

Abstract
Vascular tone control is essential in blood pressure regulation, shock, ischemia-reperfusion, inflammation, vessel injury/repair, wound healing, temperature regulation, digestion, exercise physiology, and metabolism. Here we show that a well-known growth factor, FGF2, long thought to be involved in many developmental and homeostatic processes, including growth of the tissue layers of vessel walls, functions in vascular tone control. Fgf2 knockout mice are morphologically normal and display decreased vascular smooth muscle contractility, low blood pressure and thrombocytosis. Following intra-arterial mechanical injury, FGF2-deficient vessels undergo a normal hyperplastic response. These results force us to reconsider the function of FGF2 in vascular development and homeostasis in terms of vascular tone control.
Many studies have implicated FGF2 in the growth and differentiation of a variety of tissues. It is a potent mesoderm inducer in amphibians1, and its addition to embryo explants enhances development of muscle and blood2–4. In cultured murine cardiomyocytes it induces proliferation5 and reversion of cardiac structural muscle protein isoforms to the fetal state6. Related to the vasculature, FGF2 is a potent angiogenic factor7, affects vascular morphogenesis of pre-endothelial cells of the mouse embryo8, mediates the mitogenic effects of thrombin on vascular smooth muscle cells9, transduces the hypertrophic response of vascular smooth muscle cells to various vasoconstrictors in culture10,11, and amplifies serum-stimulated vascular smooth muscle cell proliferation through both intracrine and autocrine mechanisms12. Expression data in mouse embryos shows Fgf2 expression at low levels throughout early stages of embryogenesis followed by high levels of expression in limb, face and heart/lung by embryonic day 13.5 (e13.5)13. In adult mammals FGF2 enhances cardiac neovascularization after ischemia-reperfusion injury14. It can accelerate regrowth of endothelium in balloon-catheterized rat arteries15 and neovascularization in healing wounds of diabetic mice16. Finally, overexpression of FGF2 in transgenic mice increases resistance to hypoxemic/ischemic cerebral damage17 and causes congenital bone defects18.
In order to better define the developmental and homeostatic functions of FGF2 we have targeted a null mutation in mouse Fgf2 using homologous recombination in embryonic stem (ES) cells. The resulting mice reveal a role for FGF2 in hematopoiesis, but not in the development of other tissues, including heart and skeleton. In the adult mouse a homeostatic role for FGF2 in vascular tone control was found, though a deficiency in FGF2 had no apparent effect on vessel repair following mechanical injury.
Production and characterization of the Fgf2 knockout allele
The Fgf2 gene was ablated by replacing a 0.5-kilobase (kb) portion of the gene including 121 bp of the proximal promoter region and the entire first exon with an Hprt minigene (Fig. 1a) in the HPRT-deficient E14TG2a ES cell line19. This strategy removes the first 59 amino acids, which have been shown to be involved in mitogenic activity and heparin and receptor binding20–22. Germline chimeras containing the null mutation were generated by blastocyst injection of one of the targeted cell lines (Fig. 1b). Animals homozygous for the targeted allele are viable (Fig. 1, c–e). Northern blot analysis of e13.5 embryos and several tissues from Fgf2−/− mice shows that no wild-type or truncated message containing exon 2 and 3 sequences are detectable (Fig. 1d). Western blot analysis on brain tissue demonstrates the absence of 18-, 21-and 22-kDa translational products in these mice (Fig. 1e).
Fig. 1.

Generation of FGF2-deficient mice, a, Targeting scheme for generation of an Fgf2 null allele in mouse. The upper line (wild-type allele) represents a 7.0-kb Scrcl/Hindlll fragment of the wild-type Fgf2 genomic DNA, which contains the promoter region and the first exon (filled box). The targeting construct contains a 0.7-kb short homology arm at the 5′ end and a 3.0-kb long homology arm at the 3′ end of the Hprt minigene (open box). A thymidine kinase gene (hatched box) is used for negative selection. Homologous recombination between the wild-type allele and the targeting construct deletes part of the promoter and entire first exon of the Fgf2 gene (targeted allele). Long arrows represent the PCR primers used to identify the targeted allele; arrowheads represent primers used to identify the wild-type allele. Probe A is a 0.6-kb “outside” probe and probe B is a 0.9-kb “inside” probe. B: BamHI, H: Hindlll, N: Narl, S: Sacl, X: Xbal. b, Southern blot analysis of DNA from electroporated ES clones following HAT and gancyclovir selection. A representative clone, G8, is shown. DNA was digested with different restriction enzymes and probed with either probe A or probe B. The expected bands for BamHi digestion: wild-type (WT), 5.0 kb; mutant (Mut), 7.5 kb. For Socl digestion: WT, 4.2 kb, Mut, 6.7 kb. For H/ndlll digestion: WT, 4.2 kb, Mut, 3.9 kb. c, Southern blot analysis of viable offspring born from parents heterozygous for the targeted allele. DNA isolated from tail clips was digested with Hindlll and probed with probe B. Wild-type mice (+/+) show a 4.2-kb band and mutant mice (−/−) show a 3.9-kb band. Heterozygous mice (+/−) show both the 4.2- and 3.9-kb diagnostic bands, d, Northern blot analysis of RNA isolated from E1 3.5 embryos and several adult tissues from 2-month-old mice from heterozygous matings. Probing poly(A)+ RNA (5 μg) message with a probe consisting of exon 2 and 3 of Fgf2 detects a 5.9-kb Fgf2 message in tissues from wild-type (+/+) mice but not in knockouts (−/−) even after extended exposure for 104 h (not shown). With mouse Fgf1 cDNA as probe, a 3.4-kb and a 2.1-kb mRNA are detected in adult brain, heart and kidney. With mouse Fgf5 cDNA as probe, Fgf5 is very abundantly detected (predominantly as a 2.4-kb species) in skin. The band immediately below the 2.4-kb Fgf5 mRNA is the residual β-actin signal after stripping. Hybridization with a β-actin probe served as RNA loading control. Note that expression of Fgf1 and 5 was similar in Fgf2+/+ and Fgf2−/− mice, e Western blot analysis of FGF2 protein in brain. Brain from Fgf2+/+ and Fgf2+/− mice expressed an 18-kDa FGF2 as well as two high molecular mass species of 21 and 22 kDa. All three protein species were absent from Fgf2+/+ mice.
Survey of developmental and hematopoiesis defects
Intercrossing Fgf2+/− mice yields a 1:2:1 ratio of +/+, +/− and −/− offspring. Fgf2−/− mice exhibit no discernible morphologic or behavioral defects and have a normal life span. Both sexes are fertile and fecundity is normal. Histological examination of newborn and adult Fgf2−/− mice (not shown) reveals no gross or microscopic abnormalities in any of the organs and tissues examined, notably brain, heart, aorta, portal vein, bone and skeletal muscle. Normal organogenesis, normal animal growth and normal female reproductive activity indicate that angiogenic processes are unaffected by the absence of FGF2. Northern blot analysis of other Fgfs was performed to determine whether they are upregulated to compensate for the loss of FGF2. Expression levels of Fgf1 or Fgf5 (Fig. 1d) or Fgf4 (not shown) are not altered in 13.5-day-old embryos or in adult brain, heart, skin and kidney. White and red blood cell counts are similar to those of wild-type mice. However, platelet counts are elevated (669 ± 87 × 103/mm3 in Fgf2−/− mice vs. 472 ± 68 × 103/mm3 in control mice; n = 8, P < 0.05), but platelet aggregation, assessed by in vitro aggregometry and microvascular bleeding time, is normal (Table 1). Methylcellulose culture of bone marrow cells from FGF2-deficient mice revealed a general decrease in interleukin 3 (IL-3)-induced colony formation and an increase in megakaryocyte colony-stimulating activity-induced megakaryocyte colony formation, compared with cultures of wild-type cells (Table 2). Colony proliferation in response to GM-CSF and erythropoietin, mitogens promoting granulocyte/macrophage and erythroid lineages, respectively, were not different between bone marrow cultures established from wild-type or Fgf2 knockout mice.
Table 1.
Peripheral blood cell counts from FGF2-deficient mice
| RBC (×106/ml) | WBC (×103/mm3) | Platelets (×103/mm3) | |
|---|---|---|---|
| Fgf2 (+/+) (+/−) | 9.0 ± 2.4 (16) | 4.6 ± 1.8 (13) | 505 ± 171 (12) |
| Fgf2(−/−) | 10.0 ± 1.0 (16) | 4.7 ± 1.1 (13) | 703 ± 214* (12) |
Values are means ± s.e.m. (number of mice) of peripheral blood ceils counted from the number of mice shown in parentheses. RBC, red blood cells; WBC, white blood cells. Experimental (Fgf2−/−) values differ significantly from control values
P < 0.01.
Table 2.
Total colony counts from bone marrow cultures stimulated with different mitogens
| CM-CSF (20 U/ml) | IL-3 (10 ng/ml) | EPO (2 U/ml) | PWM-SCM (10%) | |
|---|---|---|---|---|
| Fgf2 (+/+)(+/−) | 175 ± 139 (5) | 248 ± 83 (5) | 76 ± 28 (3) | 94 ± 43 (6) |
| Fgf2(−/−) | 196 ± 159 (5) | 169 ± 75** (5) | 153 ± 71 (3) | 155 ± 60* (6) |
Values are means ± s.e.m. (n) of total number of colonies containing at least 5 cells counted per individual methylcellulose culture of 2 × 105 (GM-CSF or IL-3) or 1 × 106 (EPO or PWM-SCM) bone marrow cells after 1 week in the presence of mitogens. GM-CSF, granulocyte/macrophage colony-stimulating factor; IL-3, interieukin 3; EPO, erythropoietln; PWM-SCM, pokeweed mitogen-stimulated spleen cell-conditioned medium used at 10% vol/vol with bone marrow culture medium. Experimental values differ significantly from control values by Student's t-test
P < 0.05 or
P < 0.01.
Role of FGF2 in the cardiovascular system
FGF2 is believed to play an important role in cardiovascular development and function23,24. Since the hearts and vasculature of Fgf2−/− mice have no apparent morphological defects, we next examined cardiovascular function by measuring left ventricular function and systemic blood pressure in the intact mice25. As shown in Table 3, we measured a reduced mean arterial blood pressure (MAP), systolic left intraventricular pressure (SLVP), maximum rate of ventricular contraction (+dP/dt) and relaxation (−dP/dt) in Fgf2−/− mice, whereas heart rate was normal.
Table 3.
Cardiovascular measurements in anesthetized mice
| Fgf2 (+/+) (+/−) | Fgf2(−/−) | |
|---|---|---|
| Mean arterial pressure (mmHg) | 98.9 ± 5.6 | 78.0 ± 5.9* |
| Peak systolic left ventricular pressure (mmHg) | 123.2 ± 7.8 | 98.3 ± 5.6** |
| Left ventricular +dP/dt (mmHg/s) | 8631.6 ± 746.6 | 7011.6 ± 567** |
| Left ventricular −dP/dt (mmHg/s) | −10716 ± 713.8 | −7616.5 ± 499.3* |
| Heart rate (beats/min) | 443.1 ± 14 | 424.5 ± 10.9 |
Means ± s.e.m. of cardiovascular parameters from Fgf2−/− mice (n = 8) and Fgf2+/+ or Fgf2+/− littermate controls (n = 7) anesthetized with ketamine and thiobutabarbital as described in the Methods. +dP/dt, maximal rate of intraventricular pressure rise during systole; −dP/dt, maximal rate of intraventricular pressure drop during diastole. Experimental values differ significantly from control values by ANOVA
P < 0.01 or
P < 0.05.
Arterial blood pressure is mainly determined by cardiac output and total peripheral resistance. Factors affecting the homeostatic integration of these two activities include intrinsic cardiac and vascular properties, such as contractility, and extrinsic features, such as autonomic innervation of the heart and vasculature and circulating hormone levels. Since Fgf2−/− mice show no sign of weakness, fatigue, myxedema or growth retardation, and since their thyroid and adrenal glands appear normal, it is unlikely that they have adrenocortical insufficiency or hypothyroidism, which could lead to a depressed cardiovascular condition. To determine whether the reduced cardiac performance (and subsequently a reduced blood pressure) is due to intrinsic defects in the heart as opposed to a vascular related complication, isolated work-performing heart preparations26 were employed. No significant differences were found in cardiac Starling function between Fgf2−/− mice and their paired normal controls (not shown). Therefore, the heart itself does not seem to contribute to the cardiovascular phenotype observed. Consequently, extracardiac factors, such as vascular tone or autonomic function, are involved.
FGF2 is required for normal vascular contractility
Vascular resistance is primarily determined by the structure and smooth muscle tone of peripheral resistance vessels. This has recently been exemplified by the Nos3 knockout mouse in which changes in vascular tone significantly affect blood pressure27. In Fgf2−/− mice, no obvious structural abnormalities are observed in the small arteries and arterioles of the skin, and no obvious differences in morphology or collagen content are found in their aortas and portal veins (not shown). Consequently, vascular contractility was analyzed by comparing isometric force of the aorta and portal vein from mutant and wild-type siblings. In the aorta, a tonic vascular smooth muscle, concentration-force relations for KCl depolarization and phenylephrine are similar with respect to maximum force generating capacity (force/cross-sectional area) and excitation-contraction coupling (not shown). However, in the portal vein, a phasic smooth muscle with characteristic rhythmic myogenic activity, tension-time integral measurements reveal significantly reduced spontaneous contractile activity, but normal sensitivity to increasing concentrations of the contractile agonist phenylephrine (Fig. 2, Table 4). The rate of force generation (+dF/dt) is also significantly lower in Fgf2−/− mice. These results suggest that the decreased vascular tone in Fgf2 knockout mice may be attributable to a reduction in vascular smooth muscle contractility. Differences in the structure of the contractile apparatus as well as in the kinetics of activation could also be involved.
Fig. 2.

Contractile activity of isolated portal veins, a, Representative tracing of a pair of portal veins showing isometric force vs time for the spontaneous myogenic contractions, b, Tension-time integrals for isolated portal veins stimulated by increasing concentrations of phenylephrine, a vasoconstrictor. Each data point represents the mean (± s.e.m. for five pair of mice) of the tension–time integral generated from each portal vein measured for 60 s at each concentration of phenylephrine.
Table 4.
Summary of contractile activity of freshly isolated portal veins
| Fgf2 (+/+),(+/−) | Fgf2(−/−) | |
|---|---|---|
| +df/dt/area [(mN/s)/mm2] | 29.2 ± 4.1 | 15.7 ± 3.6* |
| −dF/dt/area [(mN/s)/mm2] | −22.5 ± 2.7 | −18.2 ± 1.9 |
| T–t integral/area (mN·s/mm2) | 202.8 ± 29.2 | 132.3 ± 9.1* |
| Spontaneous frequency (contraction/min) | 3.3 ± 0.3 | 2.6 ± 0.3 |
Means ± s.e.m. of portal vein contractile activity from six pairs of mice. Each force measurement was normalized with the vessel cross-sectional area. +dF/dt, maximum rate of force development; −dF/dt, maximum rate of relaxation; T–t integral (tension–time integral), which was measured for a period of 60 s in the steady state. Experimental values differ significantly from control values by Student's t-test
P < 0.05.
Injury of the carotid artery
FGF2 is thought to play an important role in the repair of intravascular injury in animals by virtue of its ability to stimulate cell proliferation, migration and matrix remodeling. In light of this ability and of our observation that FGF2 plays a role in vascular tone control, it was of special interest to determine the arterial response to mechanical injury in Fgf2 knockout mice. A rigid bead probe was used to injure the carotid artery. This procedure generates a graded injury along the length of the carotid, resulting in simple endothelial cell denudation proximal to the aorta (Fig. 3a) and significant smooth muscle damage including medial swelling, bleeding and smooth muscle cell death distal to the aorta (Fig. 3b). In this technique, disruption of elastic layers does not routinely occur (Fig. 3, c and d); however, platelet deposition on the injured intimal surface is present (Fig. 3, a–d). Comparable carotid artery lesions are present in both wild-type and Fgf2−/− mice after 24 hours. Four weeks after injury, the carotid arterial lesions are similar in Fgf2−/− and control mice. Injured vessels from control and Fgf2−/− mice form a cellular, eccentric neointima and exhibit expanded medial and adventitial layers (Fig. 3, e–p). Reflecting the severity of injury at different positions along the carotid, injured vessels from both Fgf2+/+ and Fgf2−/− mice form larger and thicker intimal lesions distal to the aorta as compared with those proximal to the aorta (Fig. 3, e–l). Of the four Fgf2−/− mice examined at 4 weeks, three of the injured carotids exhibit lesions, whereas the fourth vessel was filled with an occluding thrombus (data not shown). Reverse transcriptase polymerase chain reaction (RT-PCR) of RNA samples from injured, wild-type carotids show that Fgf2 expression is equal to or decreased by day 2, increases approximately twofold by day 4 and is three to four times as high as that of the uninjured, bilateral carotid by day 6 after injury (Fig. 4). The presence of hyperplastic vascular tissues in repairing vessels of Fgf2−/− mice demonstrates that the lack of FGF2 has no apparent effect on early phase vascular hyperplasia.
Fig. 3.

Vascular hyperplasia following carotid injury occurs in Fgf2+/+ mice. Carotid arteries were injured in adult mice and examined for the development of hyperplastic vascular layers. a–d, Hematoxylin and eosin (H&E)-stained sections from vessels 1 day after injury from control (Fgf2+/+, Fgf2+/+ n = 4) and Fgf2−/− (n = 5) mice show a range of injury features from (a) endothelial cell denudation with platelet deposition (open arrowheads) proximal to the heart (prox) to (b) severe medial damage leading to medial hemorrhage (arrow) distal to the heart near the thyroid (dist). c, H&E-stained section of an Fgf2−/− mouse carotid showing severe medial injury, d, Serial section of c stained with an elastin/van Gieson stain (e/VG) to highlight the elastic layers, which remained intact (open arrows), e–p H&E- and e/VG-stained serial, section pairs from 4-week post-injury carotids from Fgf2−/− (n = 3) and Fgf2+/+ (n = 2) mice show hyperplastic responses involving the adventitial (*), medial (white arrow), and intimal (closed arrow heads) layers. Panels e, f, i, and j are from the same wild-type mouse showing lesions from two positions (dist and prox). Panels g, h, k, and l show lesions from the same Fgf2 knockout mouse. Panels m, n, o, and p show lesions at the middle position (mid) along the injured carotid from a wild-type (m, n) and Fgf2−/− mouse (o, p). The scale bar shown in d applies to all 1-day panels; the dimension scale in p is the same for all 4-week panels.
Fig. 4.
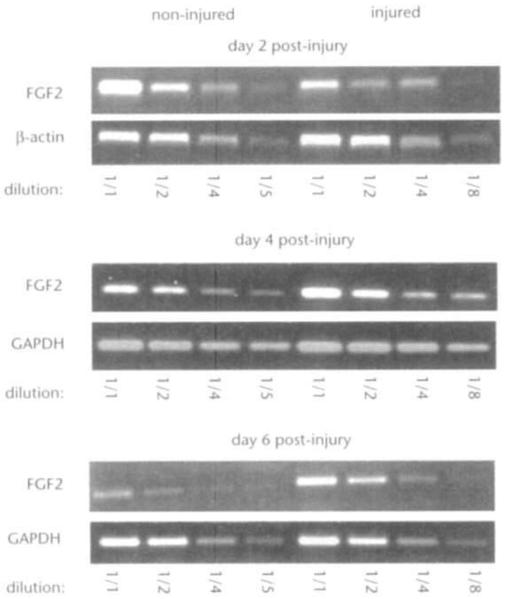
Increased expression of Fgf2 mRNA in injured carotid arteries. RTPCR of Fgf2 transcripts from wild-type injured and noninjured, bilateral control carotids at 2, 4 and 6 days after injury. Representative results from one of the two experiments performed at each time point are shown. After transcribing mRNA into cDNA, serial dilutions (as indicated) were used in the reaction to ensure linear amplification. Amplification of glyceraldehyde phosphodehydrogenase (GAPDH) or β-actin transcripts served to control for template quantity.
Discussion
Vascular tone control and hypotension
We have identified FGF2 as a molecular player in the centrally important homeostatic process of vascular tone control in the mouse. A consequence of the absence of FGF2 is a decrease in vascular tone, which may contribute to reduced mean arterial blood pressure. If so, this may be a reflection of compromised activity in resistance vessels, or in capacitance veins, because they can store as much as 60% of total blood volume. The limited ability of a subset of vascular smooth muscle to generate force may reflect altered gene expression affecting components of the contractile apparatus. Conversely, FGF2 may function as an autocrine/paracrine factor, regulating smooth muscle activity either through mediation of calcium mobilization24 or coordination of humoral vasoreactive signals28. At the present time we do not believe that calcium mobilization is affected, because of the similarity of the relative slopes of the phenylephrine dependent time-tension curves shown in Fig. 2b. Rather, we anticipate that the defect lies either in the contractile apparatus or in altered autonomic cardiovascular responsiveness.
Because of the small size of the mouse, only large vessels are accessible to physiological analysis; consequently, most studies investigating vascular tone control in the mouse look only at the largest of the vessels, the aorta. We looked at both the arterial and venous side, using both tonic (aorta) and phasic (portal vein) vessels and found decreased tonus only in the latter. The portal vein measurements were of special interest because of their functional similarity to resistance vessels, which also contain phasic smooth muscle29. Further technological refinements are needed for measuring vascular tone in true resistance vessels and total peripheral resistance in the mouse.
Vascular injury
The extensive literature on the role of FGF2 in the hyperplastic response of the vessel wall layers to luminal injury30 strongly suggests that FGF2-deficient vessels would respond to vascular injury with less growth than in wild-type control litter-mates. Mechanical injury was chosen rather than toxicity- or inflammation-based injuries because the role of FGF2 in repair of the last two named models is less well described. Unexpectedly, no obvious differences were found with respect to endothelial cell denudation, platelet deposition, smooth muscle damage, which ranged from medial edema to severe smooth muscle deletion, or repair. Since a difference caused by the absence of FGF2 would be expected only if the injury was severe enough to liberate FGF2 in wild-type animals, we examined post-injury Fgf2 expression levels in injured wild-type mice and found it to be substantially elevated. Consequently, if FGF2 plays an important role in the repair process, effects from its absence should have been detectable. Whether subtle differences occur at intermediate time points is unknown. If they do, they would probably be of minor significance and/or would be masked by other repair processes, so that at 4 weeks there is no detectable difference. In addition, we cannot rule out minor morphometric changes, though gross measurements do not indicate any differences.
Together, these results indicate that the significant major players in the hyperplastic response of the vessel layers following mechanical injury would be factors other than FGF2, such as plasminogen31, or other factors working in conjunction with FGF2. However, these results do not rule out the possibility that FGF2 may play other roles in the repairing vessel that do not involve growth. Recent reports suggest that vessel patency following injury is in part determined by a remodeling process involving permanent changes in vessel diameter. This in turn can be in part due to adventitial remodeling and vasoactivity in a manner such that intimal thickening is not stenotic unless the total diameter of the vessel is decreased, thereby resulting in a reduction in luminal area32. We have shown here that Fgf2 expression increases in the carotid artery following vessel injury, that FGF2-deficient mice respond to intravascular injury with a normal hyperplastic response, and that FGF2-deficient mice have reduced vascular tone. Together, these results suggest that FGF2 functions more to control tonus during vessel remodeling than to control growth in the vessel wall. Others have shown that vascular smooth muscle cells can modulate between proliferative and quiescent states33,34, and they can also modulate their contractility through structural muscle protein isoform changes35,36. Therefore, it is justifiable to speculate that FGF2 could be playing more of a role in regulation of muscle function during remodeling, perhaps as a contractile maintenance factor, than in the growth process or in the regulation of vessel size.
Hematopoiesis
By acting in concert with hematopoietic cytokines, FGF2 is thought to positively regulate hematopoiesis by affecting both stromal cells and early and committed hematopoietic progenitor cells37. Consistent with this notion, bone marrow cells from Fgf2 knockout mice display significantly decreased responsiveness to the most general hematopoietic cytokine, IL-3. Curiously, however, this effect is not observed in peripheral blood cell counts, which are unaffected by the absence of FGF2. There are clearly compensatory hematopoietic mechanisms that correct the deficiency in the animal. An exception to the notion of FGF2 as a positive hematopoietic regulator is megakaryocyte proliferation. Unlike the general stem cell mitogen IL-3, pokeweed mitogen-stimulated spleen cell-conditioned medium, a source of megakaryocyte-stimulating factor, stimulates megakaryocyte colony formation from bone marrow cells of Fgf2 knockout mice more than from cells from FGF2 wild-type mice. Also, unlike the case with IL-3, this effect does play itself out in the periphery by leading to an increased level of platelets. Platelet function, however, remains normal. Consequently, FGF2 in vivo appears to negatively control platelet production, perhaps by regulating pathways involved in responding to signals for increasing platelets. However, whether the effect of FGF2 on megakaryocyte function is mediated through direct or indirect means is still unclear.
FGF ligand/receptor specificity
This work presents another step in clarifying FGF ligand specificity in many developing tissues. The fact that expected developmental defects in angiogenesis or organogenesis of brain, heart or skeleton, for example, were not found suggests that our understanding of ligand/receptor specificity for the FGFs is poorly understood. We cannot at this time rule out redundant expression or gene compensation on the part of other Fgfs, though we have found no changes in expression levels of Fgfs 1, 4 or 5 in 13.5-day-old embryos or in adult brain, heart, skin and kidney, some of which are coexpressed with Fgf2, and generation of Fgf2 and Fgf5 double-knockout mice has produced no additional obvious developmental phenotypes (G. Martin, personal communication). Neither can we rule out that compensatory developmental processes are induced by alterations in the normal developmental program that result in normal adult tissue, nor the possibility that on other genetic backgrounds we may see additional phenotypes, nor the possibility that apparently normal tissues, for example, those of the skeleton and nervous system, will respond differently to stress or injury in the absence of FGF2. However, assuming for the present that some of the anticipated phenotypes are not to be found, we must then ask which FGFs are the true ligands for the expected FGF signals. Bone development provides a case in point. Autosomal dominant mutations in human FGF receptors 1–3 lead to Pfeiffer (FGFR1 and 2)38,39, Crouzon (FGFR2)39,40 and Jackson-Weiss (FGFR2)40 syndromes, and also to achondroplasia (FGFR3)41. Fgfr3 ablation in mice causes skeletal overgrowth42. These data are all consistent with the short bone phenotype of transgenic mice in which we ubiquitously overexpressed FGF2 (ref. 18). It is, therefore, surprising that the Fgf2 knockout mouse displays no noticeable skeletal defects. This strongly suggests that either FGF2 is not the important FGF for bone development, or that FGF2 acts in concert with other FGFs in skeletogenesis.
Conclusion
Identification of FGF2 as a significant regulator of vascular tone gives it the potential for playing important roles in many homeostatic processes including blood pressure regulation, shock, ischemia-reperfusion, inflammation, injury/repair situations involving the vasculature, nervous system and dermal wound healing, temperature regulation, digestion, exercise physiology, and metabolism. As our investigation of vascular injury suggests, the Fgf2 knockout mouse will be useful for enabling us to refine our understanding of FGF2 function in many of these homeostatic processes.
Methods
Generation of Fgf2 knockout mice
The targeting vector was derived from a 6.7-kb genomic clone isolated from a strain 129 mouse genomic library. From this clone, a 4-kb Xbal/BamHI fragment was subcloned into the Bluescript KS II plasmid (Stratagene, La Jolla, CA). A 3.2-kb hypoxanthine phosphoribosyl transferase (Hprt) minigene was used to replace the 0.5-kb Narl/Xbal fragment of the Fgf2 genomic DNA, which contained a portion of the promoter region and the first exon of the Fgf2 gene. A thymidine kinase gene was then inserted at the 3' end of the genomic DNA as a negative selection marker. E14TG2a ES cells (1 × 107) were electroporated with the targeting vector at the final concentration of 5 nM. ES cells were maintained continuously in medium containing 1 × HAT (10−1 M hypoxanthine, 4 × 10−7 M aminopterin, 1.6 × 10−5 M thymidine, Sigma Chemical Co.) 24 h after electroporation. Gancyclovir (2 mM) was added to the culture 48 h after the electroporation for 3–5 days to select for the loss of the Tk gene. Double-resistant colonies were genotyped for the targeted allele by PCR analysis using the following primers: upper primer: 5'-AGG AGG CAA GTG GAA AAC GAA; Lower primer: 5'- CCC AGA AAG CGA AGG AAC AAA. This primer pair amplified a 1299 bp fragment that spanned the 5' recombination junction. Genotyping of the original targeted cells was determined by PCR, and confirmed by Southern blotting.
Correctly targeted E14TG2a ES cells were microinjected into C57BL/6 strain blastocysts. Blastocysts were then transferred to pseudopregnant foster mothers and allowed to develop to full term. Chimeric mice were bred to strain Black Swiss mice to test for germline transmission. To genotype the knockout mice, the primer pair initially used to identify the targeted ES clones were used to identify the targeted allele. The following primer pair was used to amplify the wild-type allele: Upper primer, 5'- CCA GAA CAC CGA CCC ACA C; lower primer, 5'- CCA CTT CGC CGA CCG TAT T. This primer pair amplified a 185-bp fragment within the first intron of the Fgf2 gene.
Southern, northern and western blot analyses
For Southern blot analysis, DNA from ES cells or mouse tails was digested with several restriction enzymes, resolved in 0.8% agarose gels, transferred to Hybond nylon membranes (Amersham, Arlington Heights, Illinois) and hybridized to 32P-labeled probes (Fig. 1). For northern blot analysis, 2.5–5 μg polyadenylated RNA was isolated from various tissues and separated on a 1% agarose gel and blotted to a nylon membrane. The membrane was then sequentially probed for mouse Fgf 1, 2 and 5 mRNAs. Mouse Fgf 1 and 5 cDNAs were used as probes. A probe consisting of exons 2 and 3 of the mouse Fgf2 gene was generated by PCR (upper primer, 5'-TC AAA CTA CAA CTC CAA GCA; lower primer, 5'-TCA GCT CTT AGC AGA CAT TGG AAG AAA) and used to detect Fgf2 mRNA. The membrane was also hybridized to a β-actin cDNA as the sample loading control. For western blot analysis, brain tissue was removed and homogenized in detergent solution containing 50 mM Tris (pH 8.0), 62.5 mM EDTA, 0.4% deoxycholic acid, 1% NP 40, 0.5 μg/ml leupeptin, 0.5 μg/ml pepstatin, 0.5 μg/ml aprotinin, 0.2 mM PMSF and 40 μM AEBSF. FGF2 was partially purified on heparin Sepharose beads and separated in a 12% polyacrylamide electrophoresis gel. FGF2 was detected using a mouse monoclonal antibody against bovine FGF2 (Upstate Biotechnology, Lake Placid, NY).
Hematology
Whole blood was obtained by letting blood from the eye of mice under 2.5% Avertin (2.5% 2,2,2-tribromoethanol, 2.5% tertiary amyl alcohol in normal saline, Aldrich, Milwaukee, Wl) anesthesia. Blood was then diluted in normal saline for red cell counting, or added to B-D Unopettes (Becton Dickinson, Rutherford, NJ) for white cell and platelet counting. Blood cells were counted in two chambers per sample by a thin hemocytometer and a phase-contrast microscope.
Bone marrow cultures
Bone marrow cells from femurs of adult mice were collected into DMEM, pelleted and resuspended in DMEM at 2 × 106 cells/ml. Cultures of bone marrow consisting of either 2 × 105 or 1 × 106 cells in 1 ml of 0.8% methylcellulose prepared with bone marrow culture medium (DMEM + 25% fetal bovine serum) and cultured for 1 week in 35-mm dishes containing various stem cell mitogens. Conditioned medium (MEM alpha + 10% fetal bovine serum) from splenocyte cultures stimulated with pokeweed mitogen was used as a crude source of megakaryocyte colony-stimulating activity43. The total number of colonies present after 1 week in each 1 ml culture of bone marrow cells was counted separately by two investigators.
Measurement of mean arterial blood pressure (MAP) and cardiac function in intact closed-chest mice
The method was detailed by Lorenz and Robbins25. In brief, mice weighing between 25 and 35 g (129/Black Swiss mixed background) were anesthetized with an intraperitoneal (i.p.) injection of 50 μg/g body weight ketamine and 100 μg/g body weight thiobutabarbital (Inactin, Research Biochemicals International, Natick, MA). Polyethylene tubing (0.4 mm outer diameter) was inserted into the abdominal aorta from the right femoral artery and connected to a low compliance pressure transducer (COBE; Cardiovascular, Arvada, CO) for measurement of the MAP. A high-fidelity, 1.8 F Millar Mikro-Tip transducer (Model SPR-612; Millar Instruments, Houston, TX) was inserted into the right carotid artery, and its tip was carefully advanced into the left ventricle to monitor cardiac performance. MAP and intraventricular pressure signals from the COBE transducer and the Millar transducer were amplified using custom-made amplifiers, and the output was recorded and analyzed using a MacLab 4/s data acquisition system connected to a Macintosh 7100/80 computer. Average values for MAP, heart rate (HR), systolic left intraventricular pressure (SLVP) were all measured directly from the pressure wave forms. In addition, maximum and minimum dP/dt (+dP/dt and −dP/dt) were calculated from a differential tracing of left ventricular pressure. All data were analyzed by mixed two-factor analysis of variance (ANOVA) with repeated measures using SuperAnova software by Abacus Concepts (Berkeley, CA).
Analysis of blood vessel contractile properties
Mice of 8–12 weeks of age were anesthetized in a charged CO2 chamber. Segments of thoracic aorta (5–7 mm) were dissected and mounted for isometric force recording as previously described44. The portal vein was dissected from the mouse by tying 4–0 suture at the two ends of the vessel (between the hepatic bifurcation and the anterior mesenteric vein). The portal vein was cut free, and each end was secured with its thread to the myograph. From the time of the dissection the vessel was maintained in physiological salt solution (PSS). PSS contained (in millimoles/liter): 118 NaCl, 4.73 KCl, 1.2 MgCl2, 0.026 EDTA, 1.2 KH2PO4, 2.5 CaCl2, 5.5 glucose and buffered with 25 NaH2CO3; pH when bubbled with 95% O2/5% CO2 was 7.4 at 37 °C. Experiments were completed at optimal tension based on adjusting the vessels to a point where maximum peak–peak oscillations were observed. Force measurements were obtained using a Harvard Apparatus differential capacitor force transducer (South Natick, MA), which was connected to a Biopac MP100 data acquisition system, that allowed precise measurements of contractile parameters including frequency of spontaneous contractions, peak to peak values for the oscillations, rates of the development of force (+dF/dt) and relaxation (−dF/dt), and tension–time integral (T–t integral).
Mechanical injury of carotid artery and analysis of injury repair
The left or right common carotid artery of an anesthetized (Avertin; 0.5 ml/20 g) mouse was exposed through a 1- to 2-cm midline incision along the neck with dissection of the overlying inner muscle layers. Two 6–0 silk ligatures were loosely placed around the vessel proximal to the distal external/internal carotid bifurcation near the head and distal to where the carotid entered the chest. Following administration of 6 units of heparin i.p., the distal ligature was tied tightly, and the proximal ligature was pulled posteriorly to fully occlude the vessel, thereby permitting insertion of the injuring probe without blood loss. An incision was made through the upper half of the internal carotid artery between the distal bifurcation and the internal/external carotid branch point. A probe made of a bead of epon resin, larger in diameter (~0.45 mm) than the vessel (~0.3 mm near the internal/external carotid bifurcation), on a 3–0 nylon suture was inserted into the vessel and advanced toward the aorta by releasing tension on the proximal ligature. The probe was repeatedly advanced and withdrawn until the vessel “grabbed” at the probe, indicating sufficient injury to induce vasospasm. The proximal occluding ligature was replaced, and the probe removed. A permanent 7–0 silk ligature was placed immediately proximal to the vessel incision. In this configuration, blood flows from the aorta, through the injured region and into the external carotid branch. At 1 day and 4 weeks after the injury, mice were perfusion-fixed in 4% paraformaldehyde. The entire neck was dissected away from the spine and cut into three sections: a proximal section, a middle section, and a distal section. Bilateral carotid arteries were examined for the extent of the arterial injury, intimal and medial growth and adventitial lesion.
Reverse transcriptase-PCR of Fgf2 mRNA from mouse carotid
The left carotids of wild-type mice were injured as described above and harvested at 2, 4 and 6 days after injury and immediately frozen in liquid nitrogen. During dissection, care was taken to remove all of the associated granulation tissue in order to minimize the contribution of Fgf2 mRNA in the sample derived from healing connective tissue. Each carotid was homogenized in 0.8 ml of RNAzol B (Tel-Test Inc., Friendswood, TX) and the RNA extracted as per manufacturer's instructions using glycogen as a carrier during RNA precipitation. The entire RNA sample was transcribed into first strand cDNA using Superscript Reverse Transcriptase (Gibco BRL, Grand Island, NY) at 42 °C in a 20-μl reaction. Serial dilutions (1:1, 1:2, 1:4 and 1:8) of the cDNA were amplified by hot start PCR for Fgf2 (Tm = 56°C, 35 cycles) or Gapdh (Tm = 57°C, 27 cycles) transcripts. Primers used for detection of Fgf2 (upper, AGGCCACTTCAAGGCCC; lower, GAAACAGTATGGCCTTCTGTCC) and Gapdh (upper, GGCCTTGACTGTGCCGTTGAATTT; lower, ACAGCCGCATCTTCTTGTGCAGTG) sequences discriminate between products derived from cDNA and genomic DNA templates, based on size.
Acknowledgments
We thank Jeffrey Robbins, Timothy Hewett and Willie Ng for work-performing heart preparations of Fgf2 knockout mouse hearts; John Duffy and Tara Riddle of the Gene Targeting and Gene-Targeted Mouse Services of the University of Cincinnati for making the knockout mouse and for assistance with the gene targeting; grants HL 41496, HL 46826, HL 58511 (T.D.) and HL 54829 (R.J.p.); and a Marion Merrel Dow Research Foundation grant to the University of Cincinnati.
References
- 1.Kessler DS, Melton DA. Vertebrate embryonic induction: Mesodermal and neural patterning. Science. 1994;266:596–604. doi: 10.1126/science.7939714. [DOI] [PubMed] [Google Scholar]
- 2.Kimelman D, Kirschner M. Synergistic induction of mesoderm by FGF and TGF-beta and the identification of an mRNA coding for FGF in the early Xenopus embryo. Cell. 1987;51:869–877. doi: 10.1016/0092-8674(87)90110-3. [DOI] [PubMed] [Google Scholar]
- 3.Flamme I, Risau W. Induction of vasculogenesis and hematopoiesis in vitro. Development. 1992;116:435–439. doi: 10.1242/dev.116.2.435. [DOI] [PubMed] [Google Scholar]
- 4.Slack JM, Isaacs HV. The Einsteck-method: Position and structure of projections formed by implants of a ventral character. Dev. Biol. 1994;161:313–317. doi: 10.1006/dbio.1994.1031. [DOI] [PubMed] [Google Scholar]
- 5.Schneider MD, McLellan WR, Black FM, Parker TG. Growth factors, growth factor response elements, and the cardiac phenotype. Basic Res. Cardiol. 1992;87(Suppl. 2):33–48. doi: 10.1007/978-3-642-72477-0_4. [DOI] [PubMed] [Google Scholar]
- 6.Parker TG, Packer SE, Schneider MD. Peptide growth factors can provoke “fetal” contractile protein gene expression in rat cardiac myocytes. J. Clin. Invest. 1990;85:507–514. doi: 10.1172/JCI114466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Folkman J, Klagsbrun M. Vascular physiology: A family of angiogenic peptides. Nature. 1987;329:671–672. doi: 10.1038/329671a0. [DOI] [PubMed] [Google Scholar]
- 8.Doetschman T, Shull M, Kier A, Coffin JD. Embryonic stem cell model systems for vascular morphogenesis and cardiac disorders. Hypertension. 1993;22:618–629. doi: 10.1161/01.hyp.22.4.618. [DOI] [PubMed] [Google Scholar]
- 9.Weiss RH, Maduri M. The mitogenic effect of thrombin in vascular smooth muscle cells is largely due to basic fibroblast growth factor. J. Biol. Chem. 1993;268:5724–5727. [PubMed] [Google Scholar]
- 10.Ali S, Davis MG, Becker MW, Dorn GW. 2. Thromboxane A2 stimulates vascular smooth muscle hypertrophy by up-regulating the synthesis and release of endogenous basic fibroblast growth factor. J. Biol. Chem. 1993;268:17397–17403. [PubMed] [Google Scholar]
- 11.Itoh H, Mukoyama M, Pratt RE, Gibbons GH, Dzau VJ. Multiple autocrine growth factors modulate vascular smooth muscle cell growth response to an giotensin II. J. Clin. Invest. 1993;91:2268–2274. doi: 10.1172/JCI116454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davis MG, et al. Intracrine and autocrine effects of basic fibroblast growth factor in vascular smooth muscle cells. J. Mol. Cell Cardiol. 1997;29:1061–1072. doi: 10.1006/jmcc.1997.0383. [DOI] [PubMed] [Google Scholar]
- 13.Hebert JM, Basilico C, Goldfarb M, Haub O, Martin GR. Isolation of cDNAs encoding four mouse FGF family members and characterization of their expression patterns during embryogenesis. Dev. Biol. 1990;138:454–463. doi: 10.1016/0012-1606(90)90211-z. [DOI] [PubMed] [Google Scholar]
- 14.Yanagisawa Miwa A, et al. Salvage of infarcted myocardium by angiogenic action of basic fibroblast growth factor. Science. 1992;257:1401–1403. doi: 10.1126/science.1382313. [DOI] [PubMed] [Google Scholar]
- 15.Lindner V, Reidy MA. Proliferation of smooth muscle cells after vascular injury is inhibited by an antibody against basic fibroblast growth factor. Proc. Natl. Acad. Sci. USA. 1991;88:3739–3743. doi: 10.1073/pnas.88.9.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsuboi R, Shi CM, Rifkin DB, Ogawa H. A wound healing model using healing-impaired diabetic mice. J. Dermatol. 1992;19:673–675. doi: 10.1111/j.1346-8138.1992.tb03757.x. [DOI] [PubMed] [Google Scholar]
- 17.MacMillan V, et al. Mice expressing a bovine basic fibroblast growth factor trans-gene in the brain show increased resistance to hypoxemic- ischemic cerebral damage. Stroke. 1993;24:1735–1739. doi: 10.1161/01.str.24.11.1735. [DOI] [PubMed] [Google Scholar]
- 18.Coffin JD, et al. Abnormal bone growth and selective translational regulation in basic fibroblast growth factor (FGF-2) transgenic mice. Mol. Biol. Cell. 1995;6:1861–1873. doi: 10.1091/mbc.6.12.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thompson S, Clarke AR, Pow AM, Hooper ML, Melton DW. Germ line transmission and expression of a corrected HPRT gene produced by gene targeting in embryonic stem cells. Cell. 1989;56:313–321. doi: 10.1016/0092-8674(89)90905-7. [DOI] [PubMed] [Google Scholar]
- 20.Baird A, Schubert D, Ling N, Guillemin R. Receptor- and heparin-binding domains of basic fibroblast growth factor. Proc. Natl. Acad. Sci. USA. 1988;85:2324–2328. doi: 10.1073/pnas.85.7.2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arakawa T, et al. The importance of Arg40 and 45 in the mitogenic activity and structural stability of basic fibroblast growth factor: Effects of acidic amino acid substitutions. J. Protein Chem. 1995;14:263–274. doi: 10.1007/BF01886783. [DOI] [PubMed] [Google Scholar]
- 22.Faham S, Hileman RE, Fromm JR, Linhardt RJ, Rees DC. Heparin structure and interactions with basic fibroblast growth factor. Science. 1996;271:1116–1120. doi: 10.1126/science.271.5252.1116. [DOI] [PubMed] [Google Scholar]
- 23.Sugi Y, Sasse J, Lough J. Inhibition of precardiac mesoderm cell proliferation by antisense oligodeoxynucleotide complementary to fibroblast growth factor-2 (FGF-2) Dev. Biol. 1993;157:28–37. doi: 10.1006/dbio.1993.1109. [DOI] [PubMed] [Google Scholar]
- 24.Merle PL, Feige JJ, Verdetti J. Basic fibroblast growth factor activates calcium channels in neonatal rat cardiomyocytes. J. Biol. Chem. 1995;270:17361–17367. doi: 10.1074/jbc.270.29.17361. [DOI] [PubMed] [Google Scholar]
- 25.Lorenz JN, Robbins J. Measurement of intraventricular pressure and cardiac performance in the intact closed-chest anesthetized mouse. Am. J. Physiol. 1997;272:H1137–H1146. doi: 10.1152/ajpheart.1997.272.3.H1137. [DOI] [PubMed] [Google Scholar]
- 26.Grupp IL, Subramaniam A, Hewett TE, Robbins J, Grupp G. Comparison of normal, hypodynamic, and hyperdynamic mouse hearts using isolated work-performing heart preparations. Am. J. Physiol. 1993;265:H1401–H1410. doi: 10.1152/ajpheart.1993.265.4.H1401. [DOI] [PubMed] [Google Scholar]
- 27.Huang PL, et al. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377:239–242. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- 28.Cristiani C, Volpi D, Landonio A, Bertolero F. Endothelin-1-selective binding sites are downregulated by transforming growth factor-beta and upregulated by basic fibroblast growth factor in a vascular smooth muscle-derived cell line. J. Cardiovasc. Pharmacol. 1994;23:988–994. doi: 10.1097/00005344-199406000-00018. [DOI] [PubMed] [Google Scholar]
- 29.Sutter MC, Ljung B. Contractility, muscle mass and agonist sensitivity of isolated portal veins from normo- and hypertensive rats. Acta Physiol. Scand. 1977;99:484–495. doi: 10.1111/j.1748-1716.1977.tb10402.x. [DOI] [PubMed] [Google Scholar]
- 30.Schwartz SM, Liaw L. Growth control and morphogenesis in the development and pathology of arteries. J. Cardiovasc. Pharmacol. 1993;21(Suppl. 1):S31–S49. doi: 10.1097/00005344-199321001-00007. [DOI] [PubMed] [Google Scholar]
- 31.Carmeliet P, Moons L, Ploplis V, Plow E, Collen D. Impaired arterial neointima formation in mice with disruption of the plasminogen gene. J. Clin. Invest. 1997;99:200–208. doi: 10.1172/JCI119148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Currier JW, Faxon DP. Restenosis after percutaneous transluminal coronary angioplasty: Have we been aiming at the wrong target? J. Am. Coll. Cardiol. 1995;25:516–520. doi: 10.1016/0735-1097(95)98445-j. [DOI] [PubMed] [Google Scholar]
- 33.Schwartz SM, Campbell GR, Campbell JH. Replication of smooth muscle cells in vascular disease. Cire. Res. 1986;58:427–444. doi: 10.1161/01.res.58.4.427. [DOI] [PubMed] [Google Scholar]
- 34.Campbell GR, Campbell JH. The phenotypes of smooth muscle expressed in human atheroma. Ann. N.Y. Acad. Sci. 1990;598:143–58. 143–158. doi: 10.1111/j.1749-6632.1990.tb42286.x. [DOI] [PubMed] [Google Scholar]
- 35.Aikawa M, et al. Human smooth muscle myosin heavy chain isoforms as molecular markers for vascular development and atherosclerosis. Circ. Res. 1993;73:1000–1012. doi: 10.1161/01.res.73.6.1000. [DOI] [PubMed] [Google Scholar]
- 36.Aikawa M, et al. Phenotypic modulation of smooth muscle cells during progression of human atherosclerosis as determined by altered expression of myosin heavy chain isoforms. Ann. N.Y. Acad. Sci. 1995;748:578–85. 578–585. doi: 10.1111/j.1749-6632.1994.tb17365.x. [DOI] [PubMed] [Google Scholar]
- 37.Allouche M. Basic fibroblast growth factor and hematopoiesis. Leukemia. 1995;9:937–942. [PubMed] [Google Scholar]
- 38.Muenke M, et al. A common mutation in the fibroblast growth factor receptor 1 gene in Pfeiffer syndrome. Nature Genet. 1994;8:269–274. doi: 10.1038/ng1194-269. [DOI] [PubMed] [Google Scholar]
- 39.Rutland P, et al. Identical mutations in the FGFR2 gene cause both Pfeiffer and Crouzon syndrome phenotypes. Nature Genet. 1995;9:173–176. doi: 10.1038/ng0295-173. [DOI] [PubMed] [Google Scholar]
- 40.Jabs EW, et al. Jackson-Weiss and Crouzon syndromes are allelic with mutations in fibroblast growth factor receptor 2. Nature Genet. 1994;8:275–279. doi: 10.1038/ng1194-275. [DOI] [PubMed] [Google Scholar]
- 41.Rousseau F, et al. Mutations in the gene encoding fibroblast growth factor receptor- 3 in achondroplasia. Nature. 1994;371:252–254. doi: 10.1038/371252a0. [DOI] [PubMed] [Google Scholar]
- 42.Colvin JS, Bohne BA, Harding CW, McEwen DG, Ornitz DM. Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nature Genet. 1996;12:390–397. doi: 10.1038/ng0496-390. [DOI] [PubMed] [Google Scholar]
- 43.Burstein SA, Adamson JW, Thorning D, Harker LA. Characteristics of murine megakaryocytic colonies in vitro. Blood. 1979;54:169–179. [PubMed] [Google Scholar]
- 44.Lalli J, Harrer JM, Luo W, Kranias EG, Paul RJ. Targeted ablation of the phospholamban gene is associated with a marked decrease in sensitivity in aortic smooth muscle. Circ. Res. 1997;80:506–513. doi: 10.1161/01.res.80.4.506. [DOI] [PubMed] [Google Scholar]