

Abstract
Electron cryomicroscopy, or cryoEM, is an emerging technique for studying the three-dimensional structures of proteins and large macromolecular machines. Electron crystallography is a branch of cryoEM in which structures of proteins can be studied at resolutions that rival those achieved by X-ray crystallography. Electron crystallography employs two-dimensional crystals of a membrane protein embedded within a lipid bilayer. The key to a successful electron crystallographic experiment is the crystallization, or reconstitution, of the protein of interest. This unit describes ways in which protein can be expressed, purified, and reconstituted into well-ordered two-dimensional crystals. A protocol is also provided for negative stain electron microscopy as a tool for screening crystallization trials. When large and well-ordered crystals are obtained, the structures of both protein and its surrounding membrane can be determined to atomic resolution.
Keywords: electron crystallography, membrane proteins, negative stain electron microscopy, protein purification, protein solubilization
Introduction
A large proportion of the human genome encodes membrane proteins. These proteins carry out vital processes such as nutrient uptake, signal transduction, and energy generation. As a result, the vast majority of all pharmaceuticals target membrane proteins, and it is imperative to understand the structure of these proteins in order to design effective drugs. Electron crystallography is the only structural biology technique in which the membrane protein of interest is crystallized within the context of a membrane and from which atomic resolution structures can emerge detailing the structures of both protein and lipid (Reichow and Gonen, 2009).
One of the first critical steps of an electron crystallographic experiment is the generation of large and well-ordered two-dimensional (2D) crystals. Analogous to 3D protein crystallization, many different crystallization conditions must be assayed and subsequently analyzed for 2D crystal formation. Here we detail some of the most common steps involved in the reconstitution of membrane proteins into highly ordered 2D crystals. The critical steps are described starting from membrane protein expression and purification to crystal screening by electron microscopy of negatively stained preparations en route to structure determination. Additionally a stepby-step procedure for negative staining is included within the 2D crystallization screening section.
Large-Scale Production of Membrane Proteins
Large amounts of purified membrane protein are needed for crystal growth and subsequent structural analysis. It is therefore important to determine a suitable source from which the necessary protein can be obtained. While some membrane proteins are naturally abundant, most membrane proteins are expressed at low levels in their native host. Moreover, the complexity of post-translational modifications renders most native sources of membrane proteins useless for high-resolution studies, as the protein that can be obtained is heterogeneous. Therefore, heterologous overexpression is necessary in order to obtain the milligram quantities of purified homogeneous protein needed to adequately assay crystallization. Several options are available for heterologous expression of membrane proteins including bacteria, yeast, insect cells, and mammalian cells.
The choice of expression system depends on many factors including, but not limited to, the organism from which the membrane protein of interest originates, the genetic tools available for overexpression, and the compatibility of an expression host to express high amounts of a non-native protein. Escherichia coli is the most common expression host because of its rapid growth and low cost, the wide array of genetic tools available, and ease of production scale-up (Baneyx, 1999). Membrane proteins naturally expressed in prokaryotes are most suitable for heterologous expression in E. coli (Surade et al., 2006). There are a large number of expression vectors available for expression in E. coli, as well as strains that have been optimized for over-expression of membrane proteins (Miroux and Walker, 1996; Wagner et al., 2008; Nannenga and Baneyx, 2011). When working with E. coli, expression conditions should be screened and optimized. Several factors that commonly affect membrane protein yields are: host strain, expression vector, time and temperature of induction, and inducer concentration. Following expression tests, protein yield is typically analyzed using SDS-PAGE and immunoblot analysis.
While membrane proteins of eukaryotic origin have been successfully expressed in E. coli, e.g., rat neurotensin receptor and human adenosine A(2a) receptor (Tucker and Grisshammer, 1996; Weiss and Grisshammer, 2002), these proteins may require post-translational modification or chaperones for proper folding and localization that are not available in E. coli; thus, eukaryotic expression systems may be required. Several eukaryotic expression systems are available for membrane proteins including yeast, insect, and mammalian cells. The particular choice of expression system depends on budget, expertise, and the available facilities. Pichia pastoris is a yeast expression system that is becoming increasingly popular because the cells can be grown to large cell densities with relatively simple culture conditions. Moreover, the genetic tools for generating stable transfectants are becoming more powerful and widespread (Ramon and Marin, 2011). An alternative to yeast expression systems is the baculovirus/insect cell expression system. Insect cell expression requires generation of recombinant baculovirus followed by the infection of insect cells. The advantage of insect cell expression relative to bacteria and yeast systems is that the cells have all of the components for the correct trafficking, folding, and modification of mammalian proteins giving rise to the ability to express milligram quantities of non-native protein (Midgett and Madden, 2007). While expression in insect cells is generally successful, they are often used only after other expression hosts have failed to produce significant yields, because compared to yeast and bacterial expression, insect cell systems are expensive, time consuming, and labor intensive. Finally, mammalian cell expression is also available; however, the cost and maintenance of cells does not typically make it an affordable option to generate milligram quantities needed for crystallization.
Membrane Protein Solubilization
Membrane proteins are embedded in the lipid bilayer, so for structural analysis they need to be extracted from these membranes and purified prior to crystallization attempts. The extraction is accomplished with detergents, and luckily a vast array of detergents is commercially available for tests. To solubilize membrane proteins, the cells that are expressing the membrane protein of interest are collected, lysed (using a sonicator, French press, or fluidizer) and the membrane fractions are collected after centrifugation. For a detailed protocol, see Gonen et al. (2000). Once the membrane fraction has been obtained, solubilization conditions can then be optimized using any number of detergents. Non-ionic detergents, such as DM (n-decyl-β-D-maltopyranoside), DDM (n-dodecyl-β-D-maltopyranoside), OG (octyl-β-D-glucopyranoside), and C12E8 (octaethylene glycol monodecyl ether) are commonly used to extract the membrane protein from the cell membrane without unfolding or damaging the membrane proteins themselves. Above the critical micelle concentration (CMC), the detergent forms micelles, which can then disrupt the lipid bilayer and bind to the membrane protein, effectively solubilizing the membraneembedded proteins (le Maire et al., 2000). The detergent used must be able to efficiently extract the target membrane protein from the membrane and keep it solubilized for the duration of the crystallization experiment.
Typically, several detergents are screened during the solubilization assay to cover a broad range of aliphatic chain lengths and types of hydophilic head groups. In addition to screening different detergents, other conditions that should be tested are detergent concentrations (ranging from 1× to 10× CMC), temperature (4°C, 25°C, 37°C), protein concentration (ranging from 1 to 10 mg/ml), and length of solubilization. Incubation of the detergent with the isolated cell membranes can be as little as 15 min or as long as overnight, depending on the stability of the protein once in solution. The basic procedure for solubilization consists of adding detergent to the membrane suspension, incubation for a period of time, and separation of the soluble and insoluble fractions by ultracentrifugation for 30 min at ~ 180,000 × g,4°C. The resulting pellet (insoluble) and supernatant (soluble—containing the extracted membrane protein) fractions are analyzed by SDS-PAGE and immunoblotting. Successful solubilization is indicated by the presence of large quantities of the target membrane protein in the supernatant.
An immunoblot from a typical solubilization assay is presented in Figure 17.15.1. In this example, membranes at a starting concentration of 10 mg/ml were incubated with several detergents at 4°C for 1 hr and then centrifuged for 1 hr at 180,000 × g, 4°C. When the total membrane protein in lane 1 is compared to the solubilized protein in the adjacent lanes, one notes that DDM and DM were able to solubilize the largest amounts of the target protein (arrow). There are many reasons a given detergent fails to solubilize a membrane protein from the lipid bilayer or that the detergent fails to stabilize the protein sufficiently well after extraction. In either case, the protein band will be small in the supernatant fractions. Therefore, buffer conditions such as salt and pH must be screened, as well as the addition of glycerol and various lipids for added protein stability during protein solubilization.
Figure 17.15.1.

Solubilization assay and immunoblotting. Membranes containing the membrane protein of interest were incubated with eight different detergents or buffer without detergent (as control) for 1 hr at 4°C, and the insoluble material pelleted at 180,000 × g, 4°C. The supernatant from each solubilization assay was blotted using His-probe (Pierce) to detect the histidine-tagged protein (arrow). The largest bands were detected for samples solubilized with DDM and DM.
Membrane Protein Purification
For successful crystallization, whether for X-ray crystallography or electron crystallography, the protein of interest must be purified to homogeneity before assaying crystallization. Here a combination of affinity, charge, hydrophobic and size-exclusion chromatography columns can be used or any number of custom-made columns. The standard method typically involves the use of nickel-NTA resin together with an expressed histidine tag on the target protein.
Adding several histidines (typically 6His, 8His, or 10His) to the membrane protein of interest (either on its N- or C-terminus) allows for efficient purification on immobilized metal ion (e.g., Ni2+ or Co2+) affinity columns (Hochuli, 1988). Briefly, the polyhistidine tag will specifically complex with the divalent cationic metals present on the column when the protein solution passes through. Following binding (which may be as short as a few minutes to as long as overnight incubation) the column is washed several times with low levels of imidazole (up to 20 mM) to reduce nonspecific binding to the column and improve protein purity. Efficient elution of bound protein is then achieved with high levels of imidazole (up to 500 mM). Polyhistidine tags typically do not impact protein folding or function and several membrane proteins have been crystallized with these affinity tags (Williams et al., 1999; Lotz et al., 2008). However, it is important to note that certain proteins are affected by the presence of the histidine tag [e.g., human serum transferrin (Mason et al., 2002) and Hsp31 (Sastry et al., 2009]. In those cases, the polyhistine tag must be removed before crystallization (Viadiu et al., 2007). It is important to note that when membrane proteins are purified directly from native sources the use of affinity tags is not possible, and other purification methods must be used.
The final step of any purification protocol is the use of size-exclusion chromatography (SEC) prior to crystallization. By running protein size standards for column calibration, SEC can be used to determine the oligomeric state of the purified protein and can give an estimate of the size of the detergent micelle, as well as information about the structural homogeneity of the protein. This final step of purification provides a way to separate aggregated protein from the pool of properly folded protein, and allows the user to change buffers, detergents, or remove unwanted species (e.g., imidazole or high levels of salt) that may affect crystallization. An important note for the purification of membrane proteins is the addition of low levels of detergent to all buffers used in purification. Generally, the detergent concentration during purification is significantly lower than during membrane solubilization; however, it is extremely important that the detergent levels not fall below the CMC or the target protein will begin to unfold and aggregate.
The Growth of 2D Crystals—Membrane Protein Reconstitution
Two-dimensional crystals are prepared by reconstitution of the membrane protein of interest into a lipid bilayer. This typically involves mixing the detergent-solubilized protein with detergent-solubilized lipids followed by slow removal of the detergent by dialysis. The removal of detergent causes the lipids to begin to form membranes into which the protein inserts. This process must be optimized with the aim of inducing the formation of large and very well ordered crystalline sheets or vesicles (Fig. 17.15.2). A large number of factors unique to each protein affect the formation of 2D crystals. Generally, the choice of lipids, pH, and lipid-to-protein ratios are first assayed in a broad screen, which is detailed in Table 17.15.1. This broad screen will provide critical information about pH and salt conditions, which can be used in subsequent experiments to improve the reconstitution.
Figure 17.15.2.
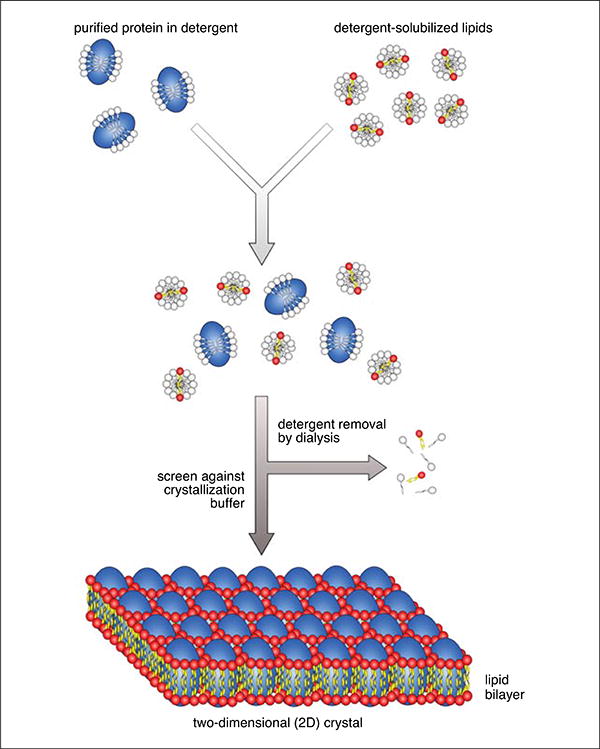
Two-dimensional crystallization of membrane proteins by slow dialysis. Purified membrane protein in a detergent solution of choice is mixed with detergent solubilized lipids. The detergent is then removed by slow dialysis, and as the detergent is removed, lipids begin to form membranes into which the protein is integrated. 2D crystals grow under certain conditions that are determined empirically. An initial broad screen is indicated in Table 17.15.1.
Table 17.15.1. The Initial Screen for Two-Dimensional Crystallization Involved Determination of Lipid Ratio, Lipid Species, and Possible pH that Favor Reconstitutiona,b,c.
| Lipid | LPRd | pH | MgCl2 |
|---|---|---|---|
| E. coli extract | 1.5 | 8 | 50 mM |
| E. coli extract | 1.0 | 8 | 50 mM |
| E. coli extract | 0.5 | 8 | 50 mM |
| E. coli extract | 1.5 | 6.5 | 50 mM |
| E. coli extract | 1.0 | 6.5 | 50 mM |
| E. coli extract | 0.5 | 6.5 | 50 mM |
| E. coli extract | 1.5 | 5 | 50 mM |
| E. coli extract | 1.0 | 5 | 50 mM |
| E. coli extract | 0.5 | 5 | 50 mM |
| E. coli extract | 1.5 | 8 | 200 mM |
| E. coli extract | 1.0 | 8 | 200 mM |
| E. coli extract | 0.5 | 8 | 200 mM |
| E. coli extract | 1.5 | 6.5 | 200 mM |
| E. coli extract | 1.0 | 6.5 | 200 mM |
| E. coli extract | 0.5 | 6.5 | 200 mM |
| E. coli extract | 1.5 | 5 | 200 mM |
| E. coli extract | 1.0 | 5 | 200 mM |
| E. coli extract | 0.5 | 5 | 200 mM |
This initial screen is the typical starting point for any electron crystallographic experiment and is very broad.
Initially screen with three different lipids: E. coli extract, DMPC, and POPC.
Test protein concentrations: 1 mg/ml is a good starting point.
LPR (lipid-to-protein ratio) is measured in mg/mg.
Choice of Lipids
Two-dimensional crystals suitable for electron diffraction have been generated using both native (Unwin, 2005) and artificial lipids (Gonen et al., 2004), as well as non-native mixed lipids (Murata et al., 2000). Both the head groups and tails of membrane lipids have been shown to interact with integral membrane proteins, (Gonen et al., 2005), so the proper lipid composition is thought to be integral for 2D crystal formation. The lipids used in 2D crystallization must be empirically determined, as there are no predetermined rules for predicting which proteins and lipid combinations will be conducive to crystallization. When artificial lipids are used, the best results have been obtained using those that best approximate the natural lipid bilayer, which has a hydrophobic core thickness of ~35 Å. Lipids with saturated or monounsaturated acyl chains are often preferred, as they approximate the natural lipid bilayer and are less susceptible to oxidation.
Lipid to Protein Ratio (LPR)
The LPR of 2D crystals must be significantly lower than what would be found in the natural membrane environment in order to promote crystal packing. This critical variable must be determined empirically for each individual protein and lipid combination. Generally, a wide screen of LPRs as outlined in Table 17.15.1 will be performed to gain a sense of the necessary LPR. If any “hits” or promising results are obtained, a narrower screen with smaller step sizes can be used to determine an optimal LPR. If no promising results are obtained, the initial screen can be repeated while varying the type of detergent or lipids.
Detergent
Detergent choice affects multiple variables in the crystallization process from the initial purification and stability of the protein in solution to the speed at which dialysis is completed and lipid bilayers form. As described previously, the detergent must be able to extract the protein from the native membrane, but must generally be nondenaturing and able to maintain the protein stability in solution. The goal of the dilution and detergent removal processes is to drop the overall detergent concentration well below the CMC to ultimately cause the lipids in the sample to form a bilayer along with the solubilized protein. It may be very difficult to lower the detergent concentration below this threshold for extremely low CMC detergents (such as Triton Χ-100 and NP40), requiring extended dialysis or dilution beyond useful protein concentrations.
Method of Detergent Removal
Detergent removal can be achieved through a variety of methods such as dialysis, simple dilution (Rémigy et al., 2003), absorption to a substrate (e.g., bio-beads; Rigaud et al., 1997), or chelation by a reagent (e.g., methyl-β-cyclodextran; Signorell et al., 2007). The majority of reported structures from 2D crystallization have used slow dialysis as the method of detergent removal (Wisedchaisri et al., 2011).
Dialysis is typically performed using dialysis buttons, which allow the screening of small (10 to 50 μl) samples and permit the user to screen a large number of crystallization parameters. The detergent-solubilized protein and lipid crystallization mixture is pipetted into the well of a dialysis button and a small piece of dialysis membrane is affixed with a rubber O-ring to seal the sample. The buttons are then placed in flasks or beakers with the desired dialysis buffer and allowed to equilibrate until the detergent is removed. Buffer is changed many times to keep the detergent concentration outside of the button low and drive the diffusion of the detergent out of the dialysis button.
Temperature
Temperature affects the dialysis rate, as well as the mobility and phase of the lipids. Generally, 2D crystallization is performed at temperatures above the lipid's transition temperature in order to keep the lipids more fluid and allow the protein freedom to laterally diffuse within the forming bilayer. Room temperature is a good starting point for crystallization screens; however, higher and lower temperatures may encourage crystallization and should be screened.
Dialysis Buffer
The type of buffer used in dialysis is one of the most important factors to screen against. Crystallization may be sensitive to the ionic strength of the dialysis buffer, and 2D crystals have been reported in solutions with salt concentrations ranging from 0 to 600 mM NaCl (Sazanov and Walker, 2000) or 1 M KCl (Frey et al., 1978). The addition of divalent cations, such as Mg+2, Ca+2, and Zn+2, has proven successful in some cases and is thought to encourage the formation of large lipid sheets. Other parameters that must be optimized within the dialysis buffer include the pH and type of buffer system used.
Negative Staining Em as a Tool for Screening 2d Crystals
At this stage, the crystallographer needs to become familiar with the electron optics and the operation of the low-dose procedures (Fujiyoshi, 1998). These are briefly summarized in Figure 17.15.3, which depicts the electron optics, as well as the electron path during low-dose procedures including the search, focus, and exposure modes.
Figure 17.15.3.
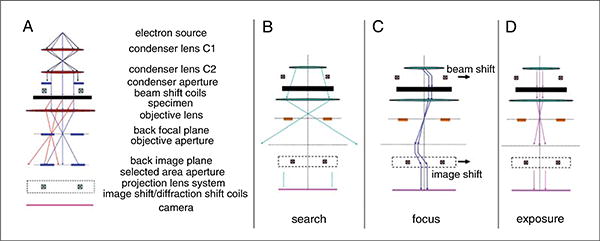
Transmission electron microscope and low dose imaging. The optical system in a TEM uses a series of lenses to focus the electron beam through the sample and onto the recording medium (A). When 2D crystals are analyzed by cryoEM the first step is to find a the crystals using search mode, a low dose setting intended to minimize electron beam induced damage to the sample (B). Once a promising area has been identified, the beam is shifted off the area of interest and focused (C). The beam is then shifted back to the sample and exposed at high doses for data collection (D). A thorough description of low dose techniques can be found in Fujiyoshi (1998).
When dialysis is complete, the crystallographer must prepare samples for negative-stain EM and assess the crystallization result as depicted in Figure 17.15.4. The first step is deciding when dialysis is complete, which is done by eye. At the start of the experiment, the solution in the dialysis button is transparent, but as the detergent is removed and the lipids begin to form vesicles, the solution becomes increasingly translucent because the membranes scatter light. When dialysis is complete, therefore, the solution inside the dialysis button appears whitish/gray (Fig. 17.15.4A, arrows). The buttons are then opened and negative-stained samples are prepared as described in detail below.
Figure 17.15.4.
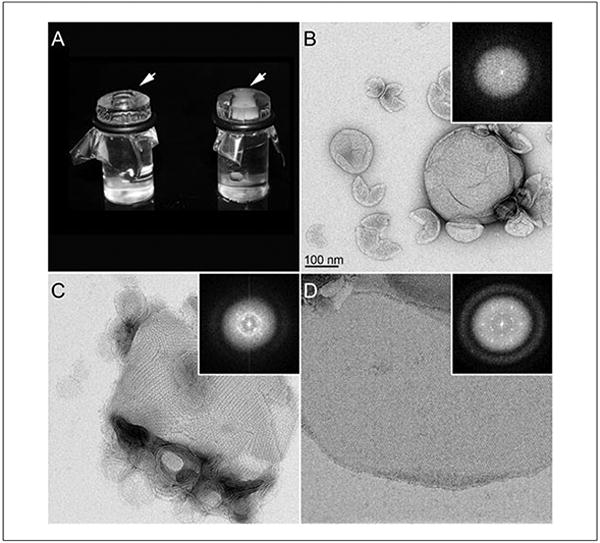
Examples of a two-dimensional crystallization assays. (A) A picture of two dialysis buttons with a well containing the protein-lipid-detergent mixture (arrows) sealed with a dialysis membrane. The button on the left is at the start of the experiment while the button on the right represents the end point. As vesicles begin to form, they scatter light, resulting in a translucent whitish/gray solution inside the well (arrow). The buttons are then opened and the solution prepared for analysis by negative-stain EM as presented in panels B-D. Proteins incorporated in to the lipid bilayer can adopt multiple conformations, with the most common being amorphous (B), small mosaic crystals (C), and large well-ordered 2D crystals (D). Inset panels are the corresponding Fourier transforms. Noncrystalline amorphous structures do not show reflections in Fourier transforms (A), while vesicles containing crystalline protein will produce reflections (C and D). Mosaic crystals (C) will lead to a Fourier transform with overlapping reflections that appear as a ring caused by multiple overlapping lattices. Well-ordered crystals as presented in (D) that produce strong and sharp reflections in Fourier transforms are ready for analysis by cryo EM for high-resolution 3D structure determination.
Procedure for Negative Staining
Preparation of uranyl formate (UF) 0.75% (staining solution)
Dissolve 37.5 mg uranyl formate in 5 ml boiling ultrapure water. Perform all subsequent steps with minimum exposure to light.
Stir for 5 min or until the uranyl formate has dissolved completely.
Add 6 μl of 5 M NaOH and stir an additional 5 min.
Filter through a 0.2-μm filter and store in the dark.
The 0.75% UF solution is stable for 3 to 4 days at room temperature. Discard if visible precipitate forms or the solution becomes discolored.
Negative stain procedure
Glow discharge a carbon-coated EM grid for 45 sec at 25 mA. The polarity can either be negative or positive depending on the sample.
Place three 50-μl drops of filtered water or buffer on a piece of clean Parafilm.
Grip the grid with a pair of anti-capillary tweezers and apply 2 μl of sample to the carbon-coated side of the grid.
Allow the sample to incubate on the grid for 20 sec.
Blot off the excess solution by gently touching the edge of the grid to filter paper.
Wash by gently touching the carbon-coated face of the grid to the first drop of wash buffer followed by blotting.
Repeat step 6 two additional times.
Place two 50-μl drops of 0.75% UF on the Parafilm.
Touch the face to the grid to the first UF drop and immediately blot as above.
Touch the face of the grid to the second drop of UF, hold in place for 20 sec, followed by blotting.
Dry the grid for ~30 sec by holding the grid next to a glass Pasteur pipet connected to an aspirator.
Store grids until EM analysis.
Steps in this procedure may be optimized for individual samples including: time and polarity of the glow discharge, amount of sample initially applied to the grid, the time the sample is allowed to soak before blotting, the number of washes, and the composition of wash buffer.
Analysis in the TEM
Negatively stained EM grids are generally examined under low magnification to first find larger membranes. Once they are found, closer examination is then done under higher magnification, usually not exceeding 50,000× to find evidence of well-ordered crystal packing (Fig. 17.15.4 B-D). In some 2D crystals, the crystalline matrix is obvious and can be identified by eye directly from the images. The existence of less obvious crystals can be confirmed by the presence of reflections in Fourier transforms. While ultimately cryoEM must be used for high-resolution 3D structure determination, data obtained from negatively stained crystals can provide important information. The unit cell size can be calculated and the space group of the crystal can be determined following data processing of negatively stained images. Overall, there are 17 possible space groups for 2D crystals as described in Table 17.15.2 and Figure 17.15.5. The reflections in the Fourier transforms also provide information on crystal size and order: large and well-ordered 2D crystals would give strong and sharp reflections, at which stage cryoEM must be used for 3D structure determination.
Table 17.15.2. The 17 Possible Space Groups in 2D Crystals.
| Plane group | 2D space group | 2D space group number | Crystal system |
|---|---|---|---|
| p1 | P1 | 1 | Oblique |
| p2 | P2 | 2 | Oblique |
| pm | P12 | 3 | Rectangle |
| pg | P121 | 4 | Rectangle |
| cm | C12 | 5 | Rectangle |
| p2mm | P222 | 6 | Rectangle |
| p2mg | P2221 | 7 | Rectangle |
| p2gg | P22121 | 8 | Rectangle |
| c2mm | C222 | 9 | Rectangle |
| p4 | P4 | 10 | Square |
| p4mm | P422 | 11 | Square |
| p4gm | P4212 | 12 | Square |
| p3 | P3 | 13 | Hexagonal |
| p3m1 | P321 | 14 | Hexagonal |
| p31m | P312 | 15 | Hexagonal |
| p6 | P6 | 16 | Hexagonal |
| p6mm | P622 | 17 | Hexagonal |
Figure 17.15.5.
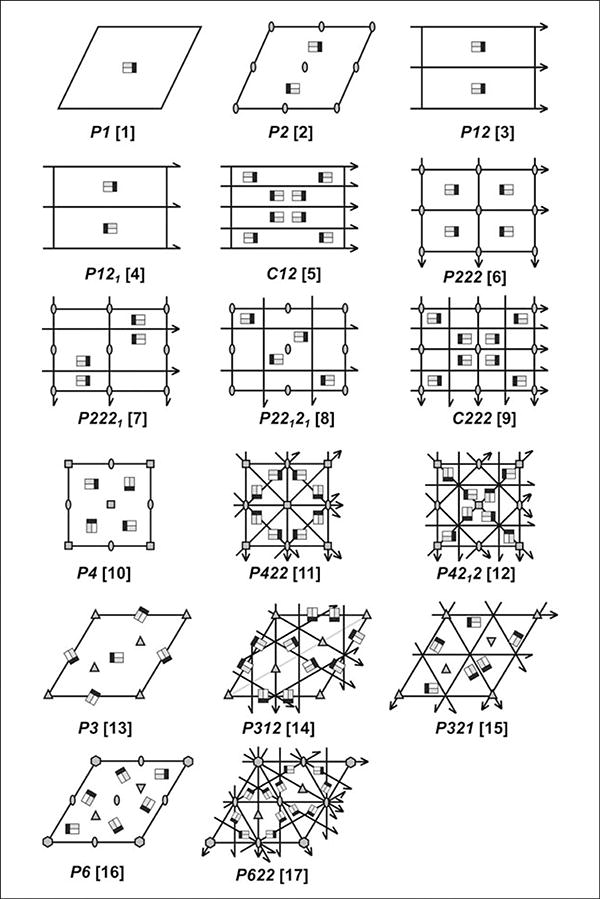
The 17 possible plane groups for 2D crystals. The black lines outline the unit cell, with full arrows indicating a 2-fold reflection and half arrows showing a 2-fold screw axis. The locations of rotations with orders of 2 (180°), 3 (120°), 4 (90°), and 6 (60°) are shown by ovals, triangles, squares, and hexagons, respectively. Typically shown as black, rotation symbols are shown filled in gray for clarity. For all panels the 2D space group number is indicated in brackets.
Conclusions
Electron crystallography is the only cryoEM technique capable of delivering atomic-resolution information for membraneembedded membrane proteins. The 2D crystals may provide information about lipidprotein interactions and shed light on processes that are otherwise inaccessible by other structural biology techniques.
Acknowledgments
We thank all members of the Gonen laboratory for helpful discussions. We thank Yifan Cheng (UCSF) for providing the cartoons for electron optics (Fig. 17.15.3). The Gonen laboratory is supported by the Howard Hughes Medical Institute.
Literature Cited
- Baneyx F. Recombinant protein expression in Escherichia coli. Curr Opin Biotechnol. 1999;10:411–421. doi: 10.1016/s0958-1669(99)00003-8. [DOI] [PubMed] [Google Scholar]
- Frey TG, Chan SHP, Schatz G. Structure and orientation of cytochrome c oxidase in crystalline membranes. J Biol Chem. 1978;253:4389–4395. [PubMed] [Google Scholar]
- Fujiyoshi Y. The structural study of membrane proteins by electron crystallography. Adv Biophys. 1998;35:25–80. doi: 10.1016/s0065-227x(98)90004-1. [DOI] [PubMed] [Google Scholar]
- Gonen T, Donaldson P, Kistler J. Galectin-3 is associated with the plasma membrane of lens fiber cells. Invest Ophthalmol Vis Sci. 2000;41:199–203. [PMC free article] [PubMed] [Google Scholar]
- Gonen T, Sliz P, Kistler J, Cheng Y, Walz T. Aquaporin-0 membrane junctions reveal the structure of a closed water pore. Nature. 2004;429:193–197. doi: 10.1038/nature02503. [DOI] [PubMed] [Google Scholar]
- Gonen T, Cheng Y, Sliz P, Hiroaki Y, Fujiyoshi Y, Harrison SC, Walz T. Lipid-protein interactions in double-layered two-dimensional AQP0 crystals. Nature. 2005;438:633–638. doi: 10.1038/nature04321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochuli E. Large-scale chromatography of recombinant proteins. J Chromatogr. 1988;444:293–302. doi: 10.1016/s0021-9673(01)94032-4. [DOI] [PubMed] [Google Scholar]
- le Maire M, Champeil P, Moller JV. Interaction of membrane proteins and lipids with solubilizing detergents. Biochim Biophys Acta. 2000;1508:86–111. doi: 10.1016/s0304-4157(00)00010-1. [DOI] [PubMed] [Google Scholar]
- Lotz M, Haase W, Kuhlbrandt W, Collinson I. Projection structure of yidC: A conserved mediator of membrane protein assembly. J Mol Biol. 2008;375:901–907. doi: 10.1016/j.jmb.2007.10.089. [DOI] [PubMed] [Google Scholar]
- Mason AB, He QY, Halbrooks PJ, Everse SJ, Gumerov DR, Kaltashov IA, Smith VC, Hewitt J, MacGillivray RT. Differential effect of a his tag at the N- and C-termini: Functional studies with recombinant human serum transferrin. Biochemistry. 2002;41:9448–9454. doi: 10.1021/bi025927l. [DOI] [PubMed] [Google Scholar]
- Midgett CR, Madden DR. Breaking the bottleneck: Eukaryotic membrane protein expression for high-resolution structural studies. J Struct Biol. 2007;160:265–274. doi: 10.1016/j.jsb.2007.07.001. [DOI] [PubMed] [Google Scholar]
- Miroux B, Walker JE. Over-production of proteins in Escherichia coli: Mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J Mol Biol. 1996;260:289–298. doi: 10.1006/jmbi.1996.0399. [DOI] [PubMed] [Google Scholar]
- Murata K, Misuoka K, Hirai T, Walz T, Agre P, Heymann JB, Engel A, Fujiyoshi Y. Structural determinants of water permeation through aquaporin-1. Nature. 2000;407:599–605. doi: 10.1038/35036519. [DOI] [PubMed] [Google Scholar]
- Nannenga BL, Baneyx F. Reprogramming chaperone pathways toimprove membrane protein expression in Escherichia coli. Protein Sci. 2011;20:1411–1420. doi: 10.1002/pro.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramon A, Marin M. Advances in the production of membrane proteins in Pichia pastoris. Biotechnol J. 2011;6:700–706. doi: 10.1002/biot.201100146. [DOI] [PubMed] [Google Scholar]
- Reichow SL, Gonen T. Lipid-protein interactions probed by electron crystallography. Curr Opin Struct Biol. 2009;19:560–565. doi: 10.1016/j.sbi.2009.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rémigy HW, Caujolle-Bert D, Suda K, Schenk A, Chami M, Engel A. Membrane protein reconstitution and crystallization by controlled dilution. FEBS Lett. 2003;555:160–169. doi: 10.1016/s0014-5793(03)01105-0. [DOI] [PubMed] [Google Scholar]
- Rigaud JL, Mosser G, Lacaperre JJ, Olofsson A, Levy D, Ranck JL. Bio-Beads: An efficient strategy for two-dimensional crystallization of membrane proteins. J Struct Biol. 1997;118:226–235. doi: 10.1006/jsbi.1997.3848. [DOI] [PubMed] [Google Scholar]
- Sastry MS, Zhou W, Baneyx F. Integrity of N- and C-termini is important for E. coli Hsp31 chaperone activity. Protein Sci. 2009;18:1439–1447. doi: 10.1002/pro.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sazanov LA, Walker JE. Cryo-electron crystallography of two sub-complexes of bovine complex I reveals the relationship between the membrane and peripheral arms. J Mol Biol. 2000;302:455–464. doi: 10.1006/jmbi.2000.4079. [DOI] [PubMed] [Google Scholar]
- Signorell GA, Kaufmann TC, Kukulski W, Engel A, Remigy HW. Controlled 2D crystallization of membrane proteins using methyl-beta-cyclodextrin. J Struct Biol. 2007;157:321–328. doi: 10.1016/j.jsb.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Surade S, Klein M, Stolt-Bergner PC, Muenke C, Roy A, Michel H. Comparative analysis and “expression space” coverage of the production of prokaryotic membrane proteins for structural genomics. Protein Sci. 2006;15:2178–2189. doi: 10.1110/ps.062312706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker J, Grisshammer R. Purification of a rat neurotensin receptor expressed in Escherichia coli. Biochem J. 1996;317:891–899. doi: 10.1042/bj3170891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J Mol Biol. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- Viadiu H, Gonen T, Walz T. Projection map of aquaporin-9 at 7 A resolution. J Mol Biol. 2007;367:80–88. doi: 10.1016/j.jmb.2006.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner S, Klepsch MM, Schlegel S, Appel A, Draheim R, Tarry M, Högbom M, van Wijk KJ, Slotboom DJ, Persson JO, de Gier JW. Tuning Escherichia coli for membrane protein overexpression. Proc Natl Acad Sci USA. 2008;105:14371–14376. doi: 10.1073/pnas.0804090105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss HM, Grisshammer R. Purification and characterization of the human adenosine A(2a) receptor functionally expressed in Escherichia coli. Eur J Biochem. 2002;269:82–92. doi: 10.1046/j.0014-2956.2002.02618.x. [DOI] [PubMed] [Google Scholar]
- Williams KA, Geldmacher-Kaufer U, Padan E, Schuldiner S, Kuhlbrandt W. Projection structure of NhaA, a secondary transporter from Escherichia coli, at 4.0 A resolution. EMBO J. 1999;18:3558–3563. doi: 10.1093/emboj/18.13.3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisedchaisri G, Reichow SL, Gonen T. Advances in structural and functional analysis of membrane proteins by electron crystallography. Structure. 2011;19:1381–1393. doi: 10.1016/j.str.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]