

Abstract
Human Mycoplasma pneumoniae (MP) pneumonia is characterized by alveolar infiltration with neutrophils and lymphocytes and lymphocyte/plasma cell infiltrates in the peri-bronchovascular area (PBVA). No mouse model has been able to mimic the pathological features seen in human MP pneumonia, such as plasma cell-rich lymphocytic infiltration in PBVA. To figure out the mechanism for inflammation by MP infection using a novel mouse model that mimics human MP pneumonia, mice were pre-immunized intraperitoneally with Th2 stimulating adjuvant, alum, alone or MP extracts with an alum, followed by intratracheal challenge with MP extracts. The toll-like receptor-2, which is the major receptor for mycoplasma cell wall lipoproteins, was strongly up-regulated in alveolar macrophages in a latter group after the pre-immunization but prior to the intratracheal challenge. Those findings demonstrated that acceleration of innate immunity by antecedent antigenic stimulation can be an important positive-feedback mechanism in lung inflammation during MP pneumonia.
Abbreviations: MP, Mycoplasma pneumoniae; MP pneumonia, Mycoplasma pneumonia; AMs, alveolar macrophage; TLR, Toll-like receptor; IT, intratracheal challenge; BAL, bronchoalveolar lavage; PBVA, peri-bronchovascular area; PVA, perivascular area; PBA, peribronchiolare area
Keywords: Mycoplasma pneumoniae pneumonia, Alveolar macrophage, Mice model, Toll-like receptor-2, Plasma cell, Mycoplasma pneumonia extracts
1. Introduction
Mycoplasma pneumoniae (MP) is a common pathogen in community acquired pneumonia. MP pneumonia can lead to acute respiratory distress syndrome [1], and is sometimes fatal. MP is an extracellular pathogen that adheres to mucosal surfaces of the respiratory and genital tracts. Mycoplasmas lack cell walls, and the cell membrane of an invading bacterium fuses with the host cell membrane to induce an immune response [2,3]. Airway diseases caused by MP include bronchiolitis, bronchitis, bronchiolitis obliterans and rarely, bronchiectasis. Recently, MP has been implicated in the pathogenesis of asthma [4]. Epithelial cells play an important role in recruiting inflammatory cells into the airways [5]. While the clinical significance of MP infection is evident, the pathogenic mechanisms for lung inflammation have not been well defined.
Cumulative information on the pathogenesis of human MP pneumonia has been gathered from pathological examination of autopsy specimens [6–12]. There have also been limited albeit important pathological reports based on studies of open lung biopsy specimens [13–17], video-assisted thoracic surgery (VATS) [18], and transbronchial lung biopsy (TBLB) [19–21]. According to these reports, the most characteristic pathological feature of human MP pneumonia is a marked plasma cell-rich lymphocytic infiltration in the peri-bronchovascular area (PBVA) [12,13,16]. Lymphocytic alveolitis has also been reported in these studies. In murine models, intranasal inoculation with alive MP has been shown to cause initial neutrophilic infiltration of the alveoli, followed by lymphocytic infiltrates thereafter. In contrast to human pathology, no murine or other animal models have exhibited prolonged plasma cell infiltration of the PBVA.
An excessive and inappropriate immune response against MP seems to be the major contributing factor in the pathogenesis of MP infection. Extrapulmonary manifestations, including arthralgia, Guillain-Barré syndrome, myocarditis, pericarditis, acute myocardial infarction, hemolytic anemia, disturbances to the coagulation mechanism, and Stevens-Johnson syndrome have been reported as complications of MP pneumonia [22]. A study has shown that peripheral blood lymphocytes respond more strongly to MP extracts among recently infected patients compared to healthy controls [23]. In addition, delayed type hypersensitivity reactions to heat-killed MP extracts are observed in skin tests of patients with MP pneumonia [24], while anergy to the tuberculin skin test has been well recognized in the early phase [25]. Alternatively, MP extracts may induce lung inflammation through up-regulation of host innate immunity. Recent studies in both mice [26] and humans [27] revealed that MP causes persistent but latent infection in the lower respiratory tracts, which may up-regulate host innate immunity.
Innate immunity against invading microbes is initiated by pathogen recognition by toll-like receptors (TLRs) followed by activation of host inflammatory responses. Among the 12 TLR family members, TLR-2, TLR-4, TLR-5 and TLR-9 have been implicated in the recognition of different bacterial components. Peptidoglycan, lipoarabinomannan, zymosan, and lipoproteins from various micro-organisms are recognized by TLR-2 [28], while lipopolysaccharide, bacterial flagellin, and bacterial DNA are recognized by TLR-4, TLR-5 and TLR-9, respectively. These TLR family members are known to activate nuclear factor κB (NF-κB) via sustained phosphorylation of p38 mitogen-activated protein kinase (MAPK). In MP pneumonia, it has been reported that TLR-2 signaling is involved in inflammatory cell activation by mycoplasma-derived lipoproteins [29]. Chu et al. demonstrated that expression of TLR-2 mRNA and protein on alveolar macrophages (AMs), and the recruitment of adaptor protein MyD88 increases after MP infection [30]. In this regard, Hayakawa et al. [31], Sekine et al. [32], and Chu et al. [33] in turn demonstrated that pre-immunization with alive MP or its extract significantly augmented the inflammatory responses after the second challenge. Thus, it is likely that subclinical, latent infection of MP in the lower respiratory tracts may up-regulate TLR-2 expression on AMs and bronchial epithelial cells augmenting MP reactivity.
In this study mice were immunized with MP extracts to mimic human MP pneumonia, and thereafter challenged with the same extracts by intratracheal exposure. We found that stimulation by MP extracts up-regulated baseline expression of TLR-2 on AMs and augmented their response to the subsequent challenge by the same extracts. Our results demonstrated that preceding or latent respiratory MP infection may trigger and synergistically augment inflammatory processes against MP extracts through up-regulation of host innate immunity.
2. Material and methods
2.1. Preparation of MP extracts
MP, ATCC29342 strain (American Type Culture Collection, Rockville, MD) was cultured in PPLO broth (Nikken Bio Medical Laboratory, Tokyo) at 37 °C under 5% CO2 for 6 day. MP was collected by centrifugation at 10,000g for 25 min, washed three times with Hanks' balanced salt solution (HBSS, Gibco, NY), and resuspended in distilled water. After undergoing homogenization 10 times for 60 s using a sonicator (Sonifile 250, Branson Ultrasonics Co, CT), the suspension was centrifuged at 10,000g for 5 min and, the supernatant was filtered, to derive “MP extracts”. The protein concentration was determined by Bio-Rad Protein Assay (Bio-Rad Laboratories, Tokyo), adjusted to 1 μg/μL with distilled water, and the aliquots were stored at −80 °C until use. Lipopolysaccharide contamination was 0.1 I.U./mL by limulus test, which showed no effect on stimulation of murine alveolar macrophages [34].
2.2. Animals and inoculation
Female BALB/c (7 week old) mice were purchased from Nippon Charles River (Tokyo), and fed with CE-2 (CLEA Japan, Inc.) in a specific pathogen free (SPF) environment. All mice were inoculated intratracheally with MP extracts with or without pre-immunization. Pre-immunization was carried out by intraperitoneal injection at 6 and 13 day prior to the intratracheal challenge (IT). Treatment models included; Model A: IT without pre-immunization; Model B: IT after twice pre-immunizing with MP extracts alone; Model C: IT after twice pre-immunizing with MP extracts plus CpG; Model D: IT after twice pre-immunizing with alum alone; and Model E: IT after twice pre-immunizing with MP extracts plus alum. As shown in Table 1, One week following the last immunization, mice were intraperitoneally anesthetized with 40 mg/kg of pentobarbiturate (Nembutal, Dainippon Sumitomo Pharma Co., Tokyo) and 80 μg/kg of medetomidine hydrochloride (Domitor®, Orion corporation, Finland), and underwent IT with 50 μg of MP extracts. Bronchoalveolar lavage fluid (BALF) and lung specimens were obtained before this process and 8, 24, 48, 96, 168 h after IT. These procedures were approved by the Institutional Experimental Animal Ethics Committee of Kyorin University.
Table 1.
Experimental schedule.
| Events | Timing | Treatments |
|---|---|---|
| Pre-immunization (intraperitoneal) | 6 and 13 days prior to intratracheal challenge | Pre-immunization with alum alone or with alum plus MP extracts |
| Intratracheal challenge | Day 0 | Intratracheal challenge with MP extracts |
| BAL analysis | 0, 8, 24, 48, 96, 168, and 336 h after intratracheal challenge | |
| Sampling for lung pathology | 0, 8, 24, 48, 96, 168, and 336 h after intratracheal challenge |
alum: a Th2 stimulating adjuvant, MP extract: Mycoplasma pneumoniae extract, derived from centrifugation of sonicated particle of alive Mycoplasma pneumoniae, described in the “Preparation of MP extracts” section above mentioned in the “Material and methods” section.
2.3. Titration of the serum antibody specific for MP
Serum antibody titers against MP were determined before and after pre-immunization by particle-agglutination test, using Serodia MycoII (Fujirebio, Tokyo).
2.4. Histopathology
After mice were euthanized, 10% formalin solution (2 mL) was instilled into the bronchial tree, and the lungs were fixed for 14 day. Paraffin embedded sections were then prepared, deparaffinized, and stained with hematoxylin–eosin (HE) or other immunohistochemical staining. Stained lung sections were evaluated for the degree of inflammation by counting cells infiltrate within the peribronchiolar and, perivascular areas, and intraalveolar spaces. All inflammatory cells were counted at 200× magnification. The number of neutrophils, lymphocytes, and macrophages were enumerated by three independent pathologists based on the cell morphology. Neutrophil infiltration within alveoli was defined as none (−), mild (+, up to 150 cells/ field), moderate (++, from 150 to 500 cells/field), or severe (+++, greater than 500 cells/field). Similarly, the degree of lymphocytic infiltration within alveoli was defined as none (−), mild (+, up to 5 cells/field), moderate (++, from 5 to 15 cells/field), or severe (+++, greater than 15 cells/field). Peri-bronchovascular infiltration by lymphocytes was defined as none (−), mild (+, up to 150 cells/field), moderate (++, from 150 to 350 cell counts/field), or severe (+++, greater than 350 cells/field), while peri-bronchovascular infiltration by plasma cells was defined as none (−), mild (+, up to 10 cells/field), moderate (++, from 10 to 50 cells/field), or severe (+++, greater than 50 cells/field).
2.5. Immunohistochemistry
CD3+ T cells and plasma cells were detected by immunohistochemically using anti-CD3 antibody (Dako, CA, USA) and anti-CD138 antibody (Dako, CA, USA), respectively. For the negative control, we used murine IgG.
The expression of RANTES, MCP-1, and TLR-2 were also analyzed immunohistochemically using polyclonal rabbit antibodies against TLR-2 (TLR-2 antibody—Aminoterminal end, ab47840), RANTES (R&D Systems, MN, USA), and MCP-1 (Hycult biotech Inc, Eindhoven area, Netherlands), respectively. For the negative controls, we used rabbit IgG (Dako, CA, USA). Color development was performed using 3-amino-9-ethyl carbazole (Dako, Glostrup, Denmark) or diaminobenzidine (Nichirei, Tokyo). The number of TLR-2/cytokine/chemokine positive lung cells were counted in each lung field (200×) by microscopy, with counts reaching up to 250/field with a total of five fields/mouse.
2.6. Cellular response to MP extracts
Spleens were minced in RPMI 1640 with 5% fetal bovine serum (FBS), and the resulting cell suspension was passed through a nylon mesh, and centrifuged at 2000g for 5 min. The cells were washed twice with phosphate-buffered saline without calcium/magnesium chloride (PBS-) and were resuspended in RPMI 1640 with 5% FBS (Iwaki, Tokyo) at a density of 106 cells/mL. Two hundred microliters of the suspension was added to each well in a flat-bottomed microtiter plates (Iwaki, Tokyo). In the same manner, bronchoalveolar lavage fluid (BALF) cells were harvested. Thereafter, 10 μg of MP extracts was added to each well followed by incubation at 37 °C for 72 h under 5% CO2. The cells were pulsed with [methyl-3H] thymidine (Moravek Biochemicals, Inc. CA) for 16 h, and uptake was measured using a liquid scintillation counter. In some experiments, AMs were purified from BALF by plastic adhesion for 30 min at 37 °C under 5% CO2, collected by gentle scraping using a cell scraper (Falcon, Ann Arbor, MI), resuspended in RPMI 1640 with 5% FBS (Iwaki), added to a 24 well plate (Falcon) at 2×104 cells/well, and incubated in the presence or absence of MP extracts (10 μg/mL). After 8 h, culture supernatants were stored at −80 °C further use.
2.7. Bronchoalveolar lavage (BAL)
BAL fluid was obtained after sacrifice through 1-cm longitudinal incision that was made to expose the trachea. BALF was obtained before and, 8 and 24 h after IT inoculation by instilling 1 mL of HBSS through a 25-gage needle inserted into the tracheal rings. A retrieved aliquot was centrifuged at 1500g for 5 min, and the supernatant was stored at −80 °C for analysis of cytokines levels in BALF. In some cases, the cell pellet was resuspended in HBSS, the density was counted by a hemocytometer, and the differentials were determined by counting 300 cells on a cyto-centrifuge slide after staining with Diff Quick (Kokusai Shiyaku Co. Ltd., Kobe).
2.8. Determination of cytokine/chemokine level
Concentrations of TNF-α, IL-1β, IL-6, MIP-2, MIP-1α, KC, MCP-1, and RANTES in BALF or culture supernatants were measured using MILLIPLEX® (# MPXMCYTO, Millipore, MA) according to manufacturer's instructions.
2.9. Real-time quantitative PCR (RT-qPCR)
Expression of TLR-2 mRNA in AMs was evaluated by real-time quantitative PCR. Because BALF cells consisted of mostly AMs in both model E (mean±SD; 96.1±2.5%) and model D (95±4%), respectively, we used total BALF cells without any purification procedure. Total RNA was extracted from alveolar macrophages using TRIzol® (Invitrgen Life Tech, CA, USA) and the RNeasy® mini kit (QIAGEN, Frankfurt, Germany), incubated with DNase I followed by reverse transcription using the SuperScript® first strand synthesis system for RT-PCR (Invitrogen). The reaction mixture included 154 ng of total RNA and random hexamers (50 ng). The mouse TLR-2 primers and probes for RT-qPCR have been previously reported [35]. TLR-2 (GenBank accession number, AF124741) primers and probes: forward primer, 5_-AAGGCATTAAGTCTCCGGAATTATC-3_; reverse primer, 5_-TCGCTTAAGTGAAGAGTCAGGTGAT-3_; probe, 5_-TCCCAAAGTCTAAAGTCGATCCGCGAC-3_. The qPCR was performed using the PCR Thermal Cycler Dice Real Time System TP800 (TaKaRa, Japan, Kyoto). The sample mixture contained 60 ng of cDNA, 100 nM of fluorogenic probe, 200 nM of primers, and Premix Ex Taq (TaKaRa). The reaction conditions included 30 s of pre-incubation at 95 °C followed by 99 cycles for 5 s at 95 °C and 40 s at 60 °C. Appropriate non-template controls were included in each PCR reaction. Relative expression levels of TLR-2 were calculated from relative levels of GAPDH (Applied Biosystems, Inc., CA).
2.10. Statistical analysis
Numerical data were evaluated for a normal distribution using the Shapiro–Wilk test and for equal variance using the Levine median test. Data were presented as the means±SD. Statistical comparisons of data were made by Student's t test. All tests were 2-sided, and p-values of less than 0.05 were considered to be statistically significant.
3. Results
3.1. Comparison of histopathological features in mice models
To establish the most appropriate mouse model that would mimic human MP pneumonia, we first carried out a review of the previous literature on the histopathology of human MP pneumonia as shown in Table 2. The major pathological findings reported in human MP pneumonia are neutrophilic and lymphocytic infiltration in the alveolar spaces and lymphocytic and plasmacytic inflammation in the PBVA. Neutophils/lymphocyte alveolitis has been variably reported among cases where specimens were obtained at autopsy [6–12], or from open [13–17], and video assisted [18], transbronchial biopsies [19–21]. Lymphocytic and plasmacytic infiltration of the PBVA would therefore constitute the most characteristic finding in human MP pneumonia. However, this pathological finding has not been demonstrated in murine models, perhaps reflecting the inadequacy of murine models in mimicking human MP pneumonia precisely. In the present study, we designed five different mouse models. As shown in Table 3, models C and E were associated with neutrophil and lymphocyte infiltration in the alveolar spaces and as well as inflammation in the PBVA to the same extent. However, an increase in plasma cells in the PBVA, was observed in only model E and D. The subsequent analyses were therefore performed using model E, with model D used as the control. As alum is known to augment Th2 responses in host immunity, plasma cell infiltration in PBVA may be associated with not only pre-immunization but also an acceleration in Th2 responses prior to IT with MP extracts.
Table 2.
Summary of the literature cases regarding pathological features of MP pneumonia in human and animal models.
| Category | No. of reports/cases | Alveolar |
PBVA |
Reference | ||
|---|---|---|---|---|---|---|
| Number of cases positive for infiltration with |
||||||
| Neu | Lym | Lym | Plasma | |||
| Autopsy | 7/16 | 9 | 3 | 9 | 11 | [6–12] |
| Open lung biopsy and video-assisted thoracic surgery | 6/13 | 3 | 0 | 11 | 9 | [13–17,18] |
| Transbronchial lung biopsy | 6/7 | 0 | 1 | 4 | 2 | [19–21] |
| Murine model | 11/11 | 6 | 3 | 10 | 0 | [26,31,33,36–43] |
| Day 1–3 (n) | 5 | 3 | 6 | |||
| Day 4–7 (n) | 1 | 1 | 3 | |||
| Day 8– (n) | 1 | 1 | 3 | |||
| Hamster model | 4 | [44–47] | ||||
| Guinia-pig model | 1 | [48] | ||||
| Day 1–3 (n) | 1 | |||||
| Day 4–7 (n) | 2 | |||||
| Day 8– (n) | 4 | |||||
Neu: neutrophil, Lym: lymphocyte, Plasma: plasma cell, Alveolar: alveolar area, PBVA: peri-bronchovascular area.
(n): number of positive cases at various phases (early: day 1–3, middle: day 4–7, late stage: day 8–, in each report. If the one mouse model had lymphocytes infiltration in the alveolar area from an early to middle stage, this was counted as early (n=1) and middle stage (n=1).
Table 3.
Pathological findings of mouse lung inoculated with MP extracts.
| Models |
Pre-immunization with |
Intratracheal challenge with |
Infiltration alveolar |
Infiltration PBVA |
Other findings | ||||
|---|---|---|---|---|---|---|---|---|---|
| MP extracts | Alum | CpG | Mp extracts | Neua | Lymb | Lymc | Plasmad | ||
| A | − | − | − | + | + | + | − | − | |
| B | + | − | − | + | ++ | + | +/− | − | |
| C | + | − | + | + | +++ | +++ | +++ | − | |
| D | − | + | − | + | ++ | ++ | ++ | + | Alveolitis |
| Intraalveolar hemorrhage | |||||||||
| E | + | + | − | + | +++ | +++ | +++ | +++ | Hyperemia in small arteries |
| Hyperplasia of type II alveolar epithelial cells | |||||||||
Pre-immunization was intraperitoneally performed once a week for twice.
CpG: a Th-1 stimulating adjuvant, alum: a Th2 stimulating adjuvant, Neu: neutrophil, Lym: lymphocyte, Plasma: plasma cell, Alveolar: alveolar area, PBVA: peri-bronchovascular area, MP extract: Mycoplasma pneumoniae extract.
All mice were inoculated by IT with MP extracts with or without pre-immunization. Pre-immunization was carried out intraperitoneally on 6 and 13 day prior to IT. Models include; Model A: without pre-immunization. Model B: following two IP immunizations with MP extracts alone. Model C: following two IP immunizations with MP extracts plus CpG. Model D: following two IP immunizations with aluminum adjuvant alone. Model E: following two IP immunizations with MP extracts plus aluminum adjuvant.
The degree of infiltration of neutrophils in the alveolar spaces was defined as none (−), mild (+, up to 150 cells/filed), moderate (++, from 150 to 500 cells/field), or severe (+++, greater than 500 cells/field).
The degree of infiltration of lymphocytes in the alveolar spaces was defined as none (−), mild (+, up to 5 cells/field), moderate (++, from 5 to 15 cells/field), severe (+++, greater than 15 cells/field).
Peribronchovascular infiltration of lymphocytes was defined as none (−), mild (+, up to 150 cells/field), moderate (++, from 150 to 350 cell counts/filed), or severe (+++, greater than 350 cells/field).
Peribronchovascular infiltration with plasma cells was defined as none (−), mild (+, up to 10 cells/field), moderate (++, from 10 to 50 cells/field), severe (+++, greater than 50 cells/field).
3.2. Pulmonary alveolar inflammation after IT inoculation
To evaluate the effect of IP with MP extracts on acquired immunity, we measured the serum antibody titer against MP 6 days after the final IP inoculation. The mean titer was found to be 417±319 in mice immunized with MP extracts plus alum, whereas it was not detected in mice with alum alone (detection limit was 40×) (Fig. 1). This demonstrated the development of humoral immunity against MP occurring after two IP immunizations with MP extracts plus alum. We evaluated the histopathological features at 6 days after the last IP immunization with and without MP extracts. Neither neutrophil nor lymphocyte infiltration was observed in the alveoli in both cases, whereas this was observed in BALF samples from both mice (data not shown). Moreover, the morphology of AMs was not distinguishable between them. The histopathological changes were also compared sequentially between models E and D at 0, 8, 24, 48, 96, and 168 h after IT. Although IT initiated inflammation in both models E and D, it was more predominant in model E compared to model D. There was marked neutrophil infiltration but less impressive macrophage and lymphocyte involvement during the first 24 h. In fact, neutrophil infiltration was seen in the alveolar spaces as early as 8 h and peaked at 24 h in both models E and D (Fig. 2A and C). Meanwhile, AMs were decreased between 8–24 h. From 24–48 h, the number of neutrophils gradually became decreased in the alveolar spaces, while lymphocyte numbers continued to increase (Fig. 2B and C), peaking at 96 h in both models. Thereafter, a decrease in lymphocytes was noted in both model D and E. These findings showed that IT induced an early neutrophilic infiltrate within the alveolar spaces followed by a later lymphocytic infiltrate.
Fig. 1.
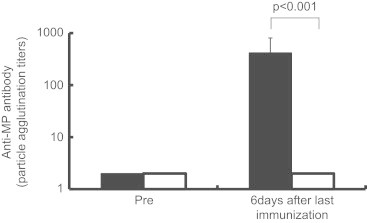
Comparison of serum antibody titers against MP between model D (□) and model E (■) were determined before and 6 day after last immunization with MP extracts. Evaluated by particle-agglutination test, Serodia MycoII (Fujirebio, Tokyo).
Fig. 2.
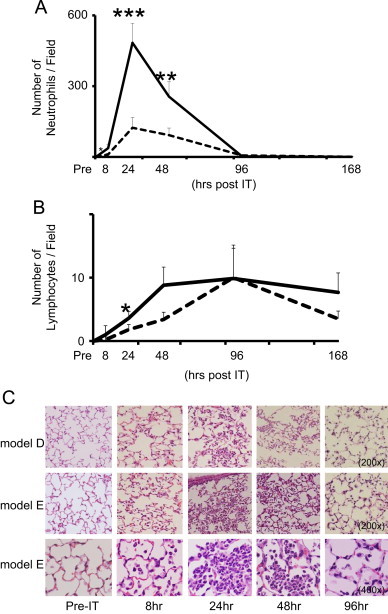
The time kinetics of neutrophils and lymphocytes infiltration into the alveolar spaces and in the lung of model D (- - -) and model E (—). Neutrophils (A) and lymphocytes (B) counts in the alveolar spaces in five independent fields on HE-stained lung sections. Averages were plotted for pre-IT, 8, 24, 48, 96, and 168 h post-IT. Statistical significance between model D and E for p<0.05, p<0.01, p<0.001 were expressed as *, **, ***, respectively. (C) Representative photomicrographs of lung tissue inflammation at pre-IT, 8, 24, 48, and 96 h post-IT in model D (the upper panels, 200×) and E (the middle, 200× and lower panels, 400×) as described in the “Materials and methods” section.
3.3. Lymphocyte infiltration in the PBVA
At 48 h, mild to moderate lymphocyte infiltration was observed in the PBVA in both models (Fig. 3A–D), although more predominantly in model E. By 168 h, these infiltrates continued to persist and increase in model E, but were less remarkable and scant in model D.
Fig. 3.
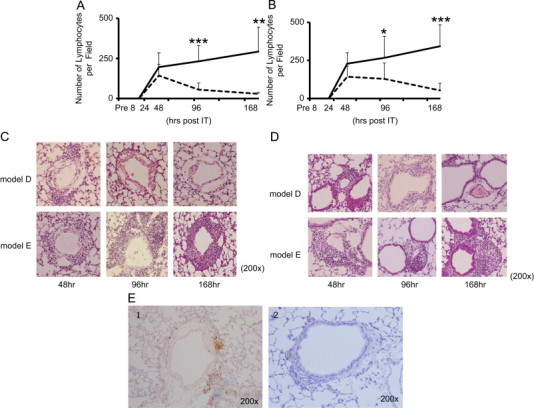
The time kinetics of lymphocyte infiltration in the peribronchiolar (A) and perivascular (B) area. Histopathological examination of lung tissue. Lung tissues were resected and stained with hematoxylin and eosin at pre-IT, 8, 24, 48, 96, and 168 h post-IT. Lymphocytes infiltrating the perivascular and peribronchiolare areas were enumerated in 10 fields at each point (200×) in model D (- - -) and model E (—). Values are expressed as means±standard deviation. p-values of p<0.05, p<0.01, p<0.001 are denoted as *, **, ***, respectively. Representative photomicrographs of perivascular (C) and peribronchiolar (D) areas in model D (upper panels) and E (lower panels) at 48 (the left), 96 (the middle), and 168 (the right) hours post-IT. CD3+ positive T cells were detected by immunohistochemical analysis using anti-CD3 antibody (E-1) (Dako, CA, USA). Negative control is noted as E-2.
3.4. Plasma cell infiltration in the PBVA
As shown in Fig. 4A–D, plasma cell infiltration, as detected by immunohistochemistry using anti-CD138 antibodies, were observed at 96 h in both the perivascular peribronchiolar areas in model E and less impressively in model D. Thereafter, infiltrates were noted to decline and began to disappear in model D as early as 168 h (Fig. 4A–D). Conversely, plasma cells increased dramatically between 96 and 168 h in model E and persisted until 336 h (data not shown). These results suggest that pre-immunization with MP extracts up-regulates plasma cell recruitment within the PBVA.
Fig. 4.
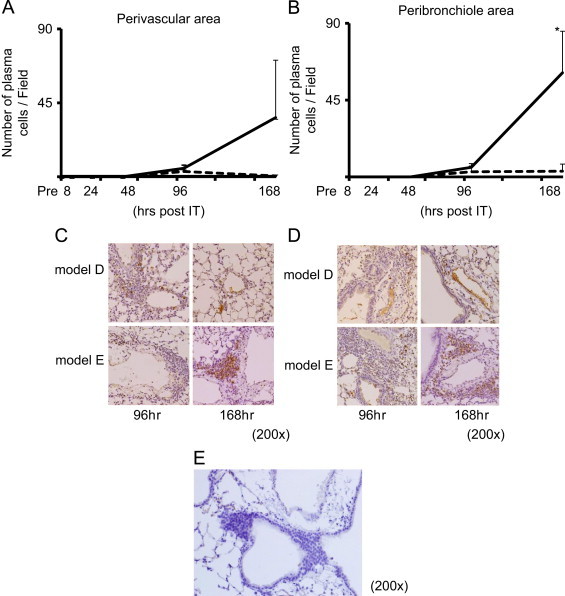
The time kinetics of plasma cell infiltration in perivascular (A) and peribronchiolar (B) areas. Lung tissue specimens were obtained from model D (- - -) and model E (—) at pre-IT, 8 h, 24 h, and 48 h post-IT, and stained immunohistochemically using anti-CD138 antibody as described in the “Materials and methods” section. Each experiment included three to five mice at each time point. All CD138 positive cells in the perivascular (C) and peribronchiolar areas (D) were counted in 5 fields at each time point (200×). Negative control is noted as (E). p-values of p<0.05 denoted as *.
3.5. Sequential changes in BALF cells
To confirm the aforementioned time kinetics of the inflammatory responses induced by IT, BALF cells were sequentially analyzed. Neutrophil counts demonstrated an increase as early as 8 h, peaked at 24 h, and decreased to baseline levels by 96 h, as consistent with histopathological findings. The number of neutrophils at 8 and 24 h were 4.0- and 4.7-folds greater in model E (9.9×105 and 2.0×107 cells/lung, respectively) than in model D (2.5×105 and 4.2×106 cells/lung, respectively (Fig. 5A). An increase in lymphocytes was observed as early as 24 h in only model E, reaching the maximum at 48 h before gradually declining to levels that were still detectable at 336 h. In contrast, an obvious increase in lymphocytes in model D was not observed throughout the whole course (Fig. 5B). We confirmed that these increased lymphocytes were consisted of CD3+/CD19− and CD3-/CD19− cells using flowcytometry (Supplementary Fig. 2). These results suggested that pre-immunization with MP extracts is a crucial process in the long-term lymphocyte alveolitis model.
Fig. 5.
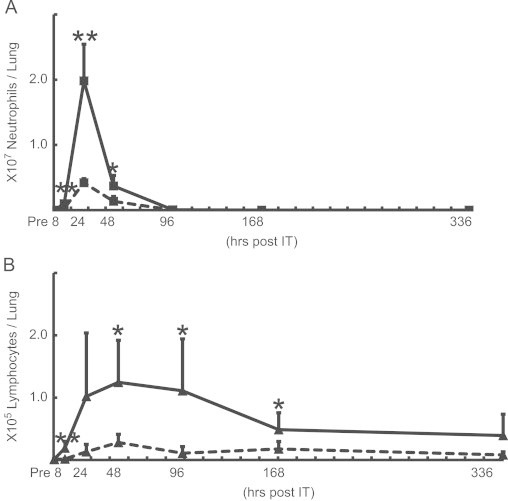
The time kinetics of neutrophils (A) and lymphocytes (B) in BALF from model D (- - -) and model E (—) at pre-IT, 8, 24, 48, 96, 168, and 336 h post-IT. The number of cells expressed as the means±SD in BALF from 8 mice at each point. p-values of p<0.05, p<0.01 are denoted as *, **, respectively.
3.6. Reactivity of BALF lymphocytes to MP extracts in vitro
To evaluate the effect of pre-immunization on the host cellular immunity, we investigated the specific response of BALF cells to MP extracts in vitro at 96 h after IT in models E and D. The stimulation indices were 0.5 and 0.7 for model E and D, respectively (Fig. 6A), which demonstrated a lack of response of BALF cells toward MP extracts. This indicated that infiltrated lymphocytes did not recognize MP extracts. In contrast, the stimulation index of splenic lymphocytes was 1.76 in model E and 0.95 in model D (Fig. 6B). These data suggested that IP immunization of MP extracts induced antigen-specific cellular immunity systemically, but not in the lung. Consequently, lymphocyte alveolitis may not be caused by MP extracts specific proliferation.
Fig. 6.
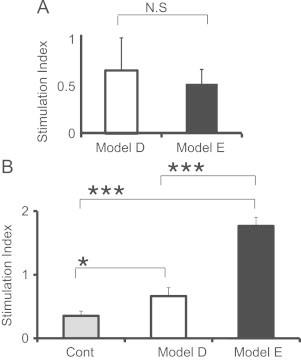
Comparison of stimulation indices (S.I.) of BALF cells (A) and splenic cells (B) in response to MP extracts between model D (□) and model E (■). Cells were obtained at 96 h post-IT (A), 6 days after the last IP (B) on each experiment respectively. The S.I. was calculated as the ratio of 3H-thymidine uptake of cells incubated with MP extracts to that incubated without MP extracts. Non immunized mice group was noted as “Cont” in (B).
3.7. Chemokine and cytokine increase in BALF after IT inoculation
Various chemokines are believed to be responsible in inducing lymphocytic alveolitis, as well as initial neutrophilic infiltration. An analysis of BALF after IT revealed that the expression of chemokines and inflammatory cytokines was up-regulated in model E. Cytokine/chemokines levels in models D and E were also analyzed at 8 and 24 h post-IT. KC, IL-6, TNF-α, and MIP-2 were all detected at 8 h in both models, but only RANTES was significantly higher in model E than model D (data not shown). At 24 h, IL-6, MCP-1, MIP-2, and RANTES were higher in model E than in model D, while IL-1β and KC levels were similar in both (Fig. 7). As both MCP-1 and RANTES are known to be a potent lymphocyte and neutrophil chemo-attractants, the increase was consistent with the histopathological features observed during the 8–96 h period after IT.
Fig. 7.
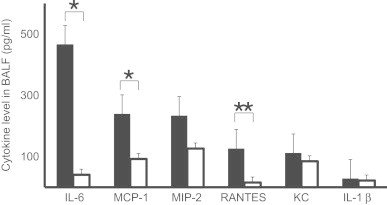
Concentration of cytokines and chemokines in BALF at 24 h post-IT in model D (□) and E (■). Each value was the mean data of 5–8 mice. p-values of p<0.05, p<0.01 are denoted as *, **, respectively.
3.8. AMs as a possible source for inflammatory cytokines/chemokines
As AMs play a central roles in host innate immunity in the lung, we evaluated the chemokine/cytokine production from AMs after stimulation with MP extracts in vivo and in vitro. At 48 h post-IT, MCP-1 and RANTES were strongly detected in AMs in model E compared to model D. In contrast, MCP-1 and RANTES were weakly stained in other lung cells such as bronchial epithelial cells and alveolar type II cells, as these chemokines were mainly produced in AMs (Fig. 8A). We also evaluated in vitro AMs cytokine production in the presence or absence of MP extracts. AMs were obtained from BALF at 6 day after two IP immunizations with MP extracts plus alum or with alum alone. When we compared the concentration of cytokines in the culture supernatants, the production of IL-1β, IL-6, MIP-2, and RANTES after 8 h incubation, was strikingly greater in the former than the latter (Fig. 8B), indicating that pre-immunization with MP extracts augmented the potential reactivity of AMs to the extracts. Taken together, it is likely that AMs primed with two IP immunizations with MP extracts constituted the major effector cells in facilitating the initial neutrophilic/lymphocytice infiltration after IT.
Fig. 8.
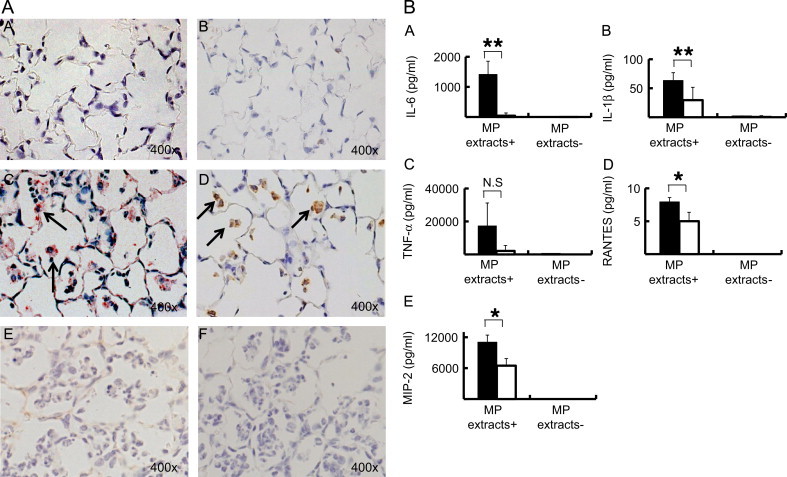
(A) The immunohistochemistry of lung sections obtained from model D (A and B) and E (C and D). The sections were stained with anti-MCP-1 (A and C) or anti-RANTES (B and D) antibodies. Arrows: positive stained AMs. Negative controls for MCP-1 and RANTES were noted as (E and F), respectively. (B) The concentration of IL-6, IL-1β, TNF-α, RANTES, and MIP-2 in culture supernatants of AMs incubated with and without MP extracts in vitro. AMs were obtained from BALF 6 days after two immunizations with alum alone (□) or with alum plus MP extracts (■). p-values of p<0.05, p<0.01 are denoted as *, **, respectively.
3.9. Up-regulation of TLR-2 expression on AMs
Previous studies have demonstrated that mycoplasma cell wall antigens activate MAPK-NF-κB signaling in AMs through TLR-2. Thus, we evaluated the expression of TLR-2 on AMs at 6 day after two IP immunizations with MP extracts plus alum or alum alone. The number of TLR-2 positive AMs in the alveolar spaces 4.6 times greater in model E (86.9%) than in model D (18.5%) (Fig. 9A and B), whereas the number of TLR-2 positive epithelial cells was even between model D and E. The expression of mRNA in BALF cells obtained from model E were 3.0 times higher than in model D (Fig. 9C). To elucidate that TLR-2 signaling is involved in cytokine production, we performed an inhibition assay using a MAPK inhibitor, SP600125 for JNK (c-Jun N-terminal kinase) enzymes. The inhibitor completely abrogated the production of IL-6 by AM stimulated with MP extracts in vitro (Fig. 9D).
Fig. 9.
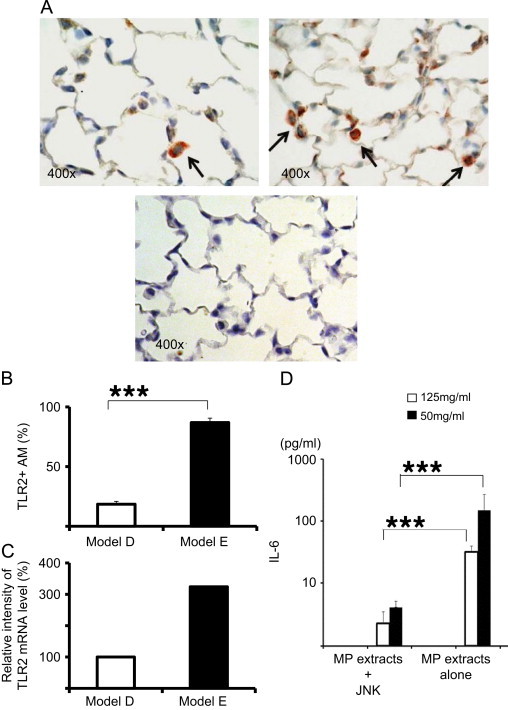
(A) Immunohistochemistry of TLR-2 positive cells in lung sections from model D (the left panel) and E (the right panel). Negative control was noted in the lower panel. (B) The percentage of TLR-2 positive AMs in lung sections from mice at 6 days after two immunizations with alum alone (□) or with alum plus MP extracts (■). (C) Relative intensity of TLR-2 mRNA level extracted from AMs in BALF from mice at 6 days after two immunizations with alum alone (□) or with alum plus MP extracts (■). (D) Inhibition assay using MAPK inhibitor, SP600125 for JNK (c-Jun N-terminal kinase) enzymes. The production of IL-6 by AM stimulated with MP extracts (12.5 mg/mL and 50 mg/mL) co-incubated with or without SP600125 (5 μM) in vitro (D).
4. Discussion
The aim of this study was to reproduce the inflammatory processes seen in human MP pneumonia using a novel mouse MP pneumonia model. None of the previously reported murine models [26,31,33,36–43] or other animal models [44–48] described persistent plasma cell infiltration in the PBVA. Thus, this is the first report that describes a murine model with longstanding plasma cell infiltration in the PBVA.
This study also described the chronological sequence of inflammatory events in MP pneumonia both quantitatively and qualitatively. We confirmed that neutrophil infiltration precedes lymphocyte infiltration in both lung sections and BALF, which is consistent with previous studies [33,36,40]. The peaked cell density was 4-fold higher in model E than D, suggesting that the effect of pre-immunization with MP extracts was a potent promoter of neutrophil infiltration. Similarly, in model E, lymphocyte infiltration peaked at 48 h, lasting up to 336 h, an effect that was not observed in model D. Our study differs from previous murine models in that we chose to utilize intraperitoneal pre-immunization with MP extracts plus alum prior to IT with MP extracts to augment the reactivity of immune cells to the extracts. Surprisingly, IT with mycoplasma cell free extracts alone was found to be sufficient to induce marked infiltration of neutrophils and lymphocytice alveolitis with prolonged lymphocyte/plasma cell inflammation in the PBVA. Moreover, the pathological observation revealed that recruitment of lymphocytes into alveolar spaces after IT in protocol D resulted in peak lymphocyte numbers equal to those seen in protocol E. The result suggests that at least a major portion of the recruited cells are not antigen-specific. This is supported by the very low stimulation index (<2) reported in Fig. 6. Although previous studies have demonstrated that inoculation of living MP induces inflammation in the lower respiratory tract in mice [33,36,37], this study suggests that an exaggerated host immune response to MP antigens may be involved in the inflammation in human MP pneumonia. Serodia MycoII is a kit which detect MP specific antibody, especially in IgM antibody. The Adjuvant “alum” was known for Th2-inducing adjuvant. Furthermore, we confirmed that using Th-1 inducing adjuvant “CpG” in our model C did not cause plasma cell infiltration in the PBVA (Table 3). Those findings suggested that Th2 reaction was essential to induce the plasma cell infiltration in the lung, which implies the possible involvement of Th2 responses in the process of human MP pneumonia.
Another aim of this study was to determine the role of innate immunity in MP pneumonia. We demonstrated that the first process is possibly up-regulation of TLR-2 expression on AMs that subsequently induces cytokine/chemokine production in response to subsequent challenges with the same MP extracts. This concept is supported by the following data. Firstly, pre-immunization with MP extracts up-regulated TLR-2 expression on AMs. Secondly, AMs from mice immunized with MP extracts plus alum produced higher amounts of RANTES and MCP-1 than mice immunized with alum alone. Recent studies have focused on innate immunity mediated by TLRs on macrophages or epithelial cells. Among the 12 TLRs, TLR-2 signaling is the major pathway for inflammation with MP pneumonia [30]. Studies have identified three lipoproteins/lipopeptides extracted from MP as ligands for TLR-2 [29]. These ligands up-regulate the expression of TLR-2, activate the MAPK-NF-κB signaling pathway, and augment the production of TNF-α, IL-6, and IL-1β. In TLR-2−/− mice, alive MP failed to stimulate MyD88 NF-κβp65 activation. Moreover, antibodies to TLR-2 blocked an increase in IL-6 in BALF after intranasal inoculation [30], and its production was completely diminished in TLR-2 KO mice. It has further been demonstrated that activation of the TLR-2 pathway is essential for inflammation in response to MP [49]. The consistent increase in cytokine/chemokine production from AMs in model E but not model D was due to up-regulated expression of TLR-2 on AMs.
Previous studies reported that MCP-1 was the most potent activator of T lymphocytes [50], and that RANTES plays an important role in effector CD8+ T cell transmigration from alveolar capillaries into the pulmonary interstitium [51]. Many cell types have been reported to produce RANTES including activated T lymphocytes, bronchial cells, fibroblasts, macrophages, and endothelial cells [52]. As shown in our study, AMs from mice pre-immunized with MP extracts plus alum are likely to be potent inducers of RANTES and MCP-1. Other factor such as vascular endothelial growth factor (VEGF) from small airway epithelial cells [53] and endothelial cells [54] might be a possible explanation for long-lasting characteristics of MP pneumonia through angiogenesis and microvascular remodeling.
It is still unknown why TLR-2 expression on AMs is up-regulated after intraperitoneal immunization of MP extracts plus alum. It is unlikely that circulating MP extracts directly stimulate AMs or their precursors in the alveolar spaces. In this regard, Fan et al. [55] reported that TLR-2 expression on AMs was up-regulated through TLR-4 by shock-activated neutrophils in vivo and in vitro. Neutrophil NADPH oxidase-derived oxidants signaling mediates the TLR-2–TLR-4 cross talk both in endothelial cells and in AMs, which results in the activation of positive feedback signals against invading pathogens.
5. Conclusion
The present study highlights distinct reaction to IT between mice with and without pre-immunization of MP extracts. Our results suggest that subclinical or preceding MP infection may greatly influence the degree of inflammation in MP pneumonia. Unraveling the mechanisms by which innate immunity regulates cytokine/chemokine expression will significantly improve our understanding of the pathogenesis of MP and will help to develop novel therapeutic strategies to control mycoplasma associated infections in humans.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
TS and both KN conducted the design of the study and had a major role in drafting the manuscript. TS carried out the experiments and the preliminary data analyses. NM helped in conducting the pretreatments and performing the experiments. YF, KI and TO participated in the pathological evaluation. DK, HW, HI, HT, SK and HG participated in the design of the study and interpretation of the experimental findings. All authors read and approved the final manuscript.
Acknowledgments
This work was supported by Grant-in-aid from Scientific Research (19590911). We are very grateful to Dr. Fusayo Adachi. Institute of Medical Science, The University of Tokyo, for their valuable contribution and to Akiko Kitazawa, Kyorin University School for providing technical assistance.
Contributor Information
Takeshi Saraya, Email: sara@yd5.so-net.ne.jp.
Koh Nakata, Email: radical@med.niigata-u.ac.jp.
Kazuhide Nakagaki, Email: nakagaki@nvlu.ac.jp.
Natsuki Motoi, Email: motoi@med.niigata-u.ac.jp.
Kuniko Iihara, Email: iimy.kuniko@nifty.ne.jp.
Yasunori Fujioka, Email: yasunori.fujioka@nikko-kinen.or.jp.
Teruaki Oka, Email: toka123@mbd.ocn.ne.jp.
Daisuke Kurai, Email: kuraida@aol.com.
Hiroo Wada, Email: wadagh@ks.kyorin-u.ac.jp.
Haruyuki Ishii, Email: h141@ks.kyorin-u.ac.jp.
Haruhiko Taguchi, Email: taguchi@ks.kyorin-u.ac.jp.
Shigeru Kamiya, Email: skamiya@ks.kyorin-u.ac.jp.
Hajime Goto, Email: h510@ks.kyorin-u.ac.jp.
Appendix A. Supplementary material
Fig. S1. Cells in lymphocyte gate were 35.8% and 57.6% for CD3 positive and CD3 negative respectively. Among CD3 positive cells, CD4-/CD8- cells were predominant. CD4 per CD8 ratio was 0.02. Fig. S2. Small percentages of CD3 positive/CD19 negative cells were observed.
References
- 1.Chan E.D., Welsh C.H. Fulminant Mycoplasma pneumoniae pneumonia. Western Journal of Medicine. 1995;162:133–142. [PMC free article] [PubMed] [Google Scholar]
- 2.Rottem S. Interaction of mycoplasmas with host cells. Physiological Reviews. 2003;83:417–432. doi: 10.1152/physrev.00030.2002. [DOI] [PubMed] [Google Scholar]
- 3.Dimitrov D.S., Franzoso G., Salman M., Blumenthal R., Tarshis M., Barile M.F. Mycoplasma fermentans (incognitus strain) cells are able to fuse with T lymphocytes. Clinical Infectious Diseases. 1993;17(Suppl. 1):S305–308. doi: 10.1093/clinids/17.supplement_1.s305. [DOI] [PubMed] [Google Scholar]
- 4.Yano T., Ichikawa Y., Komatu S., Arai S., Oizumi K. Association of Mycoplasma pneumoniae antigen with initial onset of bronchial asthma. American Journal of Respiratory and Critical Care Medicine. 1994;149:1348–1353. doi: 10.1164/ajrccm.149.5.8173777. [DOI] [PubMed] [Google Scholar]
- 5.Dakhama A., Kraft M., Martin R.J., Gelfand E.W. Induction of regulated upon activation, normal T cells expressed and secreted (RANTES) and transforming growth factor-beta 1 in airway epithelial cells by Mycoplasma pneumoniae. American Journal of Respiratory Cell and Molecular Biology. 2003;29:344–351. doi: 10.1165/rcmb.2002-0291OC. [DOI] [PubMed] [Google Scholar]
- 6.Benisch B.M., Fayemi A., Gerber M.A., Axelrod J. Mycoplasmal pneumonia in a patient with rheumatic heart disease. American Journal of Clinical Pathology. 1972;58:343–348. doi: 10.1093/ajcp/58.3.343. [DOI] [PubMed] [Google Scholar]
- 7.Halal F., Brochu P., Delage G., Lamarre A., Rivard G. Severe disseminated lung disease and bronchiectasis probably due to Mycoplasma pneumoniae. Canadian Medical Association Journal. 1977;117:1055–1056. [PMC free article] [PubMed] [Google Scholar]
- 8.Kaufman J.M., Cuvelier C.A., Van der Straeten M. Mycoplasma pneumonia with fulminant evolution into diffuse interstitial fibrosis. Thorax. 1980;35:140–144. doi: 10.1136/thx.35.2.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koletsky R.J., Weinstein A.J. Fulminant Mycoplasma pneumoniae infection. Report of a fatal case, and a review of the literature. American Review of Respiratory Disease. 1980;122:491–496. doi: 10.1164/arrd.1980.122.3.491. [DOI] [PubMed] [Google Scholar]
- 10.Maisel J.C., Babbitt L.H., John T.J. Fatal Mycoplasma pneumoniae infection with isolation of organisms from lung. Journal of the American Medical Association. 1967;202:287–290. [PubMed] [Google Scholar]
- 11.Meyers B.R., Hirschman S.Z. Fatal infections associated with Mycoplasma pneumoniae: discussion of three cases with necropsy findings. Mount Sinai Journal of Medicine. 1972;39:258–264. [PubMed] [Google Scholar]
- 12.Parker F., Jr, Jolliffe L.S., Finland M. Primary atypical pneumonia; report of eight cases with autopsies. Archives of Pathology (Chicago) 1947;44:581–608. [PubMed] [Google Scholar]
- 13.Coultas D.B., Samet J.M., Butler C. Bronchiolitis obliterans due to Mycoplasma pneumoniae. Western Journal of Medicine. 1986;144:471–474. [PMC free article] [PubMed] [Google Scholar]
- 14.Ebnother M., Schoenenberger R.A., Perruchoud A.P., Soler M., Gudat F., Dalquen P. Severe bronchiolitis in acute Mycoplasma pneumoniae infection. Virchows Archiv. 2001;439:818–822. doi: 10.1007/s004280100473. [DOI] [PubMed] [Google Scholar]
- 15.Llibre J.M., Urban A., Garcia E., Carrasco M.A., Murcia C. Bronchiolitis obliterans organizing pneumonia associated with acute Mycoplasma pneumoniae infection. Clinical Infectious Diseases. 1997;25:1340–1342. doi: 10.1086/516124. [DOI] [PubMed] [Google Scholar]
- 16.Rollins S., Colby T., Clayton F. Open lung biopsy in Mycoplasma pneumoniae pneumonia. Archives of Pathology and Laboratory Medicine. 1986;110:34–41. [PubMed] [Google Scholar]
- 17.Wachowski O., Demirakca S., Muller K.M., Scheurlen W. Mycoplasma pneumoniae associated organising pneumonia in a 10 year old boy. Archives of Disease in Childhood. 2003;88:270–272. doi: 10.1136/adc.88.3.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chan E.D., Kalayanamit T., Lynch D.A., Tuder R., Arndt P., Winn R. Mycoplasma pneumoniae-associated bronchiolitis causing severe restrictive lung disease in adults: report of three cases and literature review. Chest. 1999;115:1188–1194. doi: 10.1378/chest.115.4.1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ganick D.J., Wolfson J., Gilbert E.F., Joo P. Mycoplasma infection in the immunosuppressed leukemic patient. Archives of Pathology and Laboratory Medicine. 1980;104:535–536. [PubMed] [Google Scholar]
- 20.Nakajima M., Kubota Y., Miyashita N., Kishimoto T., Kobashi Y., Niki Y. [An adult case of pneumonia due to Mycoplasma pneumoniae and Chlamydia psittaci] Kansenshogaku Zasshi. 1996;70:87–92. doi: 10.11150/kansenshogakuzasshi1970.70.87. [DOI] [PubMed] [Google Scholar]
- 21.Ohmichi M., Miyazaki M., Ohchi T., Morikawa Y., Tanaka S., Sasaki H. [Fulminant Mycoplasma pneumoniae pneumonia resulting in respiratory failure and a prolonged pulmonary lesion] Nihon Kokyuki Gakkai Zasshi. 1998;36:374–380. [PubMed] [Google Scholar]
- 22.Bott L., Santos C., Thumerelle C., Mars A., Deschildre A., Catteau B. [Severe Stevens-Johnson syndrome in 4 children] Archives of Pediatrics. 2007;14:1435–1438. doi: 10.1016/j.arcped.2007.08.020. [DOI] [PubMed] [Google Scholar]
- 23.Biberfeld G., Biberfeld P., Sterner G. Cell-mediated immune response following Mycoplasma pneumoniae infection in man. I. Lymphocyte stimulation. Clinical & Experimental Immunology. 1974;17:29–41. [PMC free article] [PubMed] [Google Scholar]
- 24.Mizutani H., Kitayama T., Hayakawa A., Nagayama E. Delayed hypersensitivity in mycoplasma pneumoniae infections. Lancet. 1971;1:186–187. doi: 10.1016/s0140-6736(71)91956-8. [DOI] [PubMed] [Google Scholar]
- 25.Biberfeld G., Sterner G. Tuberculin anergy in patients with Mycoplasma pneumoniae infection. Scandinavian Journal of Infectious Diseases. 1976;8:71–73. doi: 10.3109/inf.1976.8.issue-2.02. [DOI] [PubMed] [Google Scholar]
- 26.Hardy R.D., Jafri H.S., Olsen K., Hatfield J., Iglehart J., Rogers B.B. Mycoplasma pneumoniae induces chronic respiratory infection, airway hyperreactivity, and pulmonary inflammation: a murine model of infection-associated chronic reactive airway disease. Infection and Immunity. 2002;70:649–654. doi: 10.1128/iai.70.2.649-654.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sutherland E.R., Martin R.J. Asthma and atypical bacterial infection. Chest. 2007;132:1962–1966. doi: 10.1378/chest.06-2415. [DOI] [PubMed] [Google Scholar]
- 28.Underhill D.M., Ozinsky A., Hajjar A.M., Stevens A., Wilson C.B., Bassetti M. The Toll-like receptor 2 is recruited to macrophage phagosomes and discriminates between pathogens. Nature. 1999;401:811–815. doi: 10.1038/44605. [DOI] [PubMed] [Google Scholar]
- 29.Shimizu T., Kida Y., Kuwano K. Mycoplasma pneumoniae-derived lipopeptides induce acute inflammatory responses in the lungs of mice. Infection and Immunity. 2008;76:270–277. doi: 10.1128/IAI.00955-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chu H.W., Jeyaseelan S., Rino J.G., Voelker D.R., Wexler R.B., Campbell K. TLR2 signaling is critical for Mycoplasma pneumoniae-induced airway mucin expression. Journal of Immunology. 2005;174:5713–5719. doi: 10.4049/jimmunol.174.9.5713. [DOI] [PubMed] [Google Scholar]
- 31.Hayakawa M., Taguchi H., Kamiya S., Fujioka Y., Watanabe H., Kawai S. Animal model of Mycoplasma pneumoniae infection using germfree mice. Clinical and Diagnostic Laboratory Immunology. 2002;9:669–676. doi: 10.1128/CDLI.9.3.669-676.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sekine H., Taguchi H., Watanabe H., Kawai S., Fujioka Y., Goto H. Immunological analysis and pathological examination of gnotobiotic mice monoassociated with Mycoplasma pneumoniae. Journal of Medical Microbiology. 2009;58:697–705. doi: 10.1099/jmm.0.007872-0. [DOI] [PubMed] [Google Scholar]
- 33.Chu H.W., Breed R., Rino J.G., Harbeck R.J., Sills M.R., Martin R.J. Repeated respiratory Mycoplasma pneumoniae infections in mice: effect of host genetic background. Microbes and Infection. 2006;8:1764–1772. doi: 10.1016/j.micinf.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 34.O'Sullivan M.G., Chilton F.H., Huggins E.M., Jr, McCall C.E. Lipopolysaccharide priming of alveolar macrophages for enhanced synthesis of prostanoids involves induction of a novel prostaglandin H synthase. Journal of Biological Chemistry. 1992;267:14547–14550. [PubMed] [Google Scholar]
- 35.Shimizu T., Kida Y., Kuwano K. A dipalmitoylated lipoprotein from Mycoplasma pneumoniae activates NF-kappaB through TLR1, TLR2, and TLR6. Journal of Immunology. 2005;175:4641–4646. doi: 10.4049/jimmunol.175.7.4641. [DOI] [PubMed] [Google Scholar]
- 36.Chu H.W., Campbell J.A., Rino J.G., Harbeck R.J., Martin R.J. Inhaled fluticasone propionate reduces concentration of Mycoplasma pneumoniae, inflammation, and bronchial hyperresponsiveness in lungs of mice. Journal of Infectious Diseases. 2004;189:1119–1127. doi: 10.1086/382050. [DOI] [PubMed] [Google Scholar]
- 37.Fonseca-Aten M., Rios A.M., Mejias A., Chavez-Bueno S., Katz K., Gomez A.M. Mycoplasma pneumoniae induces host-dependent pulmonary inflammation and airway obstruction in mice. American Journal of Respiratory Cell and Molecular Biology. 2005;32:201–210. doi: 10.1165/rcmb.2004-0197OC. [DOI] [PubMed] [Google Scholar]
- 38.Hardy R.D., Jafri H.S., Olsen K., Wordemann M., Hatfield J., Rogers B.B. Elevated cytokine and chemokine levels and prolonged pulmonary airflow resistance in a murine Mycoplasma pneumoniae pneumonia model: a microbiologic, histologic, immunologic, and respiratory plethysmographic profile. Infection and Immunity. 2001;69:3869–3876. doi: 10.1128/IAI.69.6.3869-3876.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kurata S., Taguchi H., Sasaki T., Fujioka Y., Kamiya S. Antimicrobial and immunomodulatory effect of clarithromycin on macrolide-resistant Mycoplasma pneumoniae. Journal of Medical Microbiology. 2010;59:693–701. doi: 10.1099/jmm.0.014191-0. [DOI] [PubMed] [Google Scholar]
- 40.Martin R.J., Chu H.W., Honour J.M., Harbeck R.J. Airway inflammation and bronchial hyperresponsiveness after Mycoplasma pneumoniae infection in a murine model. American Journal of Respiratory Cell and Molecular Biology. 2001;24:577–582. doi: 10.1165/ajrcmb.24.5.4315. [DOI] [PubMed] [Google Scholar]
- 41.Opitz O., Pietsch K., Ehlers S., Jacobs E. Cytokine gene expression in immune mice reinfected with Mycoplasma pneumoniae: the role of T cell subsets in aggravating the inflammatory response. Immunobiology. 1996;196:575–587. doi: 10.1016/s0171-2985(97)80073-3. [DOI] [PubMed] [Google Scholar]
- 42.Techasaensiri C., Tagliabue C., Cagle M., Iranpour P., Katz K., Kannan T.R. Variation in colonization, ADP-ribosylating and vacuolating cytotoxin, and pulmonary disease severity among Mycoplasma pneumoniae strains. American Journal of Respiratory and Critical Care Medicine. 2010;182:797–804. doi: 10.1164/rccm.201001-0080OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wubbel L., Jafri H.S., Olsen K., Shelton S., Barton Rogers B., Gambill G. Mycoplasma pneumoniae pneumonia in a mouse model. Journal of Infectious Diseases. 1998;178:1526–1529. doi: 10.1086/314439. [DOI] [PubMed] [Google Scholar]
- 44.Barile M.F., Chandler D.K., Yoshida H., Grabowski M.W., Razin S. Hamster challenge potency assay for evaluation of Mycoplasma pneumoniae vaccines. Infection and Immunity. 1988;56:2450–2457. doi: 10.1128/iai.56.9.2450-2457.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cimolai N., Taylor G.P., Mah D., Morrison B.J. Definition and application of a histopathological scoring scheme for an animal model of acute Mycoplasma pneumoniae pulmonary infection. Microbiology and Immunology. 1992;36:465–478. doi: 10.1111/j.1348-0421.1992.tb02045.x. [DOI] [PubMed] [Google Scholar]
- 46.Dajani A.S., Clyde W.A., Jr, Denny F.W. Experimental Infection with Mycoplasma pneumoniae (Eaton's Agent) Journal of Experimental Medicine. 1965;121:1071–1086. doi: 10.1084/jem.121.6.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fernald G.W., Clyde W.A., Jr, Bienenstock J. Immunoglobulin-containing cells in lungs of hamsters infected with Mycoplasma pneumoniae. Journal of Immunology. 1972;108:1400–1408. [PubMed] [Google Scholar]
- 48.Jacobs E., Drews M., Stuhlert A., Buttner C., Klein P.J., Kist M. Immunological reaction of guinea-pigs following intranasal Mycoplasma pneumoniae infection and immunization with the 168 kDa adherence protein. Journal of General Microbiology. 1988;134:473–479. doi: 10.1099/00221287-134-2-473. [DOI] [PubMed] [Google Scholar]
- 49.Shimizu T., Kida Y., Kuwano K. A triacylated lipoprotein from Mycoplasma genitalium activates NF-kappaB through Toll-like receptor 1 (TLR1) and TLR2. Infection and Immunity. 2008;76:3672–3678. doi: 10.1128/IAI.00257-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim J.J., Nottingham L.K., Sin J.I., Tsai A., Morrison L., Oh J. CD8 positive T cells influence antigen-specific immune responses through the expression of chemokines. Journal of Clinical Investigation. 1998;102:1112–1124. doi: 10.1172/JCI3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Galkina E., Thatte J., Dabak V., Williams M.B., Ley K., Braciale T.J. Preferential migration of effector CD8+ T cells into the interstitium of the normal lung. Journal of Clinical Investigation. 2005;115:3473–3483. doi: 10.1172/JCI24482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stellato C., Beck L.A., Gorgone G.A., Proud D., Schall T.J., Ono S.J. Expression of the chemokine RANTES by a human bronchial epithelial cell line. Modulation by cytokines and glucocorticoids. Journal of Immunology. 1995;155:410–418. [PubMed] [Google Scholar]
- 53.Thaikoottathil J.V., Martin R.J., Zdunek J., Weinberger A., Rino J.G., Chu H.W. Cigarette smoke extract reduces VEGF in primary human airway epithelial cells. European Respiratory Journal. 2009;33:835–843. doi: 10.1183/09031936.00080708. [DOI] [PubMed] [Google Scholar]
- 54.McDonald D.M. Angiogenesis and remodeling of airway vasculature in chronic inflammation. American Journal of Respiratory and Critical Care Medicine. 2001;164:S39–45. doi: 10.1164/ajrccm.164.supplement_2.2106065. [DOI] [PubMed] [Google Scholar]
- 55.Fan J., Li Y., Vodovotz Y., Billiar T.R., Wilson M.A. Neutrophil NAD(P)H oxidase is required for hemorrhagic shock-enhanced TLR2 up-regulation in alveolar macrophages in response to LPS. Shock. 2007;28:213–218. doi: 10.1097/shk.0b013e318033ec9d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Cells in lymphocyte gate were 35.8% and 57.6% for CD3 positive and CD3 negative respectively. Among CD3 positive cells, CD4-/CD8- cells were predominant. CD4 per CD8 ratio was 0.02. Fig. S2. Small percentages of CD3 positive/CD19 negative cells were observed.