

Abstract
Heterogeneous clinical responses to histone deacetylase inhibitors (HDACi) in diffuse large B-cell lymphoma (DLBCL) have prompted a need for evaluating the impact of mutations in the histone acetyl transferases (HAT) CREBBP and EP300 on HDACi treatment outcome. We identified four DLBCL cell lines; Toledo, with mutations in CREBBP and EP300, SUDHL-7 with mutation of CREBBP and wild-type (wt) EP300, RL with mutation of EP300 and wt CREBBP, and U2932 with wt CREBBP and wt EP300. Vorinostat treatment induced apoptosis significantly more rapid and profound in the CREBBP/EP300 double mutant cell line. Our results suggest that pre-treatment stratification according to HAT defects may be relevant in DLBCL.
Keywords: Histone acetyl transferase, Somatic mutation, Diffuse large B-cell lymphoma, Histone deacetylase inhibition, Vorinostat
1. Introduction
By catalyzing post-translational acetylation of lysine residues, the histone acetyl transferases (HAT) CREBBP and EP300 moderate the activities of both histone and non-histone proteins. This regulates vital cellular processes such as cell cycling and apoptosis by modulating chromatin structure and by altering the activity of transcription factors.1 The roles of CREBBP and EP300 in the pathogenesis of leukemia and malignant lymphomas have recently been highlighted.2,3 CREBBP and EP300 belong to one of three HAT families based on their catalytic domains. They are capable of acetylating the lysine residues of all 4 core histones as well as over 70 other proteins.4,5
It has been shown that acetylation of the tumor suppressor protein p53 leads to its activation, while acetylation of onco-protein BCL6 leads to its functional inactivation.6 In germinal center B-cells BCL6 inhibits transcription of p53 and p53 mediated apoptosis, which allows the cells to tolerate DNA strand breaks during immunoglobulin class switch recombination and somatic hypermutation that are necessary for normal B-cell maturation. This implies that loss-of-acetylation may contribute to lymphomagenesis by BCL6 over-expression and p53 inactivation, which will allow for the survival of germinal center cells with aberrant chromosomal breaks.7
Interestingly, acetylation of BCL6 hinders its ability to recruit histone deacetylases (HDACs) to transcription start sites; thereby hindering HDAC mediated transcriptional repression of its target genes.
The HDAC inhibitor (HDACi) vorinostat (suberoylanilidehydroxamic acid) inhibits the enzymatic activity of HDAC1, HDAC2, HDAC3 (Class I) and HDAC6 (Class II) and therefore show multiple effects of various proteins.
CREBBP mutations have been identified in DLBCL (39%) and follicular lymphoma (FL) (41%), EP300 mutations being found in 10% of the same DLBCLs and in 8.7% of FL samples, respectively.3 Due to >90% sequence similarity within the two genes, they have been suggested to be functionally homologous8; however, co-existing alterations of both CREBBP and EP300 were found in only 6/72 cases.3 For both genes, a clustering of mutations was observed in the catalytic HAT domains. HAT domain mutant proteins had lost their acetyl transferase activity, while no significant effects were observed when mutations were located outside the HAT domain. While the acetylation activity of the individual mutations was thoroughly described, the consequences for cell survival were not analyzed and is thus the focus of this study.
A direct comparison of HDACi induced apoptosis in DLBCL cells with single vs. double mutated vs. wt CREBBP/EP300 genes has so far not been reported. Because of the above mentioned physiologically important function of acetylation in the regulation of apoptosis in germinal center cells and in B-cell lymphomagenesis, we speculated whether different responses to vorinostat could be observed in B-cell tumors with and without CREBBP/EP300 mutations.
In phase I and II trials, NHL patients treated with vorinostat have exhibited heterogeneous clinical responses. Here we present experimental data suggesting that vorinostat-induced apoptosis in HAT mutant DLBCL cell lines may serve as a comparable endpoint when drawing parallels to results from these clinical trials.
2. Materials and methods
2.1. Mutation scanning
We designed a mutation screening assay based on the high-resolution melting analysis (HRM), and subsequently verified variant HRM curves by the use of denaturing gradient gel electrophoresis (DGGE) and direct sequencing that covered the genomic regions that encode the catalytic domains as described.9 Sanger sequencing of aberrant DGGE bands was performed with non-clamped primers by the service of Eurofins® (www.eurofins.de).
When the goal of identifying the concomitant EP300/CREBBP mutated DLBCL cell line (Toledo) and the EP300 mutated/CREBBP wt cell line (RL) was achieved, we drew advantage from the results by Pasqualucci and colleagues who demonstrated SUDHL-7 to harbor two loss-of-function mutations of CREBBP, and to be EP300 wt. In addition, they proved U2932 to be CREBBP/EP300 wt, both by direct sequencing of whole-genome amplified DNA. These cell lines were obtained from the authors.
2.2. Treatment with vorinostat
The cell lines were incubated in the absence or presence of vorinostat in increasing concentrations (0.1, 1.0, 2.5 μM), and DMSO for 48 h. Vorinostat was re-added to cell cultures at 24 h. Apoptosis assay by Annexin V/FITC and propidium iodide (PI) (BD Pharmingen) was performed to evaluate the effect of the dosages of vorinostat on early and late apoptosis, respectively. All apoptosis assays were performed in triplicates.
2.3. Statistics
The level of apoptosis between the cell lines was compared using one-way Analysis Of Variance (ANOVA). All P-values were two-sided. Confidence limits given in figures were 95% confidence. SPSS version 19 was used for calculations (2010, IBM, Somer, NY, USA).
3. Results
Screening of the Toledo cell line revealed a single base change of the EP300 gene (Fig. 1a,b). Furthermore, we identified a 3 bp deletion of the Toledo CREBBP gene (Fig. 1c). Screening of RL revealed a missense mutation in EP300 (Fig. 1d). The SUDHL-7 harbors two CREBBP mutations both with deleterious effects key to HAT activity as assessed by in vivo acetylation assays in mice (exon 26: Y1450C and exon 30: ΔS1680).3
Fig. 1.
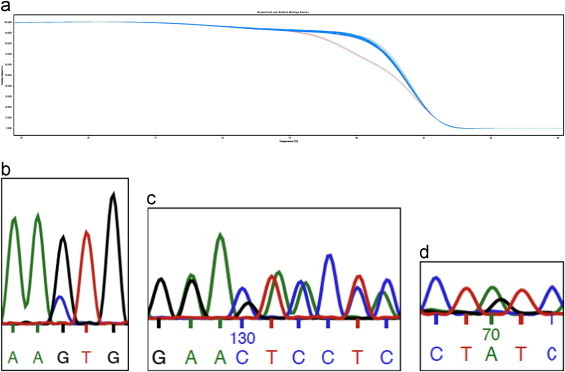
(a) An altered HRM curve for the Toledo cell line (marked by red curves) demonstrates the first screening step. (b) Direct sequencing revealed a missense mutation of the Toledo cell line in exon 28 of the EP300 gene (G–>C transversion resulting in an amino acid substitution, L1520V). (c) Direct sequencing revealed a 3 bp deletion causing a frame shift of the Toledo cell line in exon 30 of the CREBBP gene (resulting in a premature stop-codon V1742X). (d) Direct sequencing revealed a missense mutation in the RL cell line in exon 26 of the EP300 gene (A–>G transition resulting in an amino acid substitution, Y1413C). (For interpretation of the reference to color in this figure, the reader is referred to the web version of this article.)
3.1. Cell line apoptosis assays
We observed dose dependent increases in the fraction of apoptotic cells in all cell lines. Statistically significant differences in the fraction of apoptotic cells between wt, single mutant, and double mutant cell lines were observed in all but the lowest concentration of vorinostat: 1 μM vorinostat (early apoptosis: P<0.0001, late apoptosis: P<0.0001) and 2.5 μM vorinostat (early apoptosis: P=0.011, late apoptosis P<0.0001), respectively. Interestingly, the CREBBP/EP300 double mutant Toledo cell line exhibited a particular profound apoptosis, when compared to the other cell lines (Fig. 2).
Fig. 2.
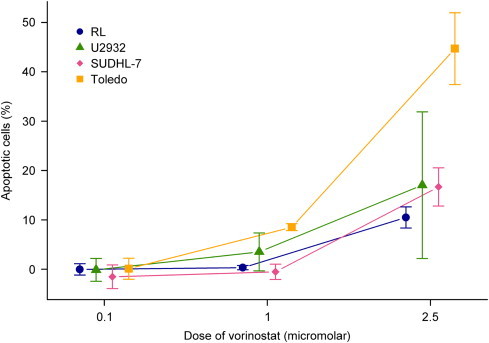
Comparison of induction of apoptosis through propidium iodide staining at 48 h of vorinostat treatment. Bars show 95% CI for the mean.
4. Discussion
Heterogeneous responses have been reported from clinical phase I and II trials of vorinostat in NHL-patients. These data strongly imply that diverse underlying molecular defects in these various tumor types determine the responses to vorinostat. Mutations in the HAT domain proteins are obvious candidates. While experimental data show that HAT mutations did not seem to affect the effectiveness of apoptosis induction by vorinostat in T-ALL cells,2 our data imply that tumors with concomitant mutations of CREBBP and EP300 may be particularly sensitive to vorinostat induced apoptosis. Given the important role of acetylation in the physiological regulation of BCL6 and p53 activity in germinal center cells, CREBBP/EP300 double mutant cells are likely to have a particularly skewed BCL6/p53-balance, leading to increased cell survival.
A plausible theory for the efficacy of HDACi in HAT mutant DLBCL is, that reestablishment of the physiological balance between acetylation and deacetylation in HAT-deficient cells by the use of HDACi, may lead to increased p53 activation and increased apoptosis. Indeed, the double-mutated Toledo cell line exhibited a swift and ultimately more profound degree of apoptosis in comparison with both wild type and single CREBBP/EP300 mutated cell lines.
Evidence of a variant induction of apoptosis to vorinostat in different DLBCL cell lines according to HAT mutational status implies that defects in CREBBP/EP300 could potentially explain the heterogeneous observations in clinical trials of vorinostat in NHL. These findings, however, need to be confirmed in future clinical studies. The efficacy of HDACi in HAT-mutation stratified DLBCL may be further optimized by testing the numerous different HDACi considering their different target specificities.
Acknowledgments
We wish to thank bioanalyst Anja Pedersen for skillful technical assistance. We also wish to thank Professor Laura Pasqualucci and Qiong Shen, Institute for Cancer Genetics Columbia University, for supplying cell lines. C.L.A. provided the conception and design of the study, acquisition of data, analysis and interpretation of data, drafting the article; F.A. provided the conception and design of the study, acquisition of data, analysis and interpretation of data, drafting the article; H.H. revised it critically for important intellectual content, and final approval of the version to be submitted; T.B. provided analysis and interpretation of the data; K.G. provided the conception and design of the study, supplied the acquisition of data, drafting of manuscript, revised it critically for important intellectual content, and final approval of the version to be submitted.
References
- 1.Brooks C.L., Gu W. The impact of acetylation and deacetylation on the p53 pathway. Protein & Cell. 2011;2:456–462. doi: 10.1007/s13238-011-1063-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mullighan C.G., Zhang J., Kasper L.H., Lerach S., Payne-Turner D., Phillips L.A. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature. 2011;471:235–239. doi: 10.1038/nature09727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pasqualucci L., Dominguez-Sola D., Chiarenza A., Fabbri G., Grunn A., Trifonov V. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011;471:189–195. doi: 10.1038/nature09730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schiltz R.L., Mizzen C.A., Vassilev A., Cook R.G., Allis C.D., Nakatani Y. Overlapping but distinct patterns of histone acetylation by the human coactivators p300 and PCAF within nucleosomal substrates. Journal of Biological Chemistry. 1999;274:1189–1192. doi: 10.1074/jbc.274.3.1189. [DOI] [PubMed] [Google Scholar]
- 5.Wang H., Walsh S.T., Parthun M.R. Expanded binding specificity of the human histone chaperone NASP. Nucleic Acids Research. 2008;36:5763–5772. doi: 10.1093/nar/gkn574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bereshchenko O.R., Gu W., Dalla-Favera R. Acetylation inactivates the transcriptional repressor BCL6. Nature Genetics. 2002;32:606–613. doi: 10.1038/ng1018. [DOI] [PubMed] [Google Scholar]
- 7.Phan R.T., Dalla-Favera R. The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature. 2004;432:635–639. doi: 10.1038/nature03147. [DOI] [PubMed] [Google Scholar]
- 8.Cohen I., Poreba E., Kamieniarz K., Schneider R. Histone modifiers in cancer: friends or foes? Genes & Cancer. 2011;2:631–647. doi: 10.1177/1947601911417176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Andersen C.L., Hasselbalch H., Grønbæk K. Lack of somatic mutations in the catalytic domains of CREBBP and EP300 genes implies a role for histone deacetylase inhibition in myeloproliferative neoplasms. Leukemia Research. 2012;36:485–487. doi: 10.1016/j.leukres.2011.11.018. [DOI] [PubMed] [Google Scholar]