic proteins and is required for normal motor function in mice. Mol Cell Biol. 2004;24:8872–8883. doi: 10.1128/MCB.24.20.8872-8883.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA, Mathews PM, Cataldo AM. The neuronal endosomal-lysosomal system in Alzheimer's disease. J Alzheimers Dis. 2001;3:97–107. doi: 10.3233/jad-2001-3114. [DOI] [PubMed] [Google Scholar]
- Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, Holtzman DM, Miller CA, Strickland DK, Ghiso J, Zlokovic BV. Clearance of Alzheimer's amyloid-ss(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest. 2000;106:1489–1499. doi: 10.1172/JCI10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara M, Petersen RC, Dickson DW, Bu G. Brain regional correlation of amyloid-beta with synapses and apolipoprotein E in nondemented individuals: potential mechanisms underlying regional vulnerability to amyloid-beta accumulation. Acta Neuropathol. 2013;125:535–547. doi: 10.1007/s00401-013-1086-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, Lin C, Yan F, Yu GQ, Rohde M, McConlogue L, Masliah E, Mucke L. TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat Med. 2001;7:612–618. doi: 10.1038/87945. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, Silverstein SC, Husemann J. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med. 2003;9:453–457. doi: 10.1038/nm838. [DOI] [PubMed] [Google Scholar]
- Xu G, Karch C, Li N, Lin N, Fromholt D, Gonzales V, Borchelt DR. Receptor-associated protein (RAP) plays a central role in modulating Abeta deposition in APP/PS1 transgenic mice. PLoS One. 2008;3:e3159. doi: 10.1371/journal.pone.0003159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Green CC, Fromholt SE, Borchelt DR. Reduction of low-density lipoprotein receptor-related protein (LRP1) in hippocampal neurons does not proportionately reduce, or otherwise alter, amyloid deposition in APPswe/PS1dE9 transgenic mice. Alzheimers Res Ther. 2012;4:12. doi: 10.1186/alzrt110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youmans KL, Leung S, Zhang J, Maus E, Baysac K, Bu G, Vassar R, Yu C, LaDu MJ. Amyloid-beta42 alters apolipoprotein E solubility in brains of mice with five familial AD mutations. J Neurosci Methods. 2011;196:51–59. doi: 10.1016/j.jneumeth.2010.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Koo EH. Biology and pathophysiology of the amyloid precursor protein. Mol Neurodegener. 2011;6:27. doi: 10.1186/1750-1326-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]