

Abstract
CD47 transduces inhibitory signals through signal-regulatory protein α (SIRPα), a plasma membrane receptor expressed by macrophages. Many cancers upregulate CD47 to evade immunosurveillance. We have recently engineered SIRPα variants that potently antagonize CD47 for use as anticancer immunotherapeutics. These high-affinity SIRPα variants synergize with antineoplastic antibodies by lowering the threshold for macrophage-mediated destruction of malignant cells.
Keywords: macrophage, cancer, antibody, CD47, SIRPa, immunotherapy, phagocytosis, immune checkpoint, molecular engineering
CD47 is a valuable target for anticancer therapy due to its function as an inhibitor of macrophage phagocytosis as well as its broad expression on a variety of human neoplasms. By binding to signal-regulatory protein α (SIRPα), a receptor expressed on the surface of macrophages, CD47 is able to transduce inhibitory signals that prevent phagocytosis (Fig. 1). We and others have recently demonstrated that blocking the interaction between CD47 and SIRPα with antibodies not only stimulates macrophages to engulf cancer cells in vitro but also exerts robust anticancer effects in vivo.1-4 We have now developed “next-generation” CD47 antagonists that bind and block human CD47 with extraordinarily high affinity.5
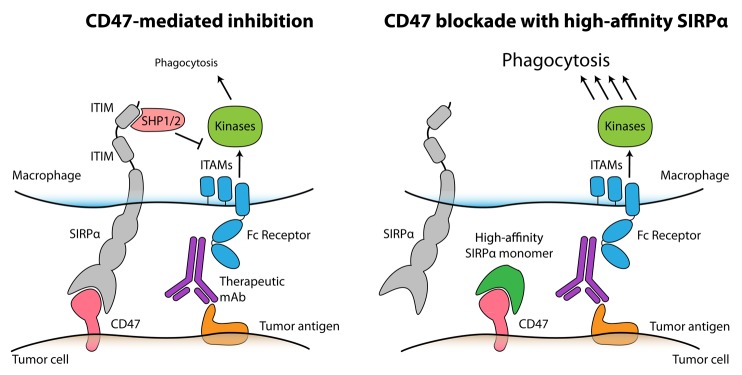
Figure 1. High-affinity SIRPα variants reduce the threshold for macrophage phagocytosis, thus enhancing the efficacy of anticancer monoclonal antibodies. Therapeutic monoclonal antibodies (mAbs) engage Fc receptors on the surface of macrophages, hence transducing positive signals via immunotyrosine-based activating motifs (ITAMs) and downstream mediators to stimulate phagocytosis. However, the phagocytic response of macrophages is limited by the expression of CD47 on tumor cells, as CD47 engages macrophage signal-regulatory protein α (SIRPα) to initiate an inhibitory signaling cascades mediated by immunotyrosine-based inhibitory motifs (ITIMs) and resulting in the activation of SHP-1 and SHP-2 phosphatases (left). The administration of high-affinity SIRPα monomers blocks CD47 on malignant cells and hence disables endogenous SIRPα signaling on macrophages. In combination with therapeutic antibodies, high-affinity SIRPα variants stimulate phagocytosis and hence mediate synergistic antitumor responses (right).
Taking a rational approach to drug design, we hypothesized that the CD47-binding N-terminal domain of SIRPα could be produced by recombinant techniques and used as a competitive CD47 antagonist. However, we found that the N-terminus of wild-type SIRPα is an ineffective CD47 antagonist and fails to stimulate phagocytosis, presumably owing to its poor binding affinity (Kd ~1 μM). Thus, we undertook a structure-based engineering strategy to improve the affinity of SIRPα for CD47. Informed by the X-ray crystal structure of human SIRPα in complex with CD47,6 we generated a combinatorial library of SIRPα variants by specifically selecting a number of amino acids for mutation. These residues appeared to be important either for the contact between SIRPα and CD47 or for the stabilization of the SIRPα hydrophobic core. Using in vitro evolution by yeast surface display, with the extracellular domain of human CD47 as a selection reagent, we isolated SIRPα variants with nine amino acid substitutions and an affinity for CD47 as low as 11.1 pM, that is, approximately 50,000-fold higher than that of wild-type SIRPα.
To evaluate the functional properties of high-affinity SIRPα variants, we developed a high-throughput assay that allows for the assessment of the phagocytic response by macrophages to cancer cells in vitro. This assay enabled us to test the ability of high-affinity SIRPα variants to modulate phagocytosis over a range of experimental conditions. In particular, we used this system to evaluate the dose-response relationship of antibodies that stimulate the phagocytic uptake of cancer cells upon opsonization, either employed alone or combined with high-affinity SIRPα variants. Indeed, high-affinity SIRPα variants increased the maximal efficacy and the potency of antineoplastic antibodies such as the CD20-targeting antibody rituximab and the epidermal growth factor receptor (EGFR)-specific antibody cetuximab.
Using xenograft murine models of human tumors, we found the phagocytosis assays were highly predictive of therapeutic responses in vivo. Thus, the co-administration of high-affinity SIRPα monomers and rituximab to lymphoma-bearing mice resulted in remarkable synergy, producing cures that persisted long after treatment discontinuation in a majority of animals. In contrast, the administration of either agent alone only caused a modest inhibition of tumor growth. Similar effects were observed when high-affinity SIRPα monomers were combined with an anti-CD52 antibody (alemtuzumab) in models of lymphoma or with an antibody specific for v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 2 (ERBB/HER2) (trastuzumab) in models of breast carcinoma. Importantly, high-affinity SIRPα monomers proved to be safe at elevated doses in a preliminary toxicity study in cynomolgus macaques, providing strong rationale for thoroughly evaluating these molecules in preparation for their translation to the clinic.
By disabling the inhibitory signals transduced by SIRPα, high-affinity SIRPα variants reduce the threshold for macrophage activation and promote phagocytic response driven by tumor-specific antibodies (Fig. 1). The degree to which the anticancer activity of a given therapeutic antibody is enhanced by CD47 blockade likely depends on multiple factors, including the levels of antigen expression on the surface of malignant cells, the isotype of its heavy chain, and the orientation assumed by the antibody upon antigen binding, which affects its ability to engage Fc receptors on immune effectors. Nonetheless, high-affinity SIRPα variants could transform antibodies with a limited ability to stimulate immune effectors into robust stimulators of phagocytic responses. Therefore, from a clinical perspective, high-affinity SIRPα variants could greatly expand the possibility of therapeutic antibodies, rescuing the potential of otherwise ineffective antibodies and extending the efficacy of successful molecules. High-affinity SIRPα monomers represent therefore a rapid, safe and effective alternative to several other approaches, including drug/toxin conjugation strategies, that have been pursued in this direction.
In the laboratory, multiple antibodies that recognize a specific combination of cell surface antigens can be used to discriminate malignant cells from the surrounding normal tissue. In the future, when additional therapeutic antibodies become available in the clinic, the same principles will be applicable to the treatment of cancer patients. These artificial, oligoclonal antibody cocktails might indeed represent a personalized approach to anticancer therapy. At least theoretically, each malignant lesion could be profiled for cell surface antigens, leading to the production of individualized cocktails of antibodies that optimally simulate tumor-specific adaptive immune responses. In this way, combinations of antibodies could be selected for their ability to optimally bind malignant cells and to induce limited toxicity to normal tissues that express only one of the antigens recognized by the cocktail. By combining these cocktails with high-affinity SIRPα variants and other strategies for the inactivation of immune checkpoints, the full promise of anticancer immunotherapy could be realized.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/oncoimmunology/article/25773
References
- 1.Majeti R, Chao MP, Alizadeh AA, Pang WW, Jaiswal S, Gibbs KD, Jr., et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. 2009;138:286–99. doi: 10.1016/j.cell.2009.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chao MP, Alizadeh AA, Tang C, Myklebust JH, Varghese B, Gill S, et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell. 2010;142:699–713. doi: 10.1016/j.cell.2010.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Willingham SB, Volkmer JP, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci U S A. 2012;109:6662–7. doi: 10.1073/pnas.1121623109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Edris B, Weiskopf K, Volkmer AK, Volkmer JP, Willingham SB, Contreras-Trujillo H, et al. Antibody therapy targeting the CD47 protein is effective in a model of aggressive metastatic leiomyosarcoma. Proc Natl Acad Sci U S A. 2012;109:6656–61. doi: 10.1073/pnas.1121629109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weiskopf K, Ring AM, Ho CC, Volkmer JP, Levin AM, Volkmer AK, et al. Engineered SIRPα variants as immunotherapeutic adjuvants to anticancer antibodies. Science. 2013;341:88–91. doi: 10.1126/science.1238856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hatherley D, Graham SC, Turner J, Harlos K, Stuart DI, Barclay AN. Paired receptor specificity explained by structures of signal regulatory proteins alone and complexed with CD47. Mol Cell. 2008;31:266–77. doi: 10.1016/j.molcel.2008.05.026. [DOI] [PubMed] [Google Scholar]