

Abstract
Bursts of action potentials are important information-bearing signals in the brain, although the neuronal specializations underlying burst generation and detection are only partially understood. In apical dendrites of neocortical pyramidal neurons, calcium spikes are known to contribute to burst generation, but a comparable understanding of basal dendritic mechanisms is lacking. Here we show that NMDA spikes in basal dendrites mediate both detection and generation of bursts through a postsynaptic mechanism. High-frequency inputs to basal dendrites markedly facilitated NMDA spike initiation compared with low-frequency activation or single inputs. Unlike conventional temporal summation effects based on voltage, however, NMDA spike facilitation depended mainly on residual glutamate bound to NMDA receptors from previous activations. Once triggered by an input burst, we found that NMDA spikes in turn reliably trigger output bursts under in vivo-like stimulus conditions. Through their unique biophysical properties, NMDA spikes are thus ideally suited to promote the propagation of bursts through the cortical network.
Introduction
Short bursts of action potentials are a key feature of spike trains observed in many brain areas, both in vivo and in vitro (Snider et al., 1998; Zhu and Connors, 1999). Bursts can be a particularly effective means for neurons to drive one another (Rancz et al., 2007) and are considered to encode either more, or more behaviorally relevant, information than individual spikes (Kepecs and Lisman, 2003). For example, in the neocortex, bursts occur more often in layer 5 pyramidal neurons (PNs) during sensory stimulation as opposed to background activation (de Kock et al., 2007) and have been shown to encode stimulus parameters, such as orientation and direction of motion, better than isolated spikes (Cattaneo et al., 1981; Martinez-Conde et al., 2000). Bursts are also the preferred stimulus for the induction of long-term potentiation of synapses in both the hippocampus and neocortex (Birtoli and Ulrich, 2004; Froemke et al., 2006).
The behavioral significance of burst firing suggests that cortical neurons must possess specialized mechanisms for burst propagation through the network. This requires mechanisms to both detect input bursts and generate output bursts in response. Enhanced sensitivity to bursts could be achieved through a variety of mechanisms, including short-term presynaptic dynamics (e.g., paired-pulse facilitation) and classical temporal summation of postsynaptic potentials (Izhikevich et al., 2003; Abbott and Regehr, 2004). On the output, burst-generation side, previous studies have shown that burst firing in PNs depends strongly on dendritic morphology (Mainen and Sejnowski, 1996) and can be driven by somatic and dendritic voltage-gated calcium and persistent sodium currents (Azouz et al., 1996; Williams and Stuart, 1999). Stimulation of layer 5 basal dendrites can also drive somatic bursts through the initiation of dendritic plateau-like potentials (Milojkovic et al., 2004), although compared with their apical counterparts, little is known regarding the biophysical mechanisms underlying burst generation in these branches or of the synaptic input patterns that would be expected to trigger them.
Notwithstanding this inventory of burst-related mechanisms, a direct biophysical link between burst detection and burst generation in pyramidal neurons has yet to be established. In apical dendrites, for example, in which the initiation of calcium spikes is the key step in burst generation, it is unclear whether synaptic input bursts play any significant role in the spike initiation process. In basal dendrites that receive the majority of inputs innervating neocortical pyramidal neurons (Larkman, 1991b), even less is known regarding mechanisms that might contribute to burst propagation. In this study, we examined the hypothesis that NMDA spikes, which are the major regenerative events in basal dendrites, could within a single biophysical mechanism provide the needed link between the input and output stages of burst propagation. Existing data, although piecemeal, indicates that NMDA spike initiation is sensitive to the frequency of synaptic activation (Schiller et al., 2000; Nevian et al., 2007; Major et al., 2008), suggesting that, on the one hand, NMDA channels are intrinsically suited to burst detection. On the other hand, the prolonged somatic depolarizations provided by NMDA spikes in basal dendrites also seem well suited to generate bursts of output spikes, similar to the role played by voltage-dependent calcium channels (VDCCs) in apical dendrites. These observations underlie the two-part hypothesis that has guided this work: (1) that NMDA spikes in PN basal dendrites are preferentially triggered by short bursts of spikes across the range of burst frequencies and durations likely to occur in vivo, and (2) once generated, NMDA spikes preferentially trigger bursts of somatic action potentials, thus “completing the circle” to ensure effective burst propagation through the cortical network. To test this hypothesis, we recorded from layer 5 pyramidal neurons in brain slices, measuring their responses to both regular spike trains with varying spike counts and interspike intervals (ISIs) as well as complex irregular spike trains taken from in vivo recordings.
Materials and Methods
Slice preparation and electrophysiological recording.
Neocortical brain slices 300 μm thick were prepared from 20- to 40-d-old Wistar rats. Whole-cell patch-clamp recordings were made from visually identified layer 5 pyramidal neurons using infrared–differential interference contrast optics. The extracellular solution contained the following (in mm): 125 NaCl, 25 NaHCO3, 25 glucose, 3 KCl, 1.25 NaH2PO4, 2 CaCl2, and 1 MgCl2, pH 7.4 (at 35–36°C). The intracellular solution contained the following (in mm): 115 K-gluconate, 20 KCl, 2 Mg-ATP, 2 Na2-ATP, 10 Na2-phosphocreatine, 0.3 GTP, 10 HEPES, and 0.2 Oregon Green 488 Bapta-1 (OGB-1), pH 7.2. In experiments in which we played back in vivo-like stimulation trains, we changed the concentration of CaCl2 (1.2 mm) and KCl (3.5 mm) to mimic the extracellular solution expected to be present in vivo. Bicuculline methiodide (1 μm) was added to the extracellular solution in all experiments. In 21 experiments, glycine was added to the extracellular solution (final concentration of 50 μm). Because no change in the shapes of EPSPs or NMDA spike initiation probability was apparent with glycine, data from these experiments was not treated separately.
All electrophysiological recordings were performed using Multi-Clamp 700A (Molecular Devices), and the data were acquired using pClamp 9 (Molecular Devices).
Generation of in vivo-like stimulation patterns.
To generate in vivo-like stimulation patterns, voltage traces acquired during in vivo recordings (see below) made at an earlier time were replayed to the cell in the slice in voltage-clamp mode. The resulting somatic current was stored and then immediately replayed to the same cell in current-clamp mode. This method ensured that the size and the temporal dynamics of the in vivo potentials were faithfully recreated.
In vivo voltage recordings.
For in vivo whole-cell voltage recordings, Wistar rats (age 30–40 d) were anesthetized with urethane (1.2 g/kg), the position of the somatosensory cortex was stereotaxically determined, and a craniotomy (0.5 × 0.5 mm) was performed. After removal of the bone, a small hole was created in the dura to allow access for the patch pipette. Recordings were performed with 7–9 MΩ resistance electrodes at 800–900 μm from pia. The intracellular solution was similar to the slice solution recordings, besides the OGB-1, which was replaced with BAPTA (0.2 mm).
Data analysis.
Data analysis was performed using Igor software (Wavemetrics) and Excel (Microsoft). To prevent spurious paired-pulse facilitation (Kim and Alger, 2001), averages and SD values of our paired-pulse responses were calculated in logarithmic scale. Spike amplitude was calculated as the difference between actual EPSP amplitude and the expected EPSP, as linearly extrapolated from subthreshold EPSPs. Spike threshold was defined as the highest subthreshold response. Burst was defined as two or more inputs within a 50 ms time window. Unless otherwise specified, statistical analysis used the unpaired Student's t test (when used in figures, *p < 0.05, ***p < 0.01).
Focal synaptic stimulation and calcium fluorescence imaging.
Focal synaptic stimulation was performed with a theta patch pipette (3–10 MΩ resistance) located in close proximity (2–5 μm) to the selected basal dendritic segment, as guided by the fluorescent image of the dendrite. The neurons were filled with a calcium-sensitive dye OGB-1, and the basal dendritic tree was imaged with a confocal imaging system (Olympus Fluoview 500) mounted on an upright BX51WI Olympus microscope equipped with a 60× (0.9 numerical aperture; Olympus) water objective. The theta stimulating electrodes were filled with Alexa Fluor 633. Stimulation duration was 0.1 ms, in a constant voltage mode.
Computer simulation.
Computer simulations were performed on OGB-1-filled and reconstructed experimental cells. Multicompartmental (45-251 dendrites, 9 segments for each compartment, average segment length of 11 μm) models were simulated in NEURON platform (time step of 0.1 ms). The values for Ri and Rm were 100 Ω/cm and 15 kΩ/cm2, respectively. Other passive biophysical parameters of the model are as follows: Eleak = −70 mV, ENa = 50 mV, EK = −87 mV, and Cm = 1.2 μF/cm−2. Voltage-gated sodium, calcium, and potassium conductances were not modeled.
Synaptic activation was modeled by simulating activation of postsynaptic AMPA and NMDA receptors (NMDARs).
Kinetics of the NMDA current was as follows:
 |
where gNMDA is the synaptic NMDA conductance, v is the membrane potential at the location of the synapse, and gMAX,NMDA is the peak NMDA conductance and the parameter that was used to plot the graphs in Figure 8.
Figure 8.

Computer simulation of the frequency-dependent amplification in basal dendrites. A, Three-dimensional reconstruction of a layer 5 pyramidal cell recorded in B–D; dendritic stimulation site is marked with a blue circle. This cell was used to create NEURON simulations shown in B–G. B, C, Comparison of actual (gray) and simulated (black) paired-pulse voltage responses to stimulations with ISI of 20 ms (B) and 200 ms (C). Experimental data were obtained in the presence of CNQX (20 μm). Close agreement between the experiment and the simulation were evident in all stimulation frequencies tested. D, Peak EPSP amplitude is plotted as a function of the stimulus intensity for paired-pulse stimulation (ISI of 20 ms; experimental data, gray squares; simulation, connected black circles) and for a single pulse (experiment, open squares; simulation, connected open circles). E, Manipulations of the dendritic voltage during single- and paired-pulse stimulations. Control curve during paired pulse (black filled circles) and single pulse (black open circles). During the paired-pulse protocol, the pan-cellular voltage was reset to the resting potential just before the onset of the second pulse (filled red circles). This manipulation increased the NMDA current needed to initiate a spike by only 2%. When the first synaptic pulse was substituted by equivalent local depolarization, NMDA spike initiation threshold increased by 46% (green). Schematic illustration of the different stimulation protocols are shown in the inset in F. F, Plot of the minimal peak NMDA conductance sufficient to initiate a dendritic spike for different ISIs. The dotted line represents the threshold for spike initiation by a single pulse. Paired-pulse stimulations with residual voltage reset to rest (red) were almost as effective as control paired-pulse stimulations (black). G, Same as in F except that AMPA conductance was included in the model. Color coding is as in F. Insets shows the somatic voltage of the “just suprathreshold” stimulation in NMDA only (F) and AMPA/NMDA conditions (G). Schematic illustration of the different stimulation protocols are shown in the inset.
AMPA synapses were modeled with an instantaneous rise time and decay time constant of 2 ms.
Double pulses were modeled by two separate AMPA (0.75 nS) and NMDAR (1.5 nS) conductance changes. The prebound glutamate effect was assumed to be mediated exclusively by double-bound NMDARs. Activation of inputs on modeled dendritic spines changed the local voltage traces by <1%; therefore, spines were not explicitly modeled in the simulations used to create the final figures.
In vivo simulation.
In vivo simulations were performed on biocytin-filled and reconstructed layer 5 pyramidal neurons (251 dendritic compartments). The passive cable parameters were similar to those used in the simulations of Figures 8 and 9 and supplemental Figure 3 (available at www.jneurosci.org as supplemental material). To realistically model somatic firing, fast sodium and potassium channels (gNa = 0.7 S/cm2, gK = 0.8 S/cm2) and slower potassium channel (gKm = 0.1 S/cm2) were inserted at the soma with dynamics similar to a previous study (Nevian et al., 2007). Excitatory synaptic connections also had similar parameters as presented above. AMPA and NMDA peak conductances were set to 0.75 and 1.5 nS, respectively. Inhibitory synapses were modeled as α-functions with a tau of 6 ms, reversal potential of −75 mV, and a peak conductance of 0.3 nS, similar to published GABAA dynamics (Poirazi et al., 2003b).
Figure 9.

Spatiotemporal conditions for spike initiation. A, Effect of spatial distribution of synapses within a single branch on initiation of NMDA spikes. A1, Example of the dendritic (red) and somatic (black) potentials after paired-pulse stimulation (ISI of 20 ms) of seven synapses located within 20 μm dendritic segment length with mean somatic distance of 256 μm from the soma. A2, Somatic and dendritic potentials after paired-pulse activation of 20 simulated synapses distributed randomly over the whole dendrite (marked in red in the inset). Note that both stimulations initiated NMDA spike at the second pulse. Dendritic voltage in both cases was measured at a distance of 256 μm from the soma. B, Left, The minimal number of synapses needed to initiate NMDA spike after focal paired-pulse (ISI of 20 ms; filled circles) or single-pulse (open circles) stimulations versus stimulation distance from the soma (n = 6 dendrites). Right, The number of synapses needed to initiate a spike for synapses distributed on the whole dendritic length (Whole dend) or the distal half of the dendrite (Distal half).
The “background” input consisted of random firing of 60 excitatory and 30 inhibitory cells. Each of these cells had 12 synaptic contacts that were mapped to random locations on the basal tree. The mean firing rate of every background cell was 1 Hz. The background activity depolarized the neuron by 7.61 ± 0.27 mV but was not strong enough to induce somatic firing.
The “signal” input consisted of eight excitatory cells, firing with an in vivo recorded firing pattern averaging 1 spikes/s (see Fig. 10D). Each signal neuron made 12 synaptic contacts with the postsynaptic neuron. These contacts were mapped to three randomly selected basal branches so that the same three branches received all the inputs from all eight signal cells.
Figure 10.

Effect of a small network of presynaptic neurons on the postsynaptic firing pattern of a layer 5 pyramidal neuron. A, Simulation setup and synaptic locations on the three-dimensional reconstruction of a layer 5 pyramidal neuron. Basal dendritic tree was contacted by 720 excitatory (open circles) and 360 inhibitory (gray circles) background inputs, firing at random with a mean firing rate of 1 Hz. The signal inputs originated from eight presynaptic neurons, each neuron having 12 synaptic contacts distributed at random locations on three postsynaptic basal branches (squares), altogether 96 inputs. B, Five-second-long dendritic (gray) and somatic (black) simulated voltage recording during activation of the presynaptic inputs by in vivo recorded firing pattern (D) showing generation of dendritic NMDA spikes (marked by pluses), somatic action potentials, and bursts (marked with an asterisk). With this firing pattern, presynaptic activity readily triggered NMDA spikes, which in turn effectively generated somatic firing. C, Similar to B but with AMPA-only synapses. The postsynaptic somatic firing rate was greatly reduced. D, Raster plot of the presynaptic activity of the eight signal neurons. The cumulative presynaptic firing rate (in bins of 100 ms) is shown by a gray curve on top. Asterisks mark presynaptic bursts. E, The percentage of the presynaptic inputs that successfully triggered postsynaptic firing as a function of the input ISI for control (black) and AMPA-only inputs (gray) conditions. High-frequency inputs significantly increased the probability for postsynaptic response in control but not in AMPA-only condition.
In summary, the neuron received 720 excitatory and 360 inhibitory background inputs and 96 additional signal inputs arriving on three basal dendrites.
Simulation runs corresponded to 10 s of real time in vivo. During each run, membrane potential of all the dendrites and the soma was analyzed, including NMDA spike occurrences and somatic action potential times.
Results
ISI dependence of NMDA spike initiation
Dendritic NMDA spikes are so called because NMDA receptor channels activation is both sufficient and mandatory for spike initiation (Schiller et al., 2000). The involvement of NMDARs predisposes NMDA spikes to be influenced by the recent history of receptor activation (Clements et al., 1992). To examine the short-term temporal dynamics of NMDA spike initiation, we used a paired-pulse protocol while varying both the ISI and stimulation intensity. In these experiments, layer 5 neocortical pyramidal neurons were filled with the fluorescent dye OGB-1 via the patch recording electrode. Their basal dendrites were imaged with a confocal microscope, and focal synaptic stimulation was delivered with an extracellular theta electrode placed in close proximity (2–5 μm) to a selected thin basal dendrite (Fig. 1A). Two pulses were given with varying stimulus intensities, with ISIs ranging from 10 to 2000 ms. This corresponded to firing frequencies ranging from background (0.5 Hz) to robust (100 Hz) stimulus-driven rates. The probability that an NMDA spike was activated by double-pulse stimulation was highly dependent on the ISI (Fig. 1B,C). At high stimulation frequencies (ISI of 10–20 ms), NMDA spikes could be initiated at low stimulation intensities and typically occurred at the second pulse. At lower frequencies (ISI of >200 ms), the stimulation intensities required to initiate a spike were substantially larger (1.6 ± 0.24-fold) than for high-frequency stimulation (p = 0.005, two-tailed paired Student's t test). Furthermore, at those higher stimulus intensities and longer ISIs, the NMDA spike was always first triggered at the first pulse (Fig. 1B), suggesting that the temporal limit of the facilitation effect had been reached. As expected, the ISI-dependent facilitation was highly dependent on activation of NMDAR channels and was completely blocked by the NMDAR blockers APV (100 μm) or MK801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate] (20 μm) (Fig. 1B) but not by the AMPAR blocker 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX); 20 μm (Fig. 1F).
Figure 1.
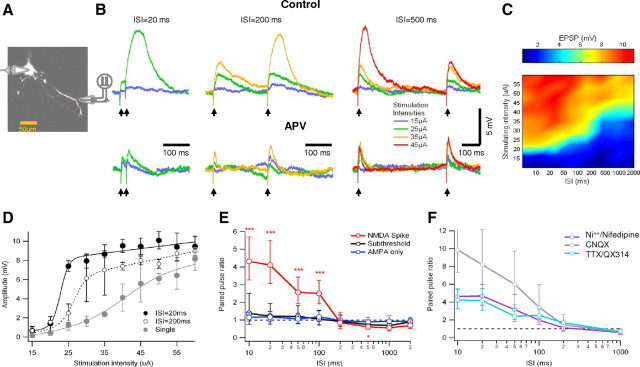
Frequency-dependent amplification in basal dendrites as shown using paired-pulse stimulation. A, Experimental setup. Whole-cell voltage-clamp recordings were performed from the soma. The cell was loaded with OGB-1 (200 μm) and was visualized using fluorescence confocal microscopy. Paired-pulse synaptic stimulation was performed via extracellular theta electrode placed under visual control near selected basal dendrites. B, Voltage responses to paired-pulse stimulations with different interstimulus intervals (ISI of 20–500 ms). Top traces were recorded in control conditions, and bottom traces were recorded in the presence of the NMDAR blocker APV (100 μm). Stimulus intensity was gradually changed at each ISI and presented in color coding (see inset). For clarity, only the lowest stimulus intensity that produced NMDA spike is shown for each ISI. Note that spikes were elicited preferentially by the second stimulus when ISIs were between 20 and 200 ms. For larger ISIs (500 ms), spikes were evoked at the first pulse. Addition of APV abolished all regenerative responses. C, Surface plot of the peak somatic EPSP amplitude evoked for all ISIs and stimulating intensities for the cell shown in B. Note the raise in voltage threshold for NMDA spike initiation as the ISI increased. D, Peak EPSP amplitude is plotted as a function of the stimulus intensity for the cell shown in B. Two ISIs (black, 20 ms; dotted line, 200 ms) and single pulse (gray) are presented. Data are shown as average ± SD. E, Summary plot of PPR as a function of ISIs (n = 10) for subthreshold responses (black), stimulations that triggered NMDA spike (red) and synaptic stimulation in the presence of APV (blue). To prevent spurious PPR, the average and SD were calculated in logarithmic scale. Note that, for stimulations that triggered NMDA spikes, strong facilitation is evident for ISIs smaller than 200 ms and paired-pulse depression for larger ISIs. The asterisks note statistical significance. F, Sodium and calcium blockers do not affect the PPR for all ISIs tested. PPR ± SD in the presence of somatic TTX or intracellular QX-314 (magenta; n = 8), the voltage-gated calcium channel blockers nifedipine and nickel (purple; n = 4), and CNQX (gray; n = 10). For clarity, only trials in which NMDA spike was present in either pulse were taken into account. The application of CNQX resulted in extremely high PPR at the short ISIs as the blocker markedly decreased the first EPSP without affecting spike amplitude.
To quantify the ISI dependence of the postsynaptic response, we calculated the paired-pulse response ratio (PPR). The PPR was calculated as [(P2 − P1)/P1], where P1 is the peak depolarization reached during the first EPSP in the pair, and P2 is the peak reached during the second synaptic response.
We found three distinct modes of paired-pulse synaptic interactions, depending on the stimulus intensity and the presence or absence of NMDA spikes. When synaptic potentials remained subthreshold for dendritic spike initiation, the PPR tended to show slight facilitation at high frequencies (122%, +71 to 45% at ISI of 20 ms). This level of facilitation was similar to that exhibited by AMPAR EPSPs alone in the presence of the NMDA antagonist APV (100 μm) (117%, +36 to 24%) (Fig. 1B,E).
At slightly higher stimulus intensities, the PPR increased substantially whenever an NMDA spike was initiated by the second pulse. The magnitude of the facilitation was highly dependent on the ISI, favoring short ISIs. On average, the second pulse was more than fourfold larger than the first pulse for ISIs of 20 ms (PPR of 411%, +140 to 104%) (Fig. 1B–E). The paired-pulse facilitation decreased for longer ISIs but was still significant at ISIs up to 100 ms. This pattern of paired-pulse facilitation was very robust and appeared in every cell in which NMDA spikes were elicited (n = 59).
After further increasing the stimulus intensity, when NMDA spikes could be elicited by the first pulse, the PPR changed again. This time a depression of the second input, expressed in both the reduced probability of NMDA spike initiation and smaller amplitude of the second pulse spike, was evident. The recovery from depression had a time constant of 1143 ± 449 ms (Fig. 1B,E).
Voltage-gated calcium and sodium channels do not contribute to frequency-dependent facilitation
Other voltage-sensitive ionic mechanisms, in addition to NMDAR channels, could in principle contribute to the frequency-dependent dendritic spike response we described. We used specific channel blockers to examine the respective role of voltage-gated sodium and calcium channels. Blockers for voltage-gated sodium channels (TTX [10 μm in an extracellular electrode located next to the cell body] and QX-314 [2(triethylamino)-N-(2,6-dimethylphenyl) acetamine] (5 mm, intracellular); n = 8) and for voltage-gated calcium channels [extracellular nickel (100 μm) and nifedipine (100 μm) (n = 5), which block T- and L-type calcium channels, respectively] did not significantly affect initiation of NMDA spikes or the PPR (Fig. 1F) (p = 0.21).
In addition, the two cells in which our stimulation elicited pure dendritic sodium spikes (Nevian et al., 2007), we found no frequency-dependent effects on the initiation of these spikes (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Thus, neither voltage-gated sodium channels or L- or T-type voltage-gated calcium channels contribute substantially to the paired-pulse facilitation effect we observed.
Activation of the NR2A subunit of the NMDAR is responsible for the paired-pulse facilitation
As we had shown above, blocking the NMDAR with APV completely abolished the frequency-dependent amplification. In layer 5 pyramidal cells, we expect to find at least two distinct populations: NMDAR with NR2A and NR2B subunits (Portera-Cailliau et al., 1996; Kew et al., 1998; Chen et al., 2000; Hsieh et al., 2002).
To investigate which of the NMDAR subtypes takes part in NMDA spike and is responsible for the frequency-dependent effects, we used specific blockers for the different subtypes of the NMDAR. Addition of NR2B blockers ifenprodil (3 μm) or Ro25-6981 [R-(R,S)-α-(4-hydroxyphenyl)-β-methyl-4-(phenylmethyl)-1-piperidine propranol] (3 μm) to the bath did not change the shape of the NMDA spike or the PPR observed under control conditions (Fig. 2A,B) (n = 11); the normalized average peak NMDA spike and area under the curve after application of the NR2B blockers were 100.4 ± 20%; (p = 0.38) and 100.15 ± 34.8% (p = 0.41, two-tailed paired Student's t test), respectively, compared with control conditions (Fig. 2C,D). In contrast, application of the NR2A blocker (R)-[(S)-1-(4-bromo-phenyl)-ethylamino]-(2,3-dioxo-1,2,3,4-tetrahydroquinoxalin-5-yl)-methyl]-phosphonic acid (NVP-AAM077) (0.4 μm; Novartis Pharma AG) completely blocked the dendritic spike, reducing the amplitude and area under curve to 5.6 ± 23%; p < 0.001 and 8.3 ± 9.6%; p < 0.001, two-tailed paired Student's t test, respectively (Fig. 2C,D) (n = 6). Additional application of APV had no apparent effect on EPSP shape or PPR (Fig. 2C,D) (n = 4).
Figure 2.

NMDA spikes are mediated by NMDAR with NR2A subunits. A, Paired-pulse (ISI of 20 ms) synaptic stimulation at an intensity sufficient to trigger an NMDA spike during control conditions (red), after bath application of the NR2B blocker ifenprodil (3 μm; green), followed by bath application of NR2A antagonist NVP-AAM077 (0.4 μm; black), and finally by APV (100 μm; blue). Stimulus intensity was identical for all traces shown. B, Maximal PPR ± SD at different ISIs, with ifenprodil (3–5 μm) or Ro25-6981 (0.5 μm; n = 7; green), and NVP-AAM077 (0.4 μm; n = 4; black). In two cells, successive application of NR2B and NR2A blockers as shown in A was given. C, D, Summary of the effect of different NR2 antagonists on NMDA spike amplitude (C) and area under the curve (D). Spikes were normalized to the average spike amplitude (or area under the curve) in control conditions, when no antagonist was present (n = 6 for NVP-AAM077 and 11 for ifenprodil or Ro25-6981). Complete NMDAR block by APV (100 μm; n = 5) is shown for comparison. Blockage of NR2B subunit receptors had little, if any, effect on dendritic spike, whereas both NVP-AAM077 and APV eliminated the spike (p < 0.001 compared with control).
Temporal summation with long stimulus trains
The paired-pulse protocol is commonly used to investigate short-term synaptic dynamics. However, a more complex pattern of responses may emerge with an increasing number of stimuli (Markram et al., 1998). To explore the temporal dynamics of NMDA spike responses with longer activation patterns, we increased the number of synaptic pulses to 10 and varied the ISI between 10 and 1000 ms (n = 6). At the highest stimulus frequencies (>50 Hz), plateau potentials (up to 500 ms in duration) and somatic action potentials were readily initiated by these prolonged trains (Fig. 3A). Similar to paired pulses, the stimulation intensity needed to evoke NMDA spikes was lowest for high-frequency inputs (Fig. 3A–C) (ISI of <200 ms). For stimulus frequencies of 10 Hz or higher, the lowest threshold for spike initiation occurred for the second or third synaptic input in 72% of the cases and almost never after the fifth (Fig. 3A,D). At these frequencies, the stimulation intensity that elicited an NMDA spike at the third pulse was only slightly lower than was needed to elicit the first spike at the second pulse (96 ± 3%; p = 0.03), suggesting that, for high-frequency inputs, presynaptic bursts of just two to three action potentials are effective in initiating NMDA spikes.
Figure 3.
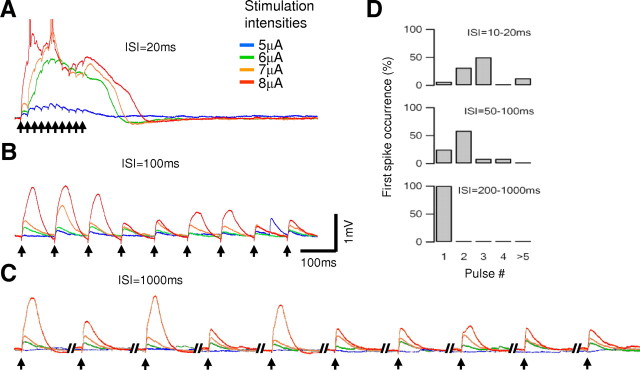
Temporal summation rules for long pulse trains. A–C, Voltage responses to long input trains (10 pulses at 4 different stimulating intensities of 5, 6, 7, and 8 μA; color coding is shown on the right) were given with an ISI of 20 ms (A), 100 ms (B), and 1000 ms (C); arrows denote timing of stimuli. Somatic action potentials are truncated. Note that, during high-frequency stimulations (ISIs of <50 ms) plateau potentials were initiated. In addition, occurrence of NMDA spikes inhibited spikes in subsequent pulses for ISI of 100 ms and especially 1000 ms. D, The stimulus pulse number in which NMDA spikes were initiated by the lowest stimulation intensity. Note that high-frequency inputs initiated spikes at the second or the third input, but for longer ISIs, the spike always occurred initially at the first pulse (n = 6).
In response to low-frequency trains (<5 Hz), NMDA spikes were always first initiated at the first pulse (Fig. 3C,D), with a threshold for spike initiation 1.58 ± 0.17 (p < 0.001) times higher than was required for spike generation at higher activation frequencies. Furthermore, temporal summation was almost absent as the ISI grew longer than 100 ms. As with paired-pulse inputs, for trains with ISIs larger than 200 ms, the occurrence of spikes early in the train tended to reduce the probability of later spikes (Fig. 3C).
Response of basal dendrites under in vivo-like conditions
The above experiments focused on the temporal parameters of NMDA spike generation in basal dendrites using well controlled stimulation patterns. To gain insight into how these dendrites transform their inputs into output spike trains under more physiological conditions, we set out to characterize the overall input–output relationship using more realistic in vivo-like activity patterns. We were especially interested in determining the contribution of local NMDA spikes to the overall input–output process, given their potential to contribute to both burst detection and burst generation. Unlike neurons in the slice preparation, neurons in vivo fire irregular patterns of action potentials and display large changes in the subthreshold membrane potential in the form of up and down states.
To mimic in vivo conditions in slice, we recorded the spontaneous activity from layer 5 pyramidal neurons in vivo. We extracted subthreshold voltage traces, which were later used to recreate in vivo-like voltage traces at the cell body by direct somatic current injection. From different recordings, we extracted the timing of action potentials, which we used to drive in vivo-like presynaptic trains through the extracellular electrode (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Despite the fact that up and down states were produced in our experiments by somatic current injections rather than by dendritic conductance changes, direct somatic stimulation mimicked the in vivo situation insofar as synaptic conductances evidently change minimally during up states (Haider et al., 2006; Waters and Helmchen, 2006).
Under down-state conditions, the somatic membrane potential was for the most part subthreshold to somatic action potential initiation. During these periods, we were able to investigate the time-dependent dynamics of NMDA spike initiation in response to irregular in vivo activation patterns. Similar to the paired-pulse stimulation protocol, inputs arriving with short ISIs reliably triggered dendritic NMDA spikes (Fig. 4A), although the time window for temporal integration was shorter for the in vivo-like inputs compared with paired-pulse stimulation (72.2 ± 7.7 ms) (Fig. 4B). As before, APV blocked the frequency-dependent amplification we observed by blocking NMDA spikes (Fig. 4A,B).
Figure 4.
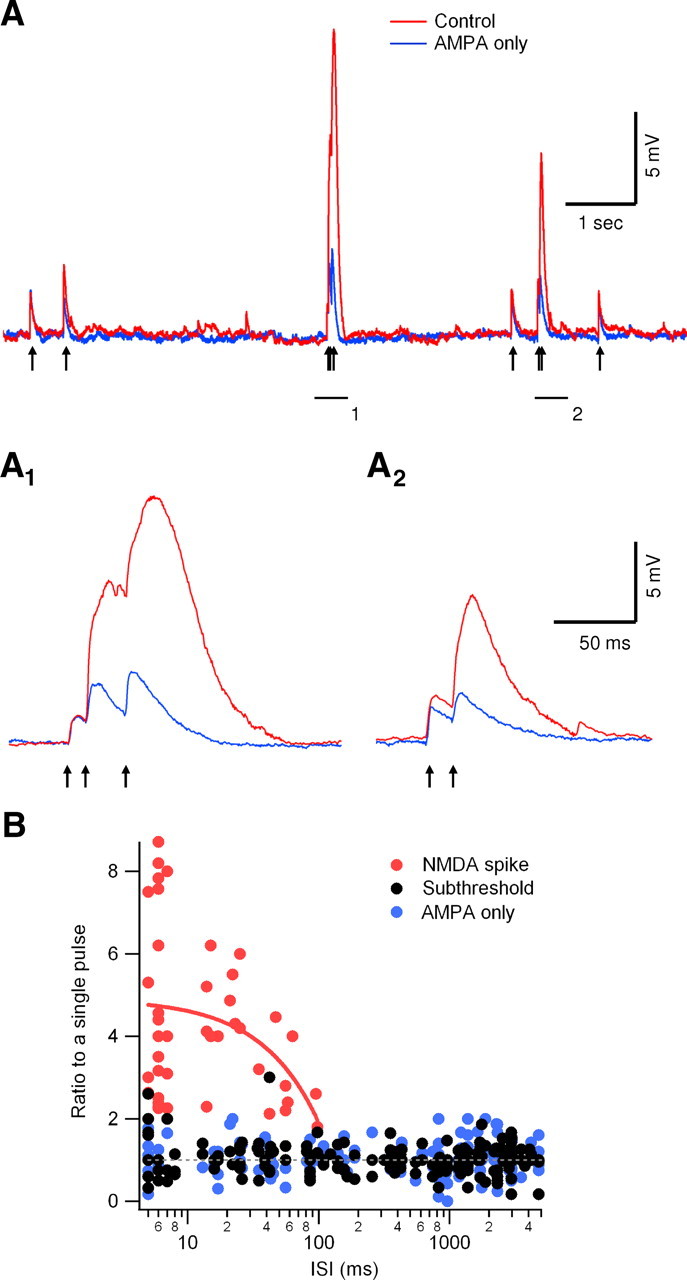
Integration of synaptic inputs driven by in vivo-like down-state stimulation patterns. A, Timing of action potentials (arrows) were extracted from a layer 5 pyramidal neuron in vivo recording and used to drive the extracellular stimulation electrode. Responses under control conditions (red) and in the presence of APV (blue; 100 μm) are shown. A1, A2, Enlargement of the two underlined regions of the trace shown in A. B, Summary plot of the ratio between NMDA spikes (red circles), subthreshold EPSPs (black circles) to a single pulse (7 cells) in control and with APV (blue circles). NMDA spike initiation was dependent on ISI; on average, the time window for spike initiation was 72.2 ± 7.7 ms (n = 7). The red line is a linear regression analysis of the ratio between NMDA spike and control prepulse amplitudes (note the logarithmic scale of the x-axis).
In up-state conditions, our in vivo-like stimulation method reliably evoked somatic postsynaptic action potentials, with NMDA spikes easily recognizable in the somatic voltage recordings (Fig. 5B, red). The overall input–output relationship under these conditions supported both the burst detection and burst generation aspects of our main hypothesis. First, high-frequency input bursts were highly efficient at evoking postsynaptic action potentials, whereas the contribution of low-frequency inputs to postsynaptic firing was negligible (Fig. 6A,B). More than 75% of high-frequency (ISI of <50 ms) presynaptic bursts triggered postsynaptic somatic firing compared with only 6% of low-frequency (ISI of >200 ms) inputs (Fig. 6A) (n = 212 and 623, high- and low-frequency inputs, respectively). Second, the output firing pattern included many bursts, with 65% of all axonal output events (n = 137) being in the form of bursts (Fig. 6B). As a result, the output ISI distribution shifted to higher frequencies compared with the input ISI distribution (Fig. 6B). Together, these findings indicate that NMDA spikes cause basal dendrites to act as “filters” that selectively transform high-frequency bursts of synaptic input into bursts of somatic action potentials, whereas low-frequency inputs to the dendritic branch tend to be filtered out. This pattern of results held true whether or not the cell was defined a priori as a burster according to its response to somatic current injections (Chagnac-Amitai et al., 1990) (see example in Fig. 5A). We found only a weak, statistically insignificant correlation between the bursting behavior of the cell during somatic current injection and the firing pattern after in vivo-like high-frequency synaptic stimulation (χ test, p = 0.11).
Figure 5.

Input–output function under in vivo-like up-state stimulation patterns. A1, Experimental setup. Whole-cell voltage-clamp recordings were performed from the soma of a layer 5 pyramidal neuron. The cell was loaded with OGB-1 (200 μm) and was visualized using fluorescence confocal microscopy. A2, Somatic voltage response to a 500-ms-long somatic current step (0.3 nA) showing the firing pattern of the neuron. A3, Synaptic stimulations were performed via extracellular theta electrode placed under visual control near selected basal dendrite. The stimulation intensity was set to initiate a local NMDA spike with paired-pulse stimulation (ISI of 20 ms). B, A 4-s-long trace recorded in vivo from a layer 5 pyramidal neuron (after truncation of action potentials; B4) was played back to the soma of the cell shown in A. Action potential firing pattern, extracted from a different in vivo recording, served to drive the stimulation of the basal dendrite (B5, arrows); asterisks mark burst firing. The postsynaptic voltage recording evoked by the in vivo-like stimulation pattern is shown in control conditions (B1) and in the presence of 100 μm APV (B2). Bursts of postsynaptic action potentials were readily evoked under control conditions (marked by asterisks; B1). In the presence of APV, the cell had to be depolarized by 8 mV above rest to initiate reliable postsynaptic firing (B3). Under these conditions, the resulting somatic output was most often in the form of a single action potential. Arrows mark the postsynaptic output.
Figure 6.

Suprathreshold input–output function after activation of a single basal branch: effect of NMDA spikes. A, The percentage of the presynaptic inputs that successfully triggered postsynaptic firing as a function of the input ISI (n = 6 cells). B, ISI distribution of postsynaptic somatic action potentials (shaded histogram; n = 287 events recorded in 6 cells) and the input ISIs (open histogram; 957 events). C, The probability of evoking a given number of postsynaptic action potentials (AP) after high-frequency (ISI of <50 ms) or low-frequency (ISI of >200 ms) presynaptic stimulation. There was a statistically significant change between the output behavior for high- and low-frequency inputs (p < 0.001, χ test). Inset, Zoom on the firing probability flowing low-frequency stimulation. D, Similar to A in the presence of 100 μm APV. Gray traces represent measurements at control resting potential. Black trace, Similar experiment assisted by 8–10 mV somatic depolarization. E, Similar to B, in the presence of 100 μm APV (n = 4 cells, output; n = 197 somatic action potentials; input, 802 stimulations). F, Similar to C in the presence of 100 μm APV and depolarization. The output pattern was not significantly different between high and low stimulation frequencies (p = 0.22, χ test).
To verify that NMDA spikes were key contributors to both the burst detection and generation processes, we repeated the experiments with bath application of APV to block NMDARs and observed a completely different output pattern. AMPA-only EPSPs failed to trigger postsynaptic firing unless up-state amplitude was increased by 5–10 mV (Fig. 5B, blue). Even under these conditions, presynaptic bursts were no more efficient as triggers of postsynaptic firing than single presynaptic action potentials. Instead, the probability that an action potential was generated was nearly flat over the range of activation frequencies tested (20% vs 26% postsynaptic probability to fire for high- and low-frequency inputs, respectively) (Fig. 6D,E). Furthermore, the postsynaptic output was now almost exclusively in the form of single action potentials; only 12.7% of the postsynaptic somatic firing events were bursts (Fig. 6E,F, 197 total postsynaptic events).
Mechanisms of burst frequency-dependent facilitation: residual voltage versus prebound NMDARs
From the above experiments, we conclude that NMDARs contribute heavily to burst frequency-dependent amplification of basal dendritic responses. How NMDARs mediate this effect is unclear, however, and could involve at least two distinct mechanisms. First, because of the slow kinetics of glutamate unbinding from NMDARs, NMDA channel activation by the first pulse can lead to a prolonged local depolarization, the voltage tail of which could help to relieve the magnesium block and increase the NMDA current in response to subsequent pulses (Clements et al., 1992; Spruston et al., 1995). Second, residual glutamate bound to NMDARs from the first pulse, including single-occupancy and double-bound NMDARs, may contribute to the frequency-dependent NMDA spike facilitation (Clements et al., 1992; Spruston et al., 1995). With this mechanism, the amplification would result from a larger number of NMDARs bound to glutamate during the second activation rather than from a nonlinearity in affinity of glutamate binding to the channel. Given that both depolarization and NMDAR activation would normally occur together, we attempted to distinguish the relative contributions of the two mechanisms because their implications for dendritic computation are different: facilitative interactions among synapses mediated by residual voltage would be expected to spread by cable propagation over larger distances in the cell, whereas effects mediated by glutamate binding would be more localized because they are tied to the activated synapses themselves.
We experimentally evaluated the contribution of residual depolarization to the burst facilitation effect by simulating, via a somatic current injection, the enduring voltage signal remaining after a single EPSP. If the first pulse during a pair enhances the effect of the second pulse through residual depolarization, an injected voltage should mimic the paired-pulse NMDAR facilitation effect.
We performed these experiments in the presence of the AMPAR blocker CNQX (20 μm) to isolate NMDAR-mediated EPSPs. This eliminated the need to inject simulated AMPAR currents at the soma, which are much larger and faster than NMDAR currents (Nevian et al., 2007), allowing us to focus specifically on the contributions of NMDARs to burst-dependent facilitation (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). We first used paired-pulse stimulation (ISI of 20 ms) under resting membrane conditions to determine the minimal stimulation intensity needed to trigger NMDA spikes (Fig. 7A, black). Single pulses at the same stimulus intensity were then applied with and without the somatic current injection. Under control paired-pulse conditions, the peak EPSPs evoked by the first pulse at threshold values for spike initiation were often <1 mV in size (0.89 ± 0.68 mV; n = 5) as recorded at the soma (Fig. 7A, gray). Based on direct dendritic recordings from basal dendrites (Nevian et al., 2007) and on computer simulations (supplemental Fig. 3, available at www.jneurosci.org as supplemental material), we estimated the local dendritic voltage under these conditions to be less than 5–7 mV. Taking into account that >70% of the somatic voltage reaches basal dendrites (Nevian et al., 2007), depolarizing the soma by 10–15 mV should be more than sufficient to mimic the residual local dendritic voltage after a single EPSP (see detailed reconstruction and computer simulation of the cells used in these experiments in Fig. 8 and supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
Figure 7.

Manipulations of the residual voltage during frequency activation. A, Stimulation of a basal dendrite located 120 μm from the soma in the presence of 20 μm CNQX. Membrane potential was controlled by somatic current injections. Stimulation intensity was increased until NMDA spike was initiated by paired-pulse stimulation protocol (ISI of 20 ms). Note that, with a single pulse at the same intensity the voltage remained subthreshold (gray, control resting potential). To simulate the effect of the residual depolarization on NMDA spike initiation, 10 mV somatic depolarization was applied during single-pulse stimulation (green). To cancel the contribution of first-pulse depolarization on NMDA spike initiation during paired-pulse stimulation, voltage was hyperpolarized by 10 mV (red). The stimulation intensity was kept constant for all traces shown. B, Summary plot of normalized peak EPSP amplitude as a function of the normalized stimulus intensity (5 cells) at control paired-pulse stimulation (ISI of 20 ms; filled black circles), single-pulse stimulation (open black circles), and single-pulse stimulation paired to 8–15 mV depolarization (green). All responses were normalized by the average NMDA spike amplitude, and stimulation intensities were normalized by the lowest intensity that triggered NMDA spike in paired-pulse stimulation protocol (ISI of 20 ms). The data were fitted by a sigmoid function (black filled for paired-pulse stimulation, black dotted for single-pulse stimulations, green line for single-pulse stimulation paired to depolarization). Somatic depolarization (8–15 mV) assisted NMDA spike initiation, but the stimulus intensity needed to initiate a spike was on average more than twofold higher than in control paired-pulse conditions. C, Same as B except that the paired-pulse stimulation was delivered in the presence of somatic hyperpolarization of 10–20 mV (red dots, red line shows the sigmoid fit). Note that the stimulation intensities needed to initiate the NMDA spike increased by only 22%.
We found that the dendritic response to a single pulse at the threshold stimulation intensity paired with somatic depolarization was not significantly different from the control EPSP generated by a single pulse at the resting potential [EPSP size increased from 0.89 ± 0.68 to 1.05 ± 0.84 mV; p = 0.48; n = 5 (Fig. 7A, green trace, B; n = 5 cells)] and was always below the threshold for dendritic spike generation. This suggests that voltage carryover between pulses is unlikely to be a major contributor to NMDA spike facilitation at these synapses.
To further investigate this issue, we counteracted the voltage persistence between pulses by hyperpolarizing the membrane potential during paired-pulse synaptic stimulation. Based on the voltage attenuation value cited above, hyperpolarizing the cell by 10–15 mV during paired-pulse synaptic activation should roughly “cancel” the local voltage at the synaptic site so that it would be close to the original resting potential at the onset of the second pulse. When we hyperpolarized the cell by 10–15 mV and tested the dendritic response to paired-pulse stimulation, we found that hyperpolarization had only a small effect on the stimulus intensity needed to initiate an NMDA spike (Fig. 7A, red trace, C) (122% compared with control intensities during the paired-pulse protocol; n = 5 cells), and, in many cases, the spikes were evident at the same intensity as in control paired-pulse stimulation (see example in Fig. 7A, red trace).
The combined findings that (1) a 10–15 mV depolarization was relatively ineffective at facilitating an NMDA spike initiated by a single pulse, and (2) a 10–15 mV hyperpolarization was relatively ineffective at impeding an NMDA spike initiated by a pair of pulses, suggest that glutamate released by the first pulse and prebound to NMDARs, rather than residual depolarization, is the primary mechanism that potentiates the response to later pulses in a stimulus frequency-dependent manner. This mechanism explains the efficacy of paired-pulse compared with single-pulse synaptic stimulation in initiating NMDA spikes (Rhodes, 2006).
Quantifying the contribution of residual voltage versus prebound glutamate: computer simulations
We used computer simulations to test our conclusions regarding the relative contributions of residual voltage versus prebound glutamate to the burst frequency-dependent amplification we had described experimentally. Compartmental simulations were performed on six three-dimensional reconstructed neurons used in the experiments; in each simulation, synapses with pure NMDA conductance were activated at the original experimental stimulation site and somatic responses were recorded (Fig. 8A). Double pulses were modeled by activating separate, equal peak conductance synapses on the first and second pulses. The compartmental model successfully reproduced the size and shape of experimentally recorded EPSPs and NMDA spikes (Fig. 8B,C).
We found that single-pulse stimulation led to an all-or-none spike when the peak NMDA conductance (i.e., the conductance that would be reached in the absence of magnesium block) was >17.9 nS (Fig. 8D–F). Paired-pulse activation decreased the amount of peak NMDA conductance needed to initiate a spike in a frequency-dependent manner. This effect was most pronounced at the shortest ISIs (9.1 nS per pulse for ISI of 10 ms), which corresponded to just over half the minimal single-pulse conductance delivered in each of the two closely spaced pulses. The minimal per-pulse conductance to achieve an NMDA spike climbed slowly as the ISI increased up to 500 ms, at which point the paired-pulse protocol required a similar NMDA peak conductance as was needed for a single isolated pulse, indicating that the temporal facilitation effect had nearly disappeared (Fig. 8D,F).
Similar to our experimental data, we used the model to manipulate the dendritic voltage during single-pulse and paired-pulse activation. When the first glutamate activation was replaced by current injection at the synaptic location, so that it exactly replicated the dendritic voltage shape after single-pulse activation (see illustration of the simulations in Fig. 8F, inset), the peak NMDA conductance needed to trigger a spike had to be increased by >60% (Fig. 8E,F, green traces). Thus, when the NMDARs activated by the first pulse were excluded from the simulation and paired-pulse facilitation rested on residual depolarization alone, initiation of NMDA spikes occurred only when NMDA currents were significantly higher than during actual paired-pulse activation.
Next we wanted to cancel out the contribution of residual voltage. We did so by activating the synapses with paired pulses, but the membrane voltage for all cellular compartments was reset to the resting potential just before the second input was applied. Canceling the residual depolarization only slightly increased the peak NMDA channel conductance required for spike initiation (for ISI of 20 ms: 9.8 vs 9.6 nS, respectively) (Fig. 8E,F, red traces). We found similar results for all ISIs up to 500 ms (Fig. 8F), with a <3% increase in the stimulus intensity needed to trigger a spike compared with the control condition.
We also investigated whether inclusion of AMPAR channels in the simulation influenced the relative contribution of voltage carryover versus prebound glutamate. We used previously reported values for the relative peak NMDA/AMPA conductances (Myme et al., 2003) (supplemental Fig. 3E, available at www.jneurosci.org as supplemental material). As expected, adding an AMPA component to the synaptic conductance reduced the minimal peak NMDA conductance needed to initiate a spike for both paired-pulse and single-pulse stimulation (8.08 and 15.96 nS, respectively) (Fig. 8G). However, the stimulation intensity was only 2.5% higher when the residual depolarization was cancelled out during paired-pulse activation and was increased by up to 61% when first-pulse NMDARs were excluded from the simulation. These results are very similar to our findings with NMDA receptors alone.
Spatial distribution of synaptic inputs to basal dendrites needed for NMDA spike initiation: simulation results
We have shown that high-frequency burst activation is effective at initiating NMDA spikes in both experiments and a model cell, in both cases when all activated synapses were nearly colocalized. The question remains as to how the burst facilitation effect we observed in basal dendrites depends on the spatial distribution of synaptic inputs to the branch. Given the difficulty of asking this question in experiments, we used our compartmental model of a layer 5 pyramidal neuron (Polsky et al., 2004; Nevian et al., 2007) to determine the number of clustered synapses needed to initiate an NMDA spike. Near a dendritic tip, we found that activation of only 5.3 ± 0.85 neighboring synapses with a double pulse (ISI of 20 ms; peak NMDA conductance of 8.06 ± 1.25 nS) or a single activation of 10.6 ± 1.11 synapses (peak NMDA conductance of 15.9 ± 1.66 nS), similar to the peak NMDA conductance values described above, was sufficient to initiate a local spike. Under these conditions, NMDA spike amplitude was 33 ± 4.94 mV at the site of stimulation and 3.8 ± 0.55 mV at the soma (Fig. 9A). As the cluster of synapses was moved to more proximal dendritic regions (50–70 μm from the soma), both the somatic NMDA spike amplitude and the number of synapses needed to activate it were increased (Fig. 9B) (28.6 ± 5.21 synapses were needed to initiate a spike, and the somatic NMDA spike amplitude was 20 ± 3.24 mV).
When synapses were distributed randomly along a basal branch [excluding the first 40 μm, which is nearly devoid of excitatory synaptic contacts (Larkman, 1991a)], 16.2 ± 2.54 and 26.6 ± 5.78 synapses were required to initiate NMDA spike with paired-pulse (ISI of 20 ms) or single-pulse activation, respectively (Fig. 9B). The corresponding somatic voltage amplitude of these spikes was intermediate between the distal and proximal focal activations (11.25 ± 3.58 mV) (Fig. 9A).
When synapses were distributed on the distal half of the branch, only 8.5 ± 2.11 synapses were needed to initiate a spike with paired-pulse activation (ISI of 20 ms) (Fig. 9B). We conclude from these simulations that tight clustering of activated synapses is not a prerequisite for NMDA spike initiation; high-frequency activation of synapses distributed along the length of the basal branch can also effectively evoke NMDA spikes.
Simulation of the input–output function of the neuron during in vivo-like stimulations: the role of NMDA spikes
To get more realistic spatiotemporal pattern of activation, we simulated a multicompartmental layer 5 pyramidal neuron that receives a signal input on top of a high tonic background activity. The signal input originated from eight presynaptic neurons. Each neuron was activated with a different in vivo recorded firing pattern so that no a priori correlations were created between the activity of the neurons. The mean firing rate of the presynaptic neurons was 0.84 ± 0.27 Hz; burst firing had a lower occurrence of 0.15 ± 0.1 Hz (Fig. 10A,D). Each of these presynaptic cells made 12 synaptic contacts that were randomly distributed on three preselected basal branches of the reconstructed neuron; activation of unitary input lead to 1.23 ± 0.45 mV somatic EPSP. These large unitary events were typical to a small subset of strong presynaptic input that are known to innervate pyramidal neurons (Markram et al., 1997; Song et al., 2005; Lefort et al., 2009).
Activation of the signal inputs lead to a marked effect on the dendritic and somatic potentials. Dendritic NMDA spikes occurred on average every 2.13 ± 1.55 s, with 81 ± 16% of them initiated by a coordinated presynaptic activity (Fig. 10B). It should be stressed that this coordinated activity was simply the result of random correlations between the firing of the different neurons at the firing rates we used. Although presynaptic bursts were not prerequisite for NMDA spike initiation, the probability of a presynaptic burst to initiate a dendritic spike was much higher than that of a single presynaptic action potential (37.5 ± 30 vs 12.5 ± 21%, respectively; p = 0.02) (Fig. 10B,D).
The probability of the neuron to fire an action potential was greatly dependent on the presence of NMDA spikes. Not only was approximately half (55 ± 19%) of the postsynaptic firing initiated during NMDA spikes, but most (77 ± 36%) of the postsynaptic burst firing occurred after a generation of a dendritic NMDA spike (Fig. 10B). Accordingly, the input–output transformation function of the modeled neuron displayed a marked efficacy of the high-frequency inputs in producing postsynaptic firing (Fig. 10E), whereas just 4% of the lone presynaptic action potentials generated somatic firing in the postsynaptic neuron, and 31% of the coordinated presynaptic activities triggered postsynaptic response (42% of the latter were bursts). Blocking NMDA spikes caused to a marked reduction in the postsynaptic firing rate (from 1.16 ± 0.46 Hz in control conditions to 0.37 ± 0.22 Hz after NMDAR blockade; p < 0.01) and postsynaptic burst firing (from 0.23 ± 0.16 Hz in control conditions to 0.05 ± 0.05 Hz after NMDAR blockade; p = 0.012) (Fig. 10C,E).
Discussion
In this study, we investigated the transformation of input signals in thin basal dendrites to output firing patterns in layer 5 pyramidal cells. We found support for the hypothesis that NMDA receptor channels contribute to both the detection and generation of bursts, suggesting that they may play a key role in promoting the propagation of spike bursts through the cortical network.
Mechanism for frequency amplification: prebound glutamate versus residual depolarization
We found that both burst detection and generation effects arose from basic physiological properties of NMDARs. As shown previously, a single synaptic input activates several NMDARs that can remain bound to glutamate with single or double occupancy for several hundred milliseconds (Clements et al., 1992; McBain and Mayer, 1994; Spruston et al., 1995; Kampa et al., 2004; Vargas-Caballero and Robinson, 2004).
Our results suggest that the frequency-dependent amplification of NMDA spike generation arises from a buildup in the number of NMDA receptor channels in the open state during the course of a burst, eventually reaching the total conductance threshold needed for local spike generation. Once this conductance threshold is reached, the depolarization that is created by the NMDA current will enable the regenerative loop that will ultimately lead to an NMDA spike. Such a buildup is possible given that a single activation does not saturate the population of postsynaptic receptors (Mainen et al., 1999). In contrast to the persistence of activated NMDARs, however, we found that carryover of depolarization from the first pulse in a burst had surprisingly little impact on the stimulus threshold at which an NMDA spike could be generated by a second pulse.
Our findings highlight a critical distinction between the essential role that voltage plays in the positive feedback loop that leads to the generation of an NMDA spike once the NMDA conductance threshold has been reached and the relatively minor role that voltage plays in setting a better or worse initial condition for spike generation. A key finding of the present study is that the tendency for NMDA channels to respond regeneratively, which was linked in previous work to the delivery of spatially concentrated inputs (Mel, 1993; Polsky et al., 2004), is also nonlinearly enhanced by temporal accumulation of peak NMDA conductance through high-frequency stimulation of unsaturated excitatory synapses.
The overriding importance of reaching the NMDA conductance threshold locally within a dendritic branch, as opposed to simply reaching a local voltage threshold, has significant functional implications. Whereas dendritic spikes in one or more branches, and backpropagating somatic action potentials, can provide widespread depolarization in a dendritic tree, the critical ingredient in NMDA spike generation is the local NMDA peak conductance, which is tied to events at locally activated synaptic sites. This stresses the ability of basal dendrites to work as independent computational compartments (Poirazi et al., 2003a,b; Polsky et al., 2004; Rhodes, 2006).
Late depression of NMDA spike responses
In addition to the frequency-dependent facilitation of the NMDA spike responses, we observed a late depression process that developed ∼500 ms after NMDA spike initiation and could last for several seconds. Activity-dependent inactivation of NMDARs has been described previously after prolonged NMDAR activation with agonists (Kyrozis et al., 1996), with strong presynaptic stimulation (Medina et al., 1999), and even after a single EPSP (Mennerick and Zorumski, 1996). It is conceivable that, during prolonged synaptic stimulation or after an NMDA spike, as in these other scenarios, NMDA receptor channels enter an activity-dependent desensitized state. Because of the efficacy of burst generation attributable to NMDA spikes, the late depression might serve as a negative feedback mechanism for restraining excessive high-frequency activity in the network.
Comparison with other forms of short-term plasticity
Frequency-dependent short-term synaptic plasticity (STP) is ubiquitous in the brain, occurring at many types of synapses (Zucker and Regehr, 2002). The best studied mechanisms mediating STP are presynaptic, most notably changes in release probability (Zucker and Regehr, 2002). Postsynaptic dendritic mechanisms can have different effects on STP; in some cases, they increase short-term facilitation (Akopian and Walsh, 2002) and in others, they reduce summation of high-frequency synaptic inputs, making synaptic signaling more linear (Magee, 1998). In this work, we have described a form of STP in basal dendrites involving the facilitation of the total postsynaptic NMDA conductance during repeated synaptic activation. This effect is mainly mediated by NMDAR channels containing NR2A subunits, with little contribution of the NR2B subunits, indicating that the NMDA spikes are mediated by synaptic receptors, with little or no contribution of extrasynaptic NMDAR channels (Tovar and Westbrook, 1999; Liu et al., 2004; Lozovaya et al., 2004; Köhr, 2006).
The postsynaptic STP effect we describe here differs in two fundamental ways from classical presynaptic STP. First, whereas the dynamics of classical STP are determined solely by the history of the specific activated synapse (but see Akopian and Walsh, 2002 for a discussion of cooperative potentiation), the degree of amplification during burst activation of NMDA receptors depends on the intensity of postsynaptic activation, changing abruptly at the transition between inputs that are subthreshold versus suprathreshold for NMDA spike initiation. Second, the degree of amplification by NMDA channels also depends on the history of activation of neighboring synapses, because NMDA spike initiation will in general depend on the coactivation of numerous synapses located within the same dendritic compartment (Polsky et al., 2004). Thus, whereas classical presynaptic STP is synapse specific and governed by the temporal activation pattern at that synapse alone, the postsynaptic form of STP we describe here is dendritic compartment specific and governed by both the spatial and temporal activation pattern of multiple contributory synapses.
Burst firing in pyramidal neurons
Burst firing is a common firing mode of cortical pyramidal neurons that is thought to be important for both neural coding and the induction of long-term plasticity processes (Birtoli and Ulrich, 2004; Froemke et al., 2006). Specialized cellular mechanisms support burst firing in CA1 and layer 5 pyramidal neurons (Azouz et al., 1996; Williams and Stuart, 1999). Prolonged depolarizing dendritic currents leading to burst firing, especially voltage-gated calcium channels, are prevalent in apical dendrites (Larkum et al., 1999), whereas basal dendrites are evidently incapable of supporting initiation of local calcium spikes (Nevian et al., 2007). Here we describe a new mechanism for burst generation in basal dendrites of layer 5 pyramidal neurons that involves the initiation of NMDA spikes. Similar to the apical dendritic spiking mechanism described previously, which can be activated by pairing of distal EPSPs and backpropagating action potentials (Larkum et al., 1999), the NMDA spike-dependent basal burst-generating mechanism we describe here also depends on coordinated spatiotemporal activation of synaptic inputs. However, burst generation in apical and basal dendrites differs in at least two ways. First, the spatial requirements for burst generation in basal dendrites are more restrictive and may be confined to a single basal branch. Hence, a single branch can act as an independent input–output information processing compartment not only at the subthreshold level but at the suprathreshold level as well (Poirazi et al., 2003a,b; Polsky et al., 2004). Second, basal dendrites impose different temporal requirements for burst generation compared with apically derived bursts. Whereas apical bursts mediated by VDCCs are initiated once a voltage threshold has been reached, regardless of the frequency of activation, NMDA spike-mediated bursts require that a threshold number of NMDA channels be fully activated by glutamate, which depends more strongly on stimulus frequency than on absolute voltage. Thus, in contrast to basal NMDA spikes, apical calcium spikes are not known to depend on bursts of synaptic input, suggesting that their role in facilitating burst propagation may be more indirect.
Conditions for NMDA spike initiation in vivo
To date, NMDA spikes have only been described under slice in vitro conditions (Schiller et al., 2000; Polsky et al., 2004; Major et al., 2008). It remains an open question under what conditions, if any, NMDA spikes occur in vivo. Modeling data presented here suggests that coactivation of only 20 synapses distributed along a basal branch, and even fewer if placed distally, can be sufficient to initiate NMDA spikes. Assuming two to three synaptic contacts per 1 μm of dendritic length (Harris et al., 1992), a thin dendrite can be contacted by hundreds of synaptic contacts, meaning that only a small percentage of the contacts on a branch must fire together to produce an NMDA spike.
Our simulations show that, in a model of a small subpopulation of presynaptic neurons that converge onto a subset of basal dendrites and fire with in vivo-like temporal patterns, NMDA spikes can be effectively generated and have a profound influence on the input–output function of the neurons. Our assumed presynaptic stimulation pattern was based on the high degree of connectivity found in some local cortical networks, such as between nearby layer 5 neocortical pyramidal neurons (Markram et al., 1997; Song et al., 2005). Recent work by Lefort et al. (2009) has also revealed high-amplitude synaptic connections in all types of neurons in the somatosensory column producing EPSPs up to 7.8 mV, which are important in driving the network to fire. It is also the case that synaptic connections between cortical neurons can form clusters of two to five closely spaced synaptic contacts per site (Rollenhagen and Lübke, 2006). Based on these anatomical and physiological data, the presynaptic innervation pattern assumption of our model, in which relatively strong unitary connections are mapped to specific branches, is a reasonable description of an important subset of the presynaptic input to pyramidal neuron basal dendrites. These types of strong inputs may be particularly well suited to contribute to active dendritic responses.
In conclusion, in a small realistic network, dendritic NMDA spikes can have a profound effect on the firing rate and the input–output transformation of the neuron. This spiking mechanism may enable the cell to reliably read presynaptic burst activity from a small number of presynaptic neurons and propagate them through the network, even during high background noise conditions.
Footnotes
This study was supported by the Israel Science Foundation, the Rappaport Foundation, National Science Foundation Collaborative Research in Computational Neuroscience Grant IIS-0613583, and National Institute of Mental Health Grant 5R01MH065918. We thank Y. Schiller for his helpful comments and discussion on this manuscript.
References
- Abbott LF, Regehr WG. Synaptic computation. Nature. 2004;431:796–803. doi: 10.1038/nature03010. [DOI] [PubMed] [Google Scholar]
- Akopian G, Walsh JP. Corticostriatal paired-pulse potentiation produced by voltage-dependent activation of NMDA receptors and L-type Ca2+ channels. J Neurophysiol. 2002;87:157–165. doi: 10.1152/jn.00115.2001. [DOI] [PubMed] [Google Scholar]
- Azouz R, Jensen MS, Yaari Y. Ionic basis of spike after-depolarization and burst generation in adult rat hippocampal CA1 pyramidal cells. J Physiol. 1996;492:211–223. doi: 10.1113/jphysiol.1996.sp021302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birtoli B, Ulrich D. Firing mode-dependent synaptic plasticity in rat neocortical pyramidal neurons. J Neurosci. 2004;24:4935–4940. doi: 10.1523/JNEUROSCI.0795-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattaneo A, Maffei L, Morrone C. Two firing patterns in the discharge of complex cells encoding different attributes of the visual stimulus. Exp Brain Res. 1981;43:115–118. doi: 10.1007/BF00238819. [DOI] [PubMed] [Google Scholar]
- Chagnac-Amitai Y, Luhmann HJ, Prince DA. Burst generating and regular spiking layer 5 pyramidal neurons of rat neocortex have different morphological features. J Comp Neurol. 1990;296:598–613. doi: 10.1002/cne.902960407. [DOI] [PubMed] [Google Scholar]
- Chen L, Cooper NG, Mower GD. Developmental changes in the expression of NMDA receptor subunits (NR1, NR2A, NR2B) in the cat visual cortex and the effects of dark rearing. Brain Res Mol Brain Res. 2000;78:196–200. doi: 10.1016/s0169-328x(00)00076-0. [DOI] [PubMed] [Google Scholar]
- Clements JD, Lester RA, Tong G, Jahr CE, Westbrook GL. The time course of glutamate in the synaptic cleft. Science. 1992;258:1498–1501. doi: 10.1126/science.1359647. [DOI] [PubMed] [Google Scholar]
- de Kock CP, Bruno RM, Spors H, Sakmann B. Layer- and cell-type-specific suprathreshold stimulus representation in rat primary somatosensory cortex. J Physiol. 2007;581:139–154. doi: 10.1113/jphysiol.2006.124321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froemke RC, Tsay IA, Raad M, Long JD, Dan Y. Contribution of individual spikes in burst-induced long-term synaptic modification. J Neurophysiol. 2006;95:1620–1629. doi: 10.1152/jn.00910.2005. [DOI] [PubMed] [Google Scholar]
- Haider B, Duque A, Hasenstaub AR, McCormick DA. Neocortical network activity in vivo is generated through a dynamic balance of excitation and inhibition. J Neurosci. 2006;26:4535–4545. doi: 10.1523/JNEUROSCI.5297-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KM, Jensen FE, Tsao B. Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: implications for the maturation of synaptic physiology and long-term potentiation. J Neurosci. 1992;12:2685–2705. doi: 10.1523/JNEUROSCI.12-07-02685.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh CY, Chen Y, Leslie FM, Metherate R. Postnatal development of NR2A and NR2B mRNA expression in rat auditory cortex and thalamus. J Assoc Res Otolaryngol. 2002;3:479–487. doi: 10.1007/s10162-002-2052-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izhikevich EM, Desai NS, Walcott EC, Hoppensteadt FC. Bursts as a unit of neural information: selective communication via resonance. Trends Neurosci. 2003;26:161–167. doi: 10.1016/S0166-2236(03)00034-1. [DOI] [PubMed] [Google Scholar]
- Kampa BM, Clements J, Jonas P, Stuart GJ. Kinetics of Mg2+ unblock of NMDA receptors: implications for spike-timing dependent synaptic plasticity. J Physiol. 2004;556:337–345. doi: 10.1113/jphysiol.2003.058842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kepecs A, Lisman J. Information encoding and computation with spikes and bursts. Network. 2003;14:103–118. [PubMed] [Google Scholar]
- Kew JN, Richards JG, Mutel V, Kemp JA. Developmental changes in NMDA receptor glycine affinity and ifenprodil sensitivity reveal three distinct populations of NMDA receptors in individual rat cortical neurons. J Neurosci. 1998;18:1935–1943. doi: 10.1523/JNEUROSCI.18-06-01935.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Alger BE. Random response fluctuations lead to spurious paired-pulse facilitation. J Neurosci. 2001;21:9608–9618. doi: 10.1523/JNEUROSCI.21-24-09608.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhr G. NMDA receptor function: subunit composition versus spatial distribution. Cell Tissue Res. 2006;326:439–446. doi: 10.1007/s00441-006-0273-6. [DOI] [PubMed] [Google Scholar]
- Kyrozis A, Albuquerque C, Gu J, MacDermott AB. Ca2+-dependent inactivation of NMDA receptors: fast kinetics and high Ca2+ sensitivity in rat dorsal horn neurons. J Physiol. 1996;495:449–463. doi: 10.1113/jphysiol.1996.sp021606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkman AU. Dendritic morphology of pyramidal neurones of the visual cortex of the rat. I. Branching patterns. J Comp Neurol. 1991a;306:307–319. doi: 10.1002/cne.903060207. [DOI] [PubMed] [Google Scholar]
- Larkman AU. Dendritic morphology of pyramidal neurones of the visual cortex of the rat. III. Spine distributions. J Comp Neurol. 1991b;306:332–343. doi: 10.1002/cne.903060209. [DOI] [PubMed] [Google Scholar]
- Larkum ME, Zhu JJ, Sakmann B. A new cellular mechanism for coupling inputs arriving at different cortical layers. Nature. 1999;398:338–341. doi: 10.1038/18686. [DOI] [PubMed] [Google Scholar]
- Lefort S, Tomm C, Floyd Sarria JC, Petersen CC. The excitatory neuronal network of the C2 barrel column in mouse primary somatosensory cortex. Neuron. 2009;61:301–316. doi: 10.1016/j.neuron.2008.12.020. [DOI] [PubMed] [Google Scholar]
- Liu XB, Murray KD, Jones EG. Switching of NMDA receptor 2A and 2B subunits at thalamic and cortical synapses during early postnatal development. J Neurosci. 2004;24:8885–8895. doi: 10.1523/JNEUROSCI.2476-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozovaya NA, Grebenyuk SE, Tsintsadze TSh, Feng B, Monaghan DT, Krishtal OA. Extrasynaptic NR2B and NR2D subunits of NMDA receptors shape “superslow” afterburst EPSC in rat hippocampus. J Physiol. 2004;558:451–463. doi: 10.1113/jphysiol.2004.063792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC. Dendritic hyperpolarization-activated currents modify the integrative properties of hippocampal CA1 pyramidal neurons. J Neurosci. 1998;18:7613–7624. doi: 10.1523/JNEUROSCI.18-19-07613.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainen ZF, Sejnowski TJ. Influence of dendritic structure on firing pattern in model neocortical neurons. Nature. 1996;382:363–366. doi: 10.1038/382363a0. [DOI] [PubMed] [Google Scholar]
- Mainen ZF, Malinow R, Svoboda K. Synaptic calcium transients in single spines indicate that NMDA receptors are not saturated. Nature. 1999;399:151–155. doi: 10.1038/20187. [DOI] [PubMed] [Google Scholar]
- Major G, Polsky A, Denk W, Schiller J, Tank DW. Spatio-temporally graded NMDA spike/plateau potentials in basal dendrites of neocortical pyramidal neurons. J Neurophysiol. 2008;99:2584–2601. doi: 10.1152/jn.00011.2008. [DOI] [PubMed] [Google Scholar]
- Markram H, Lübke J, Frotscher M, Roth A, Sakmann B. Physiology and anatomy of synaptic connections between thick tufted pyramidal neurones in the developing rat neocortex. J Physiol. 1997;500:409–440. doi: 10.1113/jphysiol.1997.sp022031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Gupta A, Uziel A, Wang Y, Tsodyks M. Information processing with frequency-dependent synaptic connections. Neurobiol Learn Mem. 1998;70:101–112. doi: 10.1006/nlme.1998.3841. [DOI] [PubMed] [Google Scholar]
- Martinez-Conde S, Macknik SL, Hubel DH. Microsaccadic eye movements and firing of single cells in the striate cortex of macaque monkeys. Nat Neurosci. 2000;3:251–258. doi: 10.1038/72961. [DOI] [PubMed] [Google Scholar]
- McBain CJ, Mayer ML. N-methyl-d-aspartic acid receptor structure and function. Physiol Rev. 1994;74:723–760. doi: 10.1152/physrev.1994.74.3.723. [DOI] [PubMed] [Google Scholar]
- Medina I, Leinekugel X, Ben-Ari Y. Calcium-dependent inactivation of the monosynaptic NMDA EPSCs in rat hippocampal neurons in culture. Eur J Neurosci. 1999;11:2422–2430. doi: 10.1046/j.1460-9568.1999.00664.x. [DOI] [PubMed] [Google Scholar]
- Mel BW. Synaptic integration in an excitable dendritic tree. J Neurophysiol. 1993;70:1086–1101. doi: 10.1152/jn.1993.70.3.1086. [DOI] [PubMed] [Google Scholar]
- Mennerick S, Zorumski CF. Postsynaptic modulation of NMDA synaptic currents in rat hippocampal microcultures by paired-pulse stimulation. J Physiol. 1996;490:405–417. doi: 10.1113/jphysiol.1996.sp021154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milojkovic BA, Radojicic MS, Goldman-Rakic PS, Antic SD. Burst generation in rat pyramidal neurones by regenerative potentials elicited in a restricted part of the basilar dendritic tree. J Physiol. 2004;558:193–211. doi: 10.1113/jphysiol.2004.061416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myme CI, Sugino K, Turrigiano GG, Nelson SB. The NMDA-to-AMPA ratio at synapses onto layer 2/3 pyramidal neurons is conserved across prefrontal and visual cortices. J Neurophysiol. 2003;90:771–779. doi: 10.1152/jn.00070.2003. [DOI] [PubMed] [Google Scholar]
- Nevian T, Larkum ME, Polsky A, Schiller J. Properties of basal dendrites of layer 5 pyramidal neurons: a direct patch-clamp recording study. Nat Neurosci. 2007;10:206–214. doi: 10.1038/nn1826. [DOI] [PubMed] [Google Scholar]
- Poirazi P, Brannon T, Mel BW. Arithmetic of subthreshold synaptic summation in a model CA1 pyramidal cell. Neuron. 2003a;37:977–987. doi: 10.1016/s0896-6273(03)00148-x. [DOI] [PubMed] [Google Scholar]
- Poirazi P, Brannon T, Mel BW. Pyramidal neuron as two-layer neural network. Neuron. 2003b;37:989–999. doi: 10.1016/s0896-6273(03)00149-1. [DOI] [PubMed] [Google Scholar]
- Polsky A, Mel BW, Schiller J. Computational subunits in thin dendrites of pyramidal cells. Nat Neurosci. 2004;7:621–627. doi: 10.1038/nn1253. [DOI] [PubMed] [Google Scholar]
- Portera-Cailliau C, Price DL, Martin LJ. N-methyl-D-aspartate receptor proteins NR2A and NR2B are differentially distributed in the developing rat central nervous system as revealed by subunit-specific antibodies. J Neurochem. 1996;66:692–700. doi: 10.1046/j.1471-4159.1996.66020692.x. [DOI] [PubMed] [Google Scholar]
- Rancz EA, Ishikawa T, Duguid I, Chadderton P, Mahon S, Häusser M. High-fidelity transmission of sensory information by single cerebellar mossy fibre boutons. Nature. 2007;450:1245–1248. doi: 10.1038/nature05995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes P. The properties and implications of NMDA spikes in neocortical pyramidal cells. J Neurosci. 2006;26:6704–6715. doi: 10.1523/JNEUROSCI.3791-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollenhagen A, Lübke JH. The morphology of excitatory central synapses: from structure to function. Cell Tissue Res. 2006;326:221–237. doi: 10.1007/s00441-006-0288-z. [DOI] [PubMed] [Google Scholar]
- Schiller J, Major G, Koester HJ, Schiller Y. NMDA spikes in basal dendrites of cortical pyramidal neurons. Nature. 2000;404:285–289. doi: 10.1038/35005094. [DOI] [PubMed] [Google Scholar]
- Snider RK, Kabara JF, Roig BR, Bonds AB. Burst firing and modulation of functional connectivity in cat striate cortex. J Neurophysiol. 1998;80:730–744. doi: 10.1152/jn.1998.80.2.730. [DOI] [PubMed] [Google Scholar]
- Song S, Sjöström PJ, Reigl M, Nelson S, Chklovskii DB. Highly nonrandom features of synaptic connectivity in local cortical circuits. PLoS Biol. 2005;3:e68. doi: 10.1371/journal.pbio.0030068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruston N, Jonas P, Sakmann B. Dendritic glutamate receptor channels in rat hippocampal CA3 and CA1 pyramidal neurons. J Physiol. 1995;482:325–352. doi: 10.1113/jphysiol.1995.sp020521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci. 1999;19:4180–4188. doi: 10.1523/JNEUROSCI.19-10-04180.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas-Caballero M, Robinson HP. Fast and slow voltage-dependent dynamics of magnesium block in the NMDA receptor: the asymmetric trapping block model. J Neurosci. 2004;24:6171–6180. doi: 10.1523/JNEUROSCI.1380-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters J, Helmchen F. Background synaptic activity is sparse in neocortex. J Neurosci. 2006;26:8267–8277. doi: 10.1523/JNEUROSCI.2152-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SR, Stuart GJ. Mechanisms and consequences of action potential burst firing in rat neocortical pyramidal neurons. J Physiol. 1999;521:467–482. doi: 10.1111/j.1469-7793.1999.00467.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JJ, Connors BW. Intrinsic firing patterns and whisker-evoked synaptic responses of neurons in the rat barrel cortex. J Neurophysiol. 1999;81:1171–1183. doi: 10.1152/jn.1999.81.3.1171. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]