

Abstract
While in RARS-T, JAK2V617F mutation is common and associated with good prognosis, the clinical and prognostic impact of this mutation in other MDS is unknown. We collected data from 132 non-RARS-T MDS with known JAK2V617F mutation status. JAK2V617F mutation was significantly correlated with lower progression to AML (p<.0011) and better overall survival (OS, p=.011). OS difference persisted after matching on age, sex, IPSS and % marrow blast (p=.031). Thus, in MDS other than RARS-T, JAK2V617F mutation may be associated with favorable outcome.
Keywords: JAK2V617F mutation, MDS, RARS-T, Prognosis, AML progression
1. Introduction
Acquired JAK2V617F somatic mutation is a hallmark of Philadelphia negative myeloproliferative neoplasm (MPN) [1,2]. In myelodysplastic syndromes (MDS), JAK2V617F mutation is seen in less than 5% of the cases [3]. Both 2001 and 2008 WHO classifications include among myelodysplastic/myeloproliferative (MDS/MPN) overlap syndromes [4] a provisional entity, refractory anemia with ringed sideroblasts associated with sustained thrombocytosis (RARS-T) that appears to have a relatively favorable prognosis [5] especially if JAK2V617F mutation, detected in about 60% of the cases, is present [3]. Apart from RARS-T, however, the impact of JAK2V617F mutation in MDS remains unknown [6–8].
We conducted a retrospective study of MDS patients with JAK2V617F mutation, after exclusion of RARS-T cases, where patients were compared with matched unmutated MDS without JAK2V617F mutation.
2. Materials/methods
2.1. Data collection
After approval by the Clermont-Ferrand University Hospital Ethics committee, a questionnaire was sent to all French centers of the Groupe Francophone des Myélodysplasies (GFM) and of the French Intergroup of Myeloproliferative neoplasms (FIM) to collect clinical, laboratory and follow up data in MDS patients with known JAK2V617F mutation status detected in blood by real-time polymerase chain reaction. We also obtained, from the GFM registry of French MDS, data from patients where JAK2V617F mutation had been tested. In the absence of specific recommendations, JAK2V617F mutation in those patients had been assessed systematically at diagnosis in a few centers, or later in the disease course, mainly because of features with potential predictive value for JAK2V617F mutation, like thrombocytosis. Patients with chronic myelomonocytic leukemia, RARS-T and MDS following MPN were excluded [4].
2.2. Statistical analysis
Descriptive statistics were used to characterize the population on parameters collected at the time of diagnosis. Overall survival (OS) was defined from diagnosis to death or censored at latest contact with the patient. Time to acute myeloid leukemia (AML) progression was defined from diagnosis to date of AML. OS curves and OS estimates were determined using the Kaplan–Meier method. Patient characteristics and outcome according to JAK2V617F mutation status were analyzed by univariate analysis using the chi-square test for qualitative analysis and Kruskal–Wallis test for quantitative analysis and log-rank test for OS comparison.
The primary endpoint of this retrospective analysis was to assess the impact of JAK2V617F mutation on MDS. In an attempt to reduce the effect of potential bias in this cohort, propensity score matching was used. This method allows balancing of all measured relevant variables between the 2 groups (JAK2V617F positive and negative).
Covariates included in the propensity-score model were sex, hemoglobin, WBC, ANC, platelet, IPSS, karyotype, marrow blasts and age known to be clinical relevant in MDS. OS analysis was performed with a Cox model adjusted with propensity score [9]. Statistical significance was considered below .05. Statistical analysis was performed using STATA 10.0 software (StataCorp, College Station, TX, USA).
3. Results
Data from 132 MDS patients with known JAK2V617F mutation status were collected in 19 centers of the FIM and GFM, including 37 JAK2V617F positive (JAK2+) and 95 JAK2V617F negative (JAK2−) cases.
Their main clinical and laboratory features are presented in Table 1. The JAK2V617F mutation burden was available in only 7 patients, showing a median level of 16% (range, 1–50%).
Table 1.
Clinical, laboratory features and outcome in MDS with or without JAK2V617 mutation.
| JAK2V617F positive | JAK2V617F negative | p | |
|---|---|---|---|
| Demographics and hematological features | n=37 | n=95 | |
| Median age (range) | 74 (47–93) | 70.5 (38–95) | .44 |
| M/F | 18/19a | 49/46 | .87 |
| Hb g/100 ml (median, range) | 10.1 (6.3–14.2) | 9.8 (6.5–14.5) | .51 |
| MCV fl (median, range) | 103 (82–125) | 98 (77–124) | .018 |
| Median WBC G/L (range) | 6.1 (1.9–17.4) | 4.4 (.9–18.4) | .004 |
| Median ANC G/L (range) | 3.7 (.2–12.6) | 2.1 (.3–15.8) | .009 |
| Median platelet count G/L (range) | 262 (35–1108) | 156 (5–663) | .002 |
| Median % marrow blasts (range) | 2 (0–16) | 4 (0–19) | <.001 |
| Karyotype | n=33 | n=94 | |
| Normal | 17 (51%) | 59 (63%) | .52 |
| Del 5q | 4 (12%) | 5 (5%) | .36 |
| Del 20q | 1 (3%) | 3 (3%) | .62 |
| +8 | 4 (12%) | 3 (3%) | .15 |
| −7/del7q | 1 (3%) | 2 (2%) | .69 |
| complex (n≥3) | 6 (21%) | 10 (11%) | .25 |
| WHO 2008 classification | n=37 | n=95 | |
| RA | 5 (13%) | 24 (25%) | .14 |
| RARS | 8 (23%) | 9 (9%) | .11 |
| RCMD | 9 (24%) | 13 (14%) | .14 |
| RAEB-1 | 7 (19%) | 27 (28%) | .26 |
| RAEB-2 | 3 (8%) | 19 (20%) | .10 |
| MDS with isolated del(5q) | 2 (5%) | 1 (1%) | .39 |
| Unclassified | 3 (8%) | 3 (3%) | .45 |
| IPSS | n=33 | n=94 | |
| Low | 13 (39%) | 32 (34%) | .58 |
| Intermediate-1 | 17 (52%) | 44 (47%) | .64 |
| Intermediate-2 | 3 (9%) | 13 (14%) | .69 |
| High | 0 (0%) | 5 (5%) | .41 |
| Treatment | n=37 | n=80 | |
| ESA | 20 (54%) | 37 (46%) | .43 |
| Lenalidomide or thalidomide | 9 (24%) | 9 (11%) | .053 |
| Intensive chemotherapy | 4 (11%) | 6 (7%) | .67 |
| Hypomethylating agent | 3 (8%) | 5 (6%) | .89 |
| Allogeneic transplant | 1 (3%) | 0 | |
Abbreviations: M: male; F: female; MCV: mean corpuscular volume; WBC: white blood cell; ANC: absolute neutrophil count; RA: refractory anemia; RARS: refractory anemia with ring siderobalsts; CRMD: refractory cytopenia with multilineage dysplasia; RAEB-1: refractory anemia with excess of blasts-1; RAEB-2: refractory anemia with excess of blasts-2; MDS: myelodysplastic syndromes; IPSS: international prognostic scoring system; ESA: Erythropoiesis stimulating agent
Data not available.
There were no significant differences between JAK2+ and JAK2− cases in terms of age, gender, hemoglobin level, WHO 2008 classification, cytogenetic findings and IPSS. In JAK2+ patients, the white blood cells (WBC) count, absolute neutrophil count (ANC), platelet count and mean corpuscular volume (MCV) were significantly higher, while the marrow blast percentage was significantly lower than in JAK2− cases.
Treatments used (Table 1) were similar in both patient groups, especially with regards to erythropoietic stimulating agents, chemotherapy and hypomethylating agents. Seventeen JAK2+ (45%) versus 36 (37.8%) JAK2− patients were red blood cell transfusion-dependent (p=.77).
Median follow up was 44 months (range 18–349) in JAK2+ and 69 months (range 32–185) in JAK2− patients (p=.95). OS was significantly longer in JAK2+ patients (median not reached) than in JAK2− patients (median 66 months) (p=.011) (Fig. 1). The estimated 5-year OS was 86% in JAK2+ patients versus 57% in JAK2− patients. The 10 year cumulative incidence of AML progression was significantly lower in JAK2+ (20%) than in JAK2− patients (47%) (p<.001).
Fig. 1.
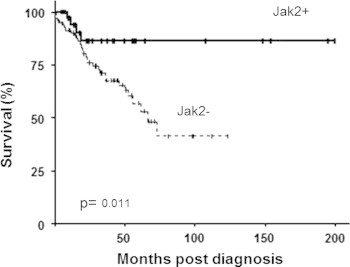
Overall survival according to JAK2V617F status in MDS.
When the analysis was matching with propensity score, OS was still significantly longer in JAK2+ patients (median not reached versus 73 months, p=.031) whereas JAK2 status had no significant prognostic value for OS in IPSS intermediate-2 and high patients (p=.18).
4. Discussion
In this series, which is to our knowledge the first to focus on MDS with JAK2V617F mutation other than RARS-T, we found JAK2+ cases to have higher WBC, ANC and platelet counts and fewer marrow blasts than JAK2− MDS, but no significant difference in cytogenetics and IPSS.
JAK2+cases had significantly lower risk of AML progression and better OS than JAK 2− patients, an OS advantage which persisted in a matched case control analysis based on age, gender, marrow blast percentage and IPSS.
Recently, in a large series of 439 MDS, Bejar et al. found no prognostic value to JAK2V617F mutation but only 13 (3%) of the patients had JAK2V617F mutation. In addition, whether some of those patients had RARS-T was not indicated [10]. Finally, only 64% of the JAK2+ cases had lower risk IPSS in Bejar et al. series, compared to 80% in our study. Indeed, we found the favorable prognosis of JAK2V617F mutation to be mainly restricted to lower risk IPSS.
Thus, we found that MDS other than RARS-T with JAK2V617F mutation were associated with laboratory features (higher WBC, ANC, platelet count) with better outcome. However, prospective studies precisely evaluating the impact of this mutation are required to confirm our findings.
Acknowledgments
We thank Françoise Boyer, Sophie Raynaud, Christian Rose, Jean-Loup Demory, Florence Nguyen-Khac, Lionel Ades, Gerard Tertian, Romain Guièze, Jean Christophe Ianotto, Dominique Bordessoule, Jean-Yves Cahn, Michaela Fontenay, Olivier Tournilhac who contributed essential patients data, Bruno Pereira to statistical advice and all GFM and FIM members.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Contributor Information
Benoit de Renzis, Email: bderenzis@chu-clermontferrand.fr.
Pierre Fenaux, Email: pierre.fenaux@avc.aphp.fr.
References
- 1.James C., Ugo V., Le Couédic J.P., Staerk J., Delhommeau F., Lacout C. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–1148. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 2.Lippert E., Boissinot M., Kralovics R., Girodon F., Dobo I., Praloran V. The JAK2-V617F mutation is frequently present at diagnosis in patients with essential thrombocythemia and polycythemia vera. Blood. 2006;108:1865–1867. doi: 10.1182/blood-2006-01-013540. [DOI] [PubMed] [Google Scholar]
- 3.Schmitt-Graeff A.H., Teo S.S., Olschewski M., Schaub F., Haxelmans S., Kirn A. JAK2V617F mutation status identifies subtypes of refractory anemia with ringed sideroblasts associated with marked thrombocytosis. Haematologica. 2008;93:34–40. doi: 10.3324/haematol.11581. [DOI] [PubMed] [Google Scholar]
- 4.Swerdlow S.H., Campo E., Harris N.L., Jaffe E.S., Pileri S.A., Stein H. 4th ed. IARC, WHO Press; 2008. WHO classification of tumours of haematopoietic and lymphoid tissues. [Google Scholar]
- 5.Shaw G.R. Ringed sideroblasts with thrombocytosis: an uncommon mixed myelodysplastic/myeloproliferative disease of older adults. British Journal of Haematology. 2005;131:180–184. doi: 10.1111/j.1365-2141.2005.05747.x. [DOI] [PubMed] [Google Scholar]
- 6.Patnaik M.M., Lasho T.L., Finke C.M., Gangat N., Caramazza D., Holtan S.G. WHO-defined ‘myelodysplastic syndrome with isolated del(5q)’ in 88 consecutive patients: survival data, leukemic transformation rates and prevalence of JAK2, MPL and IDH mutations. Leukemia. 2010;24:1283–1289. doi: 10.1038/leu.2010.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ingram W., Lea N.C., Cervera J., Germing U., Fenaux P., Cassinat B. The JAK2 V617F mutation identifies a subgroup of MDS patients with isolated deletion 5q and a proliferative bone marrow. Leukemia. 2006;20:1319–1321. doi: 10.1038/sj.leu.2404215. [DOI] [PubMed] [Google Scholar]
- 8.Sokol L., Caceres G., Rocha K., Stockero K.J., Dewald D.W., List A.F. JAK2(V617F) mutation in myelodysplastic syndrome (MDS) with del(5q) arises in genetically discordant clones. Leukemia Research. 2010;34:821–823. doi: 10.1016/j.leukres.2009.09.016. [DOI] [PubMed] [Google Scholar]
- 9.Rosenbaum P.R., Rubin D.B. The central role of the propensity score in observational studies for causal effects. Biometrika. 1983;70(1):41–55. [Google Scholar]
- 10.Bejar R., Stevenson K., Abdel-Wahab O., Galili N., Nilsson B., Garcia-Manero G. Clinical effect of point mutations in myelodysplastic syndromes. New England Journal of Medicine. 2011;364:2496–2506. doi: 10.1056/NEJMoa1013343. [DOI] [PMC free article] [PubMed] [Google Scholar]