

Abstract
Background
Estrogen-related receptors (ERRs) are orphan nuclear hormone receptors expressed in metabolically active tissues and modulate numerous homeostatic processes. ERRs do not bind the ligand estrogen, but they are able to bind the estrogen response element (ERE) embedded within the ERR response elements (ERREs) to regulate transcription of genes. Previous work has demonstrated that adult mice lacking Errβ have altered metabolism and meal patterns. To further understand the biological role of Errβ, we characterized the stress response of mice deficient for one or both alleles of Errβ.
Results
Sox2-Cre:Errβ mice lack Errβ expression in all tissues of the developing embryo. Sox2-Cre:Errβ+/lox heterozygotes were obese, had increased Npy and Agrp gene expression in the arcuate nucleus of the hypothalamus, and secreted more corticosterone in response to stress. In contrast, Sox2-Cre:Errβlox/lox homozygotes were lean and, despite increased Npy and Agrp gene expression, did not secrete more corticosterone in response to stress. Sox2-Cre:Errβ+/lox and Sox2-Cre:Errβlox/lox mice treated with the Errβ and Errγ agonist DY131 demonstrated increased corticotropin-releasing hormone (Crh) expression in the paraventricular nucleus of the hypothalamus, although corticosterone levels were not affected. Nes-Cre:Errβlox/lox mice, which selectively lack Errβ expression in the nervous system, also demonstrated elevated stress response during an acoustic startle response test and decreased expression of both Crh and corticotropin-releasing hormone receptor 2 (Crhr2).
Conclusions
Loss of Errβ affects body composition, neuropeptide levels, stress hormones, and centrally-modulated startle responses of mice. These results indicate that Errβ alters the function of the hypothalamic-pituitary-adrenocortical axis and indicates a role for Errβ in regulating stress response.
Background
ERRs are nuclear hormone receptors that regulate multiple homeostatic processes throughout life [1]. ERRs were initially identified on the basis of sequence homology to estrogen receptors (ERs) [2]. The homology between Errs and Ers is 36% in the ligand binding domain and 68% in the DNA binding domain. ERRs bind both ERR response elements (ERREs) and the closely related estrogen response elements (EREs) embedded within an ERRE sequence on DNA to modulate transcription of target genes [3-8]. Errs activate gene transcription by binding to DNA, either as a monomer, homodimer, or a heterodimer complex, which includes two different Err isoforms [1,6,7,9,10]. While their binding sites are similar to those of Ers, Errs do not bind estradiol and instead activate transcription in a ligand-independent manner, leading to their classification as orphan nuclear receptors. The three different Err genes, α, β and γ, have highly conserved ligand and DNA binding domains and thus may regulate homeostatic processes in a compensatory manner [11].
In mice, Errβ and Errγ are selectively expressed in the brain and multiple peripheral tissues [2,12-14] and share the highest degree of sequence homology [11], suggesting that they may share overlapping functions. Since Errs recognize the same response elements, they are likely to regulate overlapping subsets of target genes [11].
We have previously reported that whole-body or central nervous system-specific deletion of Errβ increases expression of Errγ and ultimately alters body composition, metabolism, meal patterns, and energy expenditure of mice [11]. Further, inhibition of Errβ or Errγ alter metabolic parameters, whole-body energy balance (e.g. body composition, food intake and neuropeptide expression), while deletion of Errβ reciprocally modulates expression of Errγ (and vice versa) suggesting that balanced expression of Errβ and Errγ is important for control of energy balance and food intake [14-18].
Alterations in glucocorticoid signaling and whole-body energy balance positively correlate with one another, with increased glucocorticoid levels resulting in increased body weight [19-21]. Errβ suppresses glucocorticoid receptor activity in neuroblastoma and kidney cells in a dose-dependent manner, suggesting that it may also regulate metabolism at least in part through modulation of the hypothalamic-pituitary-adrenal (HPA) axis [22]. The HPA axis is regulated by corticotrophin-releasing hormone (Crh) released from neurosecretory cells of the hypothalamic paraventricular nucleus. Crh stimulates release of adrenocorticotropic hormone (ACTH) from the anterior pituitary, and ACTH, in turn, triggers glucocorticoid secretion from the adrenal gland. Negative feedback from ACTH and glucocorticoid secretion ultimately modulates Crh expression in the paraventricular nucleus via glucocorticoid receptors [23]. Disrupting glucocorticoid feedback loops can alter whole-body energy balance (e.g. body weight). Glucocorticoid excess (Cushing’s disease) increases central fat deposition, whereas decreased body weight is associated with glucocorticoid insufficiency (Addison’s disease) [19-21]. In addition to these effects on metabolism, alterations in the HPA axis can also influence anxiety and stress, which increase Neuropeptide Y (Npy) secretion. Npy further augments obesity susceptibility by inducing food intake and contributing to leptin resistance [23-25].
Consequently, we propose that Errβ modulates stress responses. Since Errβ suppresses glucocorticoid receptor activity [22], we hypothesized that the HPA axis may be altered in mice that carry heterozygous or homozygous loss of function mutations of Errβ in all somatic tissues [14,26,27]. The effects of Errβ deficiency on body weight, body composition, neuropeptide levels, stress hormones, and stress responses were examined in Sox2-Cre:Errβ+/lox and Sox2-Cre:Errβlox/lox mice, in which Errβ expression is disrupted in all somatic tissues. These results indicate that Errβ modulates stress responses, at least in part through central mechanisms.
Results
Errβ gene dosage alters body weight and body composition
Sox2-Cre:Errβ+/lox heterozygous mice express one allele of Errβ, resulting in higher levels of Errβ expression relative to Sox2-Cre:Errβlox/lox homozygous mice. Alterations in energy balance are observed in mice deficient for Errβ in all embryonic tissues (Sox2-Cre:Errβlox/lox) [14]. Because Errβ is proposed to modulate energy balance in a dose-dependent manner, we characterized Sox2-Cre:Errβlox/lox and Sox2-Cre:Errβ+/lox mice to determine whether gene dosage altered development of body weight and body composition. We previously showed that Sox2-Cre:Errβlox/lox mice have decreased body weight and fat mass by nine months of age [8]. Body weight and body composition (fat mass and lean mass) were measured in Sox2-Cre:Errβlox/lox, Sox2-Cre:Errβ+/lox, and WT mice at three weeks and at nine months of age (Table 1). By three weeks, body composition differences began to emerge between the genotypes: Sox2-Cre: Errβ+/lox mice significantly increased fat mass (fat mass: F1,8 = 9.32, P = 0.05), while Sox2-Cre:Errβlox/lox mice trended toward decreased fat mass (fat mass: F1,10 = 4.95, P = 0.05) compared to WT mice. There was no difference in body weight among the genotypes at three weeks, implying that alterations in body composition arise prior to weight changes in Errβ-deficient mice.
Table 1.
Body weight and body composition, physical activity and meal patterns of wild type (WT), Sox2-Cre:Errβ+/lox, and Sox2-Cre:Errβlox/loxmice
| Genotype | Age |
Body composition |
|
Meal patterns |
||||
|---|---|---|---|---|---|---|---|---|
|
Body weight |
Fat mass |
Lean mass |
Activity |
Pellets |
Satiety ratio |
IMI |
||
| (grams) | (grams) | (grams) | (beam breaks) | (number) | (IMI/meal size) | (minutes) | ||
| WT |
3 weeks |
13.1 ± 0.5 |
2.09 ± 0.09 |
10.06 ± 0.41 |
|
|
|
|
|
Sox2-Cre:Errβ+/lox |
3 weeks |
14.0 ± 0.7 |
2.58 ± 0.16* |
10.56 ± 0.62* |
|
|
|
|
|
Sox2-Cre:Errβlox/lox |
3 weeks |
12.9 ± 0.8 |
1.64 ± 0.17 |
10.51 ± 0.61 |
|
|
|
|
| WT |
9 months |
36.5 ± 0.9 |
12.19 ± 0.65 |
23.09 ± 0.46 |
67207 ± 8601 |
128 ± 15 |
8.3 ± 0.9 |
107 ± 17 |
|
Sox2-Cre:Errβ+/lox |
9 months |
46.1 ± 3.0* |
21.43 ± 1.84* |
24.00 ± 0.63 |
93599 ± 9879 |
238 ± 35* 5.6 ± 0.7# |
102 ± 10 |
|
| Sox2-Cre:Errβlox/lox | 9 months | 28.4 ± 1.3* | 5.69 ± 0.74* | 21.64 ± 0.40* | 133741 ± 20533* | 260 ± 57* | 4.1 ± 0.7* | 67 ± 11* |
*P < 0.05 relative to WT.
#P = 0.05 relative to WT.
Activity is a measurement for the number of beam break, which represents horizontal physical activity that is parallel to the ground.
Meal patterns for Sox2-Cre:Errβlox/lox mice are adapted from [14].
At nine months of age, Sox2-Cre:Errβ+/lox mice had increased fat mass and no change in lean mass relative to WT mice (fat mass: F1,9 = 35.90, P = 0.002). However, Sox2-Cre:Errβlox/lox mice demonstrated the opposite trend in body composition, with decreases in both fat and lean mass (fat free mass) relative to WT mice (fat mass: F1,10 = 46.53, P < =0.0001; lean mass: F1,10 = 6.21, P = 0.03). Accordingly, body weight increased in Sox2-Cre:Errβ+/lox mice (F1,9 = 32.31, P = < 0.000001) and decreased in Sox2-Cre:Errβlox/lox mice (F1,10 = 32.57, P = 0.0004) relative to WT mice. Given these differences, the Sox2-Cre:Errβ+/lox mice surprisingly had a similar macrostructure of food intake as the Sox2-Cre:Errβlox/lox[14], relative to WT mice. Specifically, after consuming a meal, the duration of time that the mouse was satiated was decreased (satiety ratio), the total number of pellets consumed was increased, and the duration of time between meals (intermeal interval, IMI) was not changed for Sox2-Cre:Errβ+/lox mice, but IMI was decreased for Sox2-Cre:Errβlox/lox mice (Table 1). The difference in IMI between the genotypes may be a compensatory change due to peripheral signals modulated by the increases in both body weight and fat mass observed in the Sox2-Cre:Errβ+/lox mice.
Hypothalamic neuropeptide expression in Errβ mutant mice
In the brain, Errβ is primarily expressed in the hindbrain, whereas Errγ is expressed in both the hindbrain and hypothalamus [14,28,29]. Nuclei of the hindbrain send primary projections to the hypothalamus (e.g., nucleus tractus solitarius to the paraventricular nucleus) and the amygdala, and activity in these nuclei can modulate hypothalamic gene expression [30-32]. Furthermore, in the absence of Errβ, Errγ can modulate food intake [14]. Since Sox2-Cre:Errβ+/lox and Sox2-Cre:Errβlox/lox mice demonstrated alterations in body weight and body composition relative to WT mice, we sought to determine if hypothalamic neuropeptides known to modulate energy balance, Npy and Agrp, were differentially expressed in the brains of these mutants. Brain tissue sections of three-week-old WT, Sox2-Cre:Errβ+/lox, and Sox2-Cre:Errβlox/lox mice were hybridized with cRNA probes specific to Npy and Agrp mRNA. Npy (Figure 1a) and Agrp (Figure 1b) staining were least intense in the hypothalamus of WT brain tissues, more intense in Sox2-Cre:Errβ+/lox brain tissues, and most intense in Sox2-Cre:Errβlox/lox brain tissues. Expression of Npy and Agrp, as determined by hypothalamic ISH staining, appears to correlate inversely with Errβ expression. Increased staining expression of Npy and Agrp may contribute to the increased fat mass of three-week-old Sox2-Cre:Errβ+/lox mice; conversely, the high levels of Npy and Agrp in Sox2-Cre:Errβlox/lox mice may be a downstream response to decreased fat mass.
Figure 1.
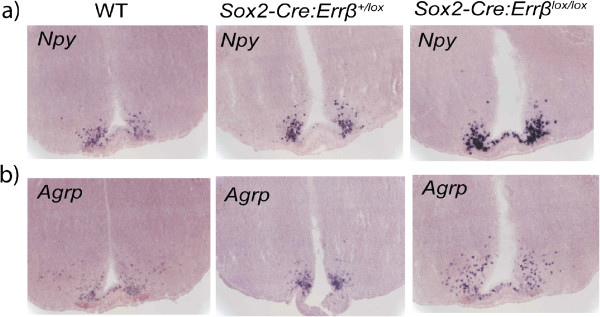
Hypothalamic neuropeptide expression in wild-type (WT), Sox2-Cre:Errβ+/lox, and Sox2-Cre:Errβlox/loxmouse brains. Brain tissues were harvested from three-week-old WT, Sox2-Cre:Errβ+/lox, and Sox2-Cre:Errβlox/lox mice; frozen tissue sections were hybridized in situ with cRNA probes to a)Npy and b)Agrp (n = 3/genotype).
Sox2-Cre:Errβlox/lox mice show elevated activity levels due to defects in vestibular system development [14,26], which likely contribute to the body weight and body composition differences observed at nine months of age. However, three-week-old Sox2-Cre:Errβ+/lox mice are not hyperactive, suggesting that activity alone does not control hypothalamic neuropeptide levels (Table 1: Sox2-Cre:Errβlox/lox vs. WT - F1,9 = 16.43, P = 0.004).
Errβ gene dosage alters expression of HPA axis components
Errβ interacts with glucocorticoid receptors in neuroblastoma and kidney cells [22,33] and may also interact in the hindbrain where Errβ is expressed [14]. Since increased Npy expression is often associated with elevated levels of glucocorticoid release, which can influence adiposity [23-25], we hypothesized that Errβ deficiency may alter stress responsiveness via glucocorticoid secretion. Therefore, stress responses of WT, Sox2-Cre:Errβ+/lox, and Sox2-Cre:Errβlox/lox mice were measured by detecting alterations in HPA axis components, Crh expression and corticosterone.
To investigate the ability of Errγ to compensate for Errβ deficiency, stress responses were investigated in the presence of synthetic agonists of Errγ. Atlhough agonists specific to individual Err isoforms are not commercially available, we were able to perform these studies using DY131, a selective agonist of both Errβ and Errγ [34]. It has been previously determined that DY131 is able to readily penetrate the blood–brain barrier, as it is both hydrophobic and has a topological surface area (TPSA) less than 70 [14]. In Sox2-Cre: Errβlox/lox null mice, DY131 would exclusively activate Errγ and that this would result in alterations in HPA axis function (e.g. Crh expression or corticosterone levels). We utilized a restraint stress paradigm to measure corticosterone serum levels during baseline, stress, and recovery phases. WT mice demonstrated increased stress-induced corticosterone levels, which returned to baseline after one hour of recovery (Figure 2a) (baseline vs. stress: F1,8 = 7.82, P = 0.03). Similar results were measured in WT mice administered DY131 (DY131 WT, baseline vs stress: F1,8 = 6.46, P = 0.03; control WT; DY131 WT, stress vs recovery: F1,11 = 8.54, P = 0.01). Sox2-Cre:Errβ+/lox mice exhibited markedly elevated corticosterone levels during stress, which may arise from altered negative feedback mechanisms that modulate corticosterone secretion (e.g. enhanced Crh secretion from the brain). Sox2-Cre:Errβ+/lox mice exhibit normal recovery to baseline one hour after the stress test (Figure 2b – control Sox2-Cre:Errβ+/lox, baseline vs stress: F1,14 = 8.62, P = 0.01). Administration of DY131 yielded similar results (DY131 Sox2-Cre:Errβ+/lox, baseline vs stress: F1,14 = 7.02, P = 0.02; control Sox2-Cre:Errβ+/lox, stress vs recovery: F1,14 = 7.14, P = 0.02; DY131 Sox2-Cre:Errβ+/lox, stress vs recovery: F1,14 = 8.83, P = 0.01).
Figure 2.
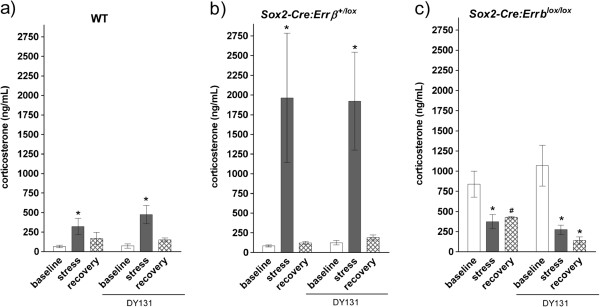
Glucocorticoid levels of wild-type (WT), Sox2-Cre:Errβ+/lox, and Sox2-Cre:Errβlox/loxmice after restraint stress. a) Baseline, stress, and recovery glucocorticoid levels were measured in serum of WT mice and after treatment with Errβ/Errγ agonist DY131 using a corticosterone radioimmunoassay. b) Baseline, stress, and recovery glucocorticoid levels were measured in serum of Sox2-Cre:Errβ+/lox mice and after treatment with Errβ/Errγ agonist DY131 using a corticosterone radioimmunoassay. c) Baseline, stress, and recovery glucocorticoid levels were measured in serum of Sox2-Cre:Errβlox/lox mice and after treatment with Errβ/Errγ agonist DY131 using a corticosterone radioimmunoassay. *P < 0.05.
In contrast, Sox2-Cre:Errβlox/lox mice had elevated baseline corticosterone levels but exhibited no increase with stress (Figure 2c – control Sox2-Cre:Errβlox/lox, baseline vs stress: F1,8 = 10.86, P = 0.02; DY131 Sox2-Cre:Errβlox/lox, baseline vs stress: F1,8 = 15.14, P = 0.01; control Sox2-Cre:Errβlox/lox; DY131 Sox2-Cre:Errβlox/lox, baseline vs recovery: F1,8 = 21.81, P = 0.01), suggesting that Errβ:Sox2-Crelox/lox mice are unable to increase corticosterone levels in response to restraint stress. In fact, expression of Crh, as determined by ISH staining, was increased in the Sox2-Cre:Errβlox/lox mice under baseline conditions, a modest increase in ISH staining was also seen in the Sox2-Cre:Errβ+/lox mice, with DY131 further increasing the ISH staining for Crh. This data suggests that Errγ may modulate expression of Crh in a manner dependent on the level of Errβ expression (Figure 3).
Figure 3.
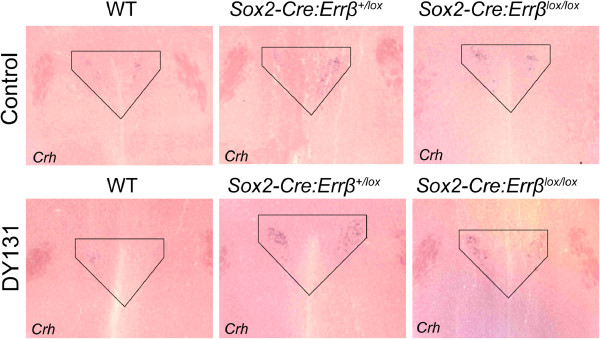
Crh expression of wild-type (WT), Sox2-Cre:Errβ+/lox, and Sox2-Cre:Errβlox/loxmice. Brain tissue of WT, Sox2-Cre:Errβ+/lox, and Sox2-Cre:Errβlox/lox mice injected with saline (top) or Errβ/Errγ agonist DY131 (bottom) were stained for Crh by ISH (n = 2/genotype).
Neural progenitor-specific deletion of Errβ alters acoustic startle response
Sox2-Cre:Errβ+/lox and Sox2-Cre:Errβlox/lox mice demonstrate differences in HPA activation, which may arise from central and/or peripheral mechanisms. In the central nervous system, Errβ expression is restricted to the hindbrain. Nes-Cre:Errβlox/lox mice lack Errβ in neural progenitor cells, effectively resulting in selective loss of Errβ expression in the hindbrain [14]. Therefore, we investigated the central role of Errβ in modulating stress responses in Nes-Cre:Errβlox/lox and WT mice using an acoustic startle test. The neuroanatomical and neurochemical basis of the acoustic startle response has been well mapped and involves neurons found in the amygdala, dorsomedial hypothalamus, and brainstem [35-39]. The amygdala elicits behavioral stress responses associated with the acoustic startle response and expresses the neuromodulators Crh and Npy [36,40]. Nes-Cre:Errβlox/lox mice have decreased Npy expression in the hindbrain [14], which may modify neural circuitry activated by physical and psychological stress and, more specifically, the acoustic startle response.
We measured PPI and the acoustic startle response to determine if Nes-Cre:Errβlox/lox mice had alterations in stress responses that arise from dysfunction of the inhibitory hindbrain circuit associated with PPI or the excitatory circuit associated with the acoustic startle response [41]. The acoustic startle response was measured after delivery of a prepulse intensity signal (0, 74, 78, 82, 86, or 90 dB) followed by the lead interval to a strong auditory stimulus. We observed a greater startle response in Nes-Cre:Errβlox/lox mice (n = 8, db120; 1081.5 ±150) compared to WT mice (n = 12, db120; 475.8 ± 27) (Figure 4a, 0db - F1,20 = 0.05, P = 0.81; 0-120db - F1,20 = 9.25, P = 0.006; 74-120db - F1,20 = 15.13, P = 0.001; 78-120db - F1,20 = 15.63, P = 0.0009; 82-120db - F1,20 = 14.04, P = 0.001; 86-120db - F1,10 = 14.17, P = 0.001; 90-120db - F1,8 = 14.98, P = 0.001). However, the amplitude of the startle response decreased in Nes-Cre:Errβlox/lox mice when the intensity of the prepulse tone increased. Crh expression was measured in the hindbrain of Nes-Cre:Errβlox/lox mice and WT mice. Indeed, Nes-Cre:Errβlox/lox mice have decreased expression of Crh and Crhr2 relative to WT (Figure 4b, F1,10 = 6.54, P = 0.03 and Figure 4c, F1,10 = 6.23, P = 0.03). These results indicate alterations in the excitatory pathway that generates a startle response, but not the inhibitory pathway arising from the pedunculopontine tegmental nucleus associated with PPI [41-43]. The increased acoustic startle response in Nes-Cre:Errβlox/lox mice may thus arise from altered activity of the excitatory pathway involving Crh and Crhr2 expression and the pontine reticular nucleus, bed nucleus of the stria terminalis, amygdala, and hypothalamus [37-39,43-48]. The hindbrain excitatory pathways, which include catecholaminergic projections to the paraventricular nucleus of the hypothalamus, increase Crh expression in the hypothalamus, suggesting that hindbrain signaling may alter the HPA-axis feedback loop [49].
Figure 4.

Acoustic startle response, pre-pulse inhibition (PPI) and Crhr2 expression for wild-type (WT) and Nes-Cre:Errβlox/loxmice. a) The force elicited by the acoustic startle response was measured from WT and Nes-Cre:Errβlox/lox mice after pre-pulse inhibition over five different acoustic intensities (74, 78, 82, 86, and 90 dB). b)Crh and c)Crhr2 levels were decreased in the hindbrain of Nes-Cre:Errβlox/lox mice, relative to WT. Data shown are mean ± SEM for each group. *P < 0.05.
Discussion
ERRs are involved with energy balance and metabolism [14-18]. Using mice globally deficient for Errβ, we have shown that Errβ modulates body composition, stress signaling, and hypothalamic neuropeptide expression (Table 2). Errβ gene dosage affected body composition and stress response with increased fat mass and corticosterone levels in Sox2-Cre:Errβ+/lox mice and decreased fat mass and corticosterone levels in Sox2-Cre:Errβlox/lox mice (Table 1 and Figure 2). Additionally, central nervous system-specific Errβ deletion alters stress associated with the acoustic startle response pathways (Figure 4).
Table 2.
Summary of phenotype difference between Sox2-Cre:Errβ+/lox, Sox2-Cre:Errβlox/loxand Nes2-Cre:Errβlox/loxmice, relative to wild type (WT)
| Phenotype | Sox2 - Cre : Errβ +/lox | Sox2 - Cre : Errβ lox/lox | Nes2 - Cre : Errβ lox/lox | |
|---|---|---|---|---|
| Body Composition | ||||
| Body weight |
↑, |
↓, |
↑ |
|
| Fat mass |
↑, |
↓, |
NC |
|
| Lean mass |
↓, |
↓, |
↑ |
|
| Hormone and Neuropeptides | ||||
| Corticosterone |
↑, |
↓, |
NA |
|
| Corticosterone (DY131) |
↑, |
↓, |
NA |
|
|
Crh expression |
↑, |
↑, |
↓ |
|
|
Crh expression (DY131) |
↑↑, |
↑↑, |
NA |
|
|
Npy expression |
↑, |
↑, |
↓ |
|
|
Agrp expression |
↑, |
↑, |
NA |
|
| Stress Behavior | ||||
| Acoustic startle response |
NA, |
NA, |
↑ |
|
| Meal patterns | ||||
| Total pellets consumed |
↑, |
↑, |
NC |
|
| Inter Meal interval (IMI) |
NC, |
↓, |
↓ |
|
| Satiety Ratio | ↓#, | ↓, | ↓ | |
↑ – increase, ↓ – decrease, NC – no change, ↑↑ – increase relative to levels with no DY131 treatment, NA – not available, #P = 0.05 relative to WT; Nes2-Cre:Errβlox/lox data adapted from [14].
Hypothalamic expression of Npy and Agrp, orexigenic factors that increase fat mass and food intake [50-52], increased in both Sox2-Cre:Errβ+/lox and Sox2-Cre:Errβlox/lox mice (Figure 1). These results suggest that increased anabolic neuropeptide expression may be due to central or peripheral mechanisms that are activated following global deletion of Errβ. Increased Npy and Agrp expression may be due to differences in leptin levels from adipose mass. Increased fat mass and lean mass were measured in Sox2-Cre:Errβ+/lox mice, although decreased fat mass and lean mass were measured in highly-active Sox2-Cre:Errβlox/lox mice at nine months of age (Table 1). Expression of Npy and leptin are coordinately regulated, as Npy blunts the effects of leptin and increased leptin levels decrease Npy expression [23,53-55]. Thus, Sox2-Cre:Errβ+/lox mice may consume more food and increase Npy expression and fat mass due to leptin resistance; Sox2-Cre:Errβlox/lox mice may increase Npy expression to compensate for decreased fat mass arising from increased physical activity (Figure 1 and Table 1). In support of this, Nes-Cre:Errβlox/lox mice have increased lean mass, no change in physical activity and have decreased Npy expression in the hindbrain [14]. Changes in body composition emerged prior to changes in body weight, suggesting that both peripheral and central signals may be altered to regulate the development of increased fat mass (Table 1).
The opposite phenotypes that are seen in the Sox2-Cre:Errβ+/lox and Sox2-Cre:Errβlox/lox mice may arise from the ability of Errβ or Errγ to regulate gene transcription as both homodimers and Errβ/Errγ heterodimers [1,6,7,9,10]. Errβ/Errγ heterodimers have been predicted to exist, but to our knowledge it has not been directly detected in vivo[1]. RIP140 is a nuclear receptor corepressor that regulates cellular metabolism [56-58]. RIP140 enhanced transcriptional activity for all three mouse Err genes [59]. Mice lacking RIP140 are lean, with increased metabolic rate and insulin sensitivity [58]. Similarly, Sox2-Cre:Errβlox/lox mice are lean with increased metabolic rate (Table 1 and [14]), and Nes-Cre:Errβlox/lox have increased lean mass, increased metabolic rate and insulin sensitivity [14]. Since deletion of both Errβ and RIP140 exhibit similar phenotypes, this suggests that increased lean mass relative to fat mass, metabolic rate and insulin sensitivity may arise from both the RIP140 corepressor and Errβ [59].
ChIP-seq analysis derived from embryonic stem cells revealed that Errβ binds the regulatory element of two genes associated with Crh activity — Corticotropin releasing hormone binding protein (Crhbp) and Corticotropin releasing hormone receptor 2 (Crhr2) — as well as one gene associated with whole-body energy balance and stress responses, Cholecystokinin B receptor (Cckbr) [60]. We hypothesize that Errβ, Crhbp and Crhr2 may modulate stress signaling by altering the biological activity of Crh in extrahypothalamic sites and/or corticosterone feedback or secretion [32,39,48,61-64]. Disruption of Errβ-dependent regulation of expression of Cckbr and/or Crhr2 may at least partially explain the abnormal meal patterns and stress behaviors (e.g. acoustic startle response, Crh expression or corticosterone levels) observed in Sox2-Cre:Errβlox/lox, Sox2-Cre:Errβ+/lox, and Nes-Cre:Errβlox/lox mice [14].
Crh is expressed in the paraventricular nucleus of the hypothalamus and initiates ACTH release from the pituitary [40,65]. Crh has since been found to be synthesized in extra-hypothalamic sites, where it also acts to modulate stress response and food intake [40,65-67]. ERR family members also modulate stress responses by regulating glucocorticoid receptor activity in muscle and neuroblastoma cell lines [22,33]. Further, Errβ and Crh are expressed in similar regions of the hindbrain [29]. Here we demonstrate that Errβ deletion modulates corticosterone levels after exposure to restraint stress, with increased levels in Sox2-Cre:Errβ+/lox mice and decreased levels in Sox2-Cre:Errβlox/lox mice relative to WT (Figure 2). Neural connections projecting to the hypothalamus from extrahypothalamic sites, such as the hindbrain, may also regulate hypothalamic Crh release and Crh expression [30,49,68-70].
Biological activity of Crh is inhibited by Crhbp, and Errβ binds to the promoter region of the Crhbp gene [60,71], which contains three ERE half sites [72]. Mice that overexpress Crhbp have increased Crh expression, potentially resulting from a compensatory response aimed at ameliorating disruptions in stress response [73]. Similarly, increased Crh expression was observed when Errβ was reduced (Sox2-Cre:Errβ+/lox) or eliminated (Sox2-Cre:Errβ+/lox) in somatic tissue, and Errγ was activated using DY131 (Figure 3). Therefore, we propose that partial or complete deletion of Errβ may alter Crh expression by modulating transcription of Crhbp or Crhr2, resulting in altered corticosterone secretion. Furthermore, Sox2:Errβlox/lox mice lack corticosterone secretion after restraint stress (Figure 2), which may result from altered Crhr2 expression (Figure 4c) and changes in negative feedback. Therefore, brain regions that express Crhr2 may show reduced Crh signaling (Figure 4b and 4c), as in the hindbrain [64].
Errβ binds to cis-regulatory regions of the Cckbr gene [60], which is expressed in the hindbrain [29,74] and the corresponding gene maps to a genomic locus of the genome associated with obesity [75]. Cckbr deficient mice (Cckbr-/-) display a similar phenotype to Sox2-Cre:Errβ+/lox mice, and have increased body weight and food intake, which may arise from changes in Cholecystokinin (Cck) signaling (e.g. satiety), and increased metabolism [74,76]. However, Cckbr-/- mice also have blunted stress responses associated with anxiety-like behavior [77] and increased Npy expression [78], which resembles the phenotype of Sox2-Cre:Errβlox/lox mice (Figure 1 and Table 1). Therefore, heterodimers of Errβ alone, or Errβ in combination with ERRγ, may regulate Cckbr transcription, thereby partially accounting for the differences in the phenotypes seen in Sox2-Cre:Errβ+/lox and Sox2-Cre:Errβlox/lox mice (Table 2). Differences in developmental compensation arising from Errβ and/or Errγ may also contribute to the phenotype differences in Sox2-Cre:Errβ+/loxand Sox2-Cre:Errβlox/lox mice.
Nes-Cre:Errβlox/lox mice show increased Errγ expression relative to WT animals [14], while mice deficient for Errγ show increased Errβ expression [17]. This suggests that homozygous mice have reciprocal patterns of Errβ and Errγ expression, potentially arising from developmental compensation and heterozygous mice may partially lack this compensation, contributing to phenotype differences. The Errβ/Errγ agonist (DY131) increased Crh expression more when Errβ expression was reduced (Sox2:Errβ+/lox mice) than when Errβ expression was absent (Sox2:Errβlox/lox mice) (Figure 3). These results suggest that the ratio of Errβ to Errγ signaling may contribute to the observed difference in Crh expression, Crhr2 expression and corticosterone secretion in the two genotypes examined.
Sox2-Cre:Errβlox/lox and Sox2-Cre:Errβ+/lox mice have alterations in the HPA axis (Figures 2 and 3). Npy, which modulates corticosterone levels [79], is altered in Sox2-Cre:Errβ+/lox and Sox2-Cre:Errβlox/lox (Figures 1 and 2). Both Crh and Npy have been implicated in modulating the acoustic startle response [32,36,40,80], which is altered in Nes-Cre:Errβlox/lox mice (Figure 4 and [14]). Given the results reported here, the phenotype differences between Sox2-Cre:Errβ+/lox and Sox2-Cre:Errβlox/lox mice may specifically arise from altered Crh expression and corticosterone levels as a result of changes in Errβ-dependent regulation of Crhbp or Crhr2 transcription, as well as through interactions of Errβ with Errγ. However, since little is known about Errβ/Errγ heterodimers or how different Err family homo and heterodimers may potentially regulate Crhbp or Crhr2 transcription deserves further investigation.
Our data suggest that central Errβ modulates stress responses, food intake and body weight, although it remains to be determined whether peripheral Errβ also modulates components of the HPA axis and acoustic startle response. Nes-Cre:Errβlox/lox mice lack Errβ in the hindbrain and have decreased expression of Crh, Crhr2 and Npy[14], suggesting that neuromodulators involved with the acoustic startle response reside in the hindbrain to modulate stress and anxiety. However, other changes in neural circuitry (e.g. altered Cckbr expression) regulating the acoustic startle response in Nes-Cre:Errβlox/lox mice are likely to exist and remain to be identified.
Conclusions
Mice heterozygous for Errβ deletion have increased fat mass and stress hormone secretion after restraint stress, while those homozygous for Errβ deletion have decreased fat mass and secrete higher baseline levels of stress hormones. These effects may be modulated by components of the HPA axis, such as Crh, Crhbp, Crhr2, Npy or Cckrb. Central Errβ signaling influences stress associated behavior (e.g. the acoustic startle response), possibly through regulation of Npy, Crh and Crhr2 expression in the hindbrain or hypothalamic projections to the amygdala [32,62,63,80]. Since the neural circuitry controlling the acoustic startle response is well-conserved between rodents and humans [36,81], these data suggest that ERRβ or ERRγ may be promising candidates for pharmacological treatment of excessive anxiety or stress levels in humans.
Methods
Animals, housing, food intake, and physical activity measurement
Sox2-Cre:Errβlox/lox, Sox2-Cre:Errβ+/lox, and wild-type (WT) (Errβlox/lox) mice were generated as previously described [26]. Briefly, Errβ mice have a conditional allele, with loxP sites flanking exon 2 of the Errβ gene that encodes the DNA binding domain (exon 2) [26]. Expression of cre recombinase will excise the loxP-flanked exon 2 from the Errβ gene. Sox2-Cre deletes Errβ from all embryonic tissues and Nestin-Cre deletes Errβ from developing neural tissue. Sox2-Cre:Errβlox/lox mice completely lack functional Errβ because both alleles have been removed. Sox2-Cre:Errβ+/lox have one wild-type allele of the Errβ gene, since the other allele has been excised by the loxP sites. These two mouse lines enable us to address possible phenotypic differences due to differences in gene dosage. Wild-type (WT) mice used for these studies were homozygous for the floxed Errβ allele. Mice were maintained on a 12:12 hour light–dark cycle in a temperature- and humidity-regulated vivarium and had ad libitum access to standard laboratory chow (2018, Harlan-Teklad, Harlan Laboratories, Frederick, MD, USA) and water at all times. Different cohorts of mice were analyzed at three weeks and nine months of age. Food intake data and physical activity levels were collected as previously described [14]. Physical activity levels were measured by detecting and counting horizontal beam breaks in a 40 cm × 40 cm × 30 cm plexiglass chamber (Digiscan, Accuscan Instruments, Columbus, OH). All experimental procedures were performed in accordance with the Johns Hopkins University School of Medicine Institutional Animal Care and Use Committee and the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
In situ hybridization assay (ISH) and quantitative real-time PCR
ISH was performed as previously described [14,82]. Briefly, digoxigenin cRNA probes to Npy and agouti-related protein (Agrp) were synthesized using the Brain Molecular Anatomy Project (BMAP) library containing sequence-verified expressed sequence tags. BMAP clones were purified using a PureLink plasmid miniprep kit per manufacturer’s protocol (Invitrogen, Carlsbad, CA, USA) and synthesized using a T3 or T7 RNA polymerase (Roche, Indianapolis, IN, USA). The riboprobe was purified using an RNA extraction kit per manufacturer’s protocol (RNeasy, Qiagen, Valencia, CA, USA). Brains were collected from mice, fresh frozen in OCT compound (Tissue Tek, Fisher Scientific, Pittsburgh, PA, USA), and cut using a cryostat into 25-μm sections onto Superfrost Plus slides (Fisher Scientific, Pittsburgh, PA, USA). Hindbrain dissection, mRNA extraction and quantitative real-time PCR was conducted as previously described [14,83]. Briefly, RNA was extracted (RNeasy, Qiagen, Valencia, CA, USA) and cDNA was synthesized using 1 μg of mRNA using Superscript II reverse transcriptase (Invitrogen) and random primers (Invitrogen). Quantitative PCR primer sequences were obtained from PrimerBank and conducted for Crh: fwd – 5’ CCTCAGCCGGTTCTGATCC 3’ and rev – 5’ GCGGAAAAAGTTAGCCGCAG 3’, Crhr2: fwd – 5’ CATCCACCACGTCCGAGAC 3’ and rev – 5’ CTCGCCAGGATTGACAAAGAA 3’ and 18S fwd – 5’ GCAATTATTCCCCATGAACG 3’ and rev- 5’ GGCCTCACTAAACCATCCAA 3’. The Ct value generated was normalized to 18S in order to obtain a ΔCt value, followed by generating the 2-ΔΔCt value by normalizing the data to control animals as previously described [84].
Restraint stress test, corticosterone radioimmunoassay, and DY131 injections
Baseline blood glucocorticoid levels were measured and mice were placed into a restraining tube (one mouse/tube) for one hour. Upon removal from the restraining tube, blood samples were collected again. Animals were then returned to their housing and blood samples were collected after a one-hour recovery period. Blood was collected in heparin-coated tubes and centrifuged at 3800 rpm for 20 min at 4°C. Corticosterone assays were performed with a radioimmunoassay kit for corticosterone per manufacturer’s directions (MP Biomedicals, Solon, OH, USA). DY131 (Tocris, Bristol, BS11, United Kingdom) at a dose of 10 μM/g body weight was injected, and data for meal patterns collected as previously described [14].
Prepulse inhibition (PPI) of acoustic startle response
Startle reactivity and PPI were measured using two startle chambers located inside a sound-attenuating chamber (San Diego Instruments, San Diego, CA, USA). Mice were placed in a Plexiglass tube within the soundproof PPI box for a five-minute acclimation period, which provides exposure to a continuous background noise (70 dB) to elicit an increase in startle amplitude [43]. Mice were then exposed for five minutes without any startle stimulus. The PPI session then began and mice were randomly exposed to the following trials: pulse alone (120 dB), no stimulus, or five prepulse combinations (a 20 ms non-startling prepulse at 74, 78, 82, 86, or 90 dB, followed by an 80 ms startle stimulus at 120 dB). The force from the startle reaction was recorded by an accelerometer with SR-LAB software (San Diego Instruments). Results were analyzed by PPI percentage, which was calculated as:
Statistical analysis
All value comparisons were made using one-way ANOVA to identify individual differences between groups, and P < 0.05 was considered significant (Statistica v.8.0, Tulsa, OK, USA).
Abbreviations
ACTH: Adrenocorticotropic hormone; Agrp: Agouti-related protein; Crh: Corticotropin-releasing hormone; Crhbp: Corticotropin releasing hormone binding protein; Crhr2: Corticotropin releasing hormone receptor 2; ERR: Estrogen-related receptor; ERRE: Estrogen-related receptor response element; HPA: Hypothalamic-pituitary-adrenal axis; IMI: Inter meal interval; ISH: In situ hybridization; Npy: Neuropeptide Y; PPI: Prepulse inhibition; WT: Wild-type.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MSB, GWW, and SB; MSB conducted all research; RDS provided technical support for measuring corticosterone levels; MSB analyzed data; MSB drafted the manuscript and MSB, GWW, and SB edited the final version. All authors read and approved the final manuscript.
Contributor Information
Mardi S Byerly, Email: mardibyerly@gmail.com.
Roy D Swanson, Email: royswanson11@gmail.com.
G William Wong, Email: gwong9@jhmi.edu.
Seth Blackshaw, Email: sblack@jhmi.edu.
Acknowledgements
MSB is supported by the National Institute of Diabetes and Digestive and Kidney Diseases training fellowship (T32DK007751), GWW by the National Institutes of Health (DK084171) and the American Heart Association (SDG2260721), and SB by a WM Keck Distinguished Young Scholar Award in Medical Research.
References
- Giguere V. Transcriptional control of energy homeostasis by the estrogen-related receptors. Endocr Rev. 2008;13:677–696. doi: 10.1210/er.2008-0017. [DOI] [PubMed] [Google Scholar]
- Giguere V, Yang N, Segui P, Evans RM. Identification of a new class of steroid hormone receptors. Nature. 1988;13:91–94. doi: 10.1038/331091a0. [DOI] [PubMed] [Google Scholar]
- Pettersson K, Svensson K, Mattsson R, Carlsson B, Ohlsson R, Berkenstam A. Expression of a novel member of estrogen response element-binding nuclear receptors is restricted to the early stages of chorion formation during mouse embryogenesis. Mech Dev. 1996;13:211–223. doi: 10.1016/0925-4773(95)00479-3. [DOI] [PubMed] [Google Scholar]
- Lu D, Kiriyama Y, Lee KY, Giguere V. Transcriptional regulation of the estrogen-inducible pS2 breast cancer marker gene by the ERR family of orphan nuclear receptors. Cancer Res. 2001;13:6755–6761. [PubMed] [Google Scholar]
- Johnston SD, Liu X, Zuo F, Eisenbraun TL, Wiley SR, Kraus RJ, Mertz JE. Estrogen-related receptor alpha 1 functionally binds as a monomer to extended half-site sequences including ones contained within estrogen-response elements. Mol Endocrinol. 1997;13:342–352. doi: 10.1210/me.11.3.342. [DOI] [PubMed] [Google Scholar]
- Vanacker JM, Pettersson K, Gustafsson JA, Laudet V. Transcriptional targets shared by estrogen receptor- related receptors (ERRs) and estrogen receptor (ER) alpha, but not by ERbeta. Embo J. 1999;13:4270–4279. doi: 10.1093/emboj/18.15.4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanacker JM, Bonnelye E, Chopin-Delannoy S, Delmarre C, Cavailles V, Laudet V. Transcriptional activities of the orphan nuclear receptor ERR alpha (estrogen receptor-related receptor-alpha) Mol Endocrinol. 1999;13:764–773. doi: 10.1210/me.13.5.764. [DOI] [PubMed] [Google Scholar]
- Deblois G, Hall JA, Perry MC, Laganiere J, Ghahremani M, Park M, Hallett M, Giguere V. Genome-wide identification of direct target genes implicates estrogen-related receptor alpha as a determinant of breast cancer heterogeneity. Cancer Res. 2009;13:6149–6157. doi: 10.1158/0008-5472.CAN-09-1251. [DOI] [PubMed] [Google Scholar]
- Huppunen J, Aarnisalo P. Dimerization modulates the activity of the orphan nuclear receptor ERRgamma. Biochem Biophys Res Commun. 2004;13:964–970. doi: 10.1016/j.bbrc.2003.12.194. [DOI] [PubMed] [Google Scholar]
- Gearhart MD, Holmbeck SM, Evans RM, Dyson HJ, Wright PE. Monomeric complex of human orphan estrogen related receptor-2 with DNA: a pseudo-dimer interface mediates extended half-site recognition. J Mol Biol. 2003;13:819–832. doi: 10.1016/S0022-2836(03)00183-9. [DOI] [PubMed] [Google Scholar]
- Giguere V. To ERR in the estrogen pathway. Trends Endocrinol Metab. 2002;13:220–225. doi: 10.1016/S1043-2760(02)00592-1. [DOI] [PubMed] [Google Scholar]
- Chen F, Zhang Q, McDonald T, Davidoff MJ, Bailey W, Bai C, Liu Q, Caskey CT. Identification of two hERR2-related novel nuclear receptors utilizing bioinformatics and inverse PCR. Gene. 1999;13:101–109. doi: 10.1016/S0378-1119(98)00619-2. [DOI] [PubMed] [Google Scholar]
- Eudy JD, Yao S, Weston MD, Ma-Edmonds M, Talmadge CB, Cheng JJ, Kimberling WJ, Sumegi J. Isolation of a gene encoding a novel member of the nuclear receptor superfamily from the critical region of Usher syndrome type IIa at 1q41. Genomics. 1998;13:382–384. doi: 10.1006/geno.1998.5345. [DOI] [PubMed] [Google Scholar]
- Byerly MS, Al Salayta M, Swanson RD, Kwon K, Peterson JM, Wei Z, Aja S, Moran TH, Blackshaw S, Wong GW. Estrogen-related receptor beta deletion modulates whole-body energy balance via estrogen-related receptor gamma and attenuates neuropeptide Y gene expression. Eur J Neurosci. 2013;13:1033–1047. doi: 10.1111/ejn.12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog B, Cardenas J, Hall RK, Villena JA, Budge PJ, Giguere V, Granner DK, Kralli A. Estrogen-related receptor alpha is a repressor of phosphoenolpyruvate carboxykinase gene transcription. J Biol Chem. 2006;13:99–106. doi: 10.1074/jbc.M509276200. [DOI] [PubMed] [Google Scholar]
- Luo J, Sladek R, Bader JA, Matthyssen A, Rossant J, Giguere V. Placental abnormalities in mouse embryos lacking the orphan nuclear receptor ERR-beta. Nature. 1997;13:778–782. doi: 10.1038/42022. [DOI] [PubMed] [Google Scholar]
- Dufour CR, Wilson BJ, Huss JM, Kelly DP, Alaynick WA, Downes M, Evans RM, Blanchette M, Giguere V. Genome-wide orchestration of cardiac functions by the orphan nuclear receptors ERRalpha and gamma. Cell Metab. 2007;13:345–356. doi: 10.1016/j.cmet.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Alaynick WA, Kondo RP, Xie W, He W, Dufour CR, Downes M, Jonker JW, Giles W, Naviaux RK, Giguere V. et al. ERRgamma directs and maintains the transition to oxidative metabolism in the postnatal heart. Cell Metab. 2007;13:13–24. doi: 10.1016/j.cmet.2007.06.007. [DOI] [PubMed] [Google Scholar]
- Miller DB, O'Callaghan JP. Neuroendocrine aspects of the response to stress. Metabolism. 2002;13:5–10. doi: 10.1053/meta.2002.33184. [DOI] [PubMed] [Google Scholar]
- Mitchell AL, Pearce SH. Autoimmune Addison disease: pathophysiology and genetic complexity. Nat Rev Endocrinol. 2012;13:306–316. doi: 10.1038/nrendo.2011.245. [DOI] [PubMed] [Google Scholar]
- Napier C, Pearce SH. Autoimmune Addison's disease. Presse Med. 2012;13:e626–e635. doi: 10.1016/j.lpm.2012.09.010. [DOI] [PubMed] [Google Scholar]
- Trapp T, Holsboer F. Nuclear orphan receptor as a repressor of glucocorticoid receptor transcriptional activity. J Biol Chem. 1996;13:9879–9882. doi: 10.1074/jbc.271.17.9879. [DOI] [PubMed] [Google Scholar]
- Bjorntorp P. Do stress reactions cause abdominal obesity and comorbidities? Obes Rev. 2001;13:73–86. doi: 10.1046/j.1467-789x.2001.00027.x. [DOI] [PubMed] [Google Scholar]
- Erickson JC, Ahima RS, Hollopeter G, Flier JS, Palmiter RD. Endocrine function of neuropeptide Y knockout mice. Regul Pept. 1997;13:199–202. doi: 10.1016/S0167-0115(97)01007-0. [DOI] [PubMed] [Google Scholar]
- Frankish HM, Dryden S, Hopkins D, Wang Q, Williams G. Neuropeptide Y, the hypothalamus, and diabetes: insights into the central control of metabolism. Peptides. 1995;13:757–771. doi: 10.1016/0196-9781(94)00200-P. [DOI] [PubMed] [Google Scholar]
- Chen J, Nathans J. Estrogen-related receptor beta/NR3B2 controls epithelial cell fate and endolymph production by the stria vascularis. Dev Cell. 2007;13:325–337. doi: 10.1016/j.devcel.2007.07.011. [DOI] [PubMed] [Google Scholar]
- Onishi A, Peng GH, Poth EM, Lee DA, Chen J, Alexis U, de Melo J, Chen S, Blackshaw S. The orphan nuclear hormone receptor ERRbeta controls rod photoreceptor survival. Proc Natl Acad Sci U S A. 2010;13:11579–11584. doi: 10.1073/pnas.1000102107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gofflot F, Chartoire N, Vasseur L, Heikkinen S, Dembele D, Le Merrer J, Auwerx J. Systematic gene expression mapping clusters nuclear receptors according to their function in the brain. Cell. 2007;13:405–418. doi: 10.1016/j.cell.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ. et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;13:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Grill HJ, Hayes MR. Hindbrain neurons as an essential hub in the neuroanatomically distributed control of energy balance. Cell Metab. 2012;13:296–309. doi: 10.1016/j.cmet.2012.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray TS, Cassell MD, Kiss JZ. Distribution of pro-opiomelanocortin-derived peptides and enkephalins in the rat central nucleus of the amygdala. Brain Res. 1984;13:354–358. doi: 10.1016/0006-8993(84)90386-X. [DOI] [PubMed] [Google Scholar]
- Yang FC, Connor J, Patel A, Doat MM, Romero MT. Neural transplants. effects On startle responses in neonatally MSG-treated rats. Physiol Behav. 2000;13:333–344. doi: 10.1016/S0031-9384(99)00256-5. [DOI] [PubMed] [Google Scholar]
- Wang SC, Myers S, Dooms C, Capon R, Muscat GE. An ERRbeta/gamma agonist modulates GRalpha expression, and glucocorticoid responsive gene expression in skeletal muscle cells. Mol Cell Endocrinol. 2010;13:146–152. doi: 10.1016/j.mce.2009.07.012. [DOI] [PubMed] [Google Scholar]
- Yu DD, Forman BM. Identification of an agonist ligand for estrogen-related receptors ERRbeta/gamma. Bioorg Med Chem Lett. 2005;13:1311–1313. doi: 10.1016/j.bmcl.2005.01.025. [DOI] [PubMed] [Google Scholar]
- Plappert CF, Pilz PK. The acoustic startle response as an effective model for elucidating the effect of genes on the neural mechanism of behavior in mice. Behav Brain Res. 2001;13:183–188. doi: 10.1016/S0166-4328(01)00299-6. [DOI] [PubMed] [Google Scholar]
- Koch M. The neurobiology of startle. Prog Neurobiol. 1999;13:107–128. doi: 10.1016/S0301-0082(98)00098-7. [DOI] [PubMed] [Google Scholar]
- Lee Y, Lopez DE, Meloni EG, Davis M. A primary acoustic startle pathway: obligatory role of cochlear root neurons and the nucleus reticularis pontis caudalis. J Neurosci. 1996;13:3775–3789. doi: 10.1523/JNEUROSCI.16-11-03775.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M, Falls WA, Campeau S, Kim M. Fear-potentiated startle: a neural and pharmacological analysis. Behav Brain Res. 1993;13:175–198. doi: 10.1016/0166-4328(93)90102-V. [DOI] [PubMed] [Google Scholar]
- Inglefield JR, Schwarzkopf SB, Kellogg CK. Alterations in behavioral responses to stressors following excitotoxin lesions of dorsomedial hypothalamic regions. Brain Res. 1994;13:151–161. doi: 10.1016/0006-8993(94)91534-2. [DOI] [PubMed] [Google Scholar]
- Liang KC, Melia KR, Campeau S, Falls WA, Miserendino MJ, Davis M. Lesions of the central nucleus of the amygdala, but not the paraventricular nucleus of the hypothalamus, block the excitatory effects of corticotropin-releasing factor on the acoustic startle reflex. J Neurosci. 1992;13:2313–2320. doi: 10.1523/JNEUROSCI.12-06-02313.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plappert CF, Pilz PK, Schnitzler HU. Factors governing prepulse inhibition and prepulse facilitation of the acoustic startle response in mice. Behav Brain Res. 2004;13:403–412. doi: 10.1016/j.bbr.2003.10.025. [DOI] [PubMed] [Google Scholar]
- Hoffman HS, Ison JR. Reflex modification in the domain of startle: I. Some empirical findings and their implications for how the nervous system processes sensory input. Psychol Rev. 1980;13:175–189. [PubMed] [Google Scholar]
- Leumann L, Sterchi D, Vollenweider F, Ludewig K, Fruh H. A neural network approach to the acoustic startle reflex and prepulse inhibition. Brain Res Bull. 2001;13:101–110. doi: 10.1016/S0361-9230(01)00607-4. [DOI] [PubMed] [Google Scholar]
- Yeomans JS, Frankland PW. The acoustic startle reflex: neurons and connections. Brain Res Brain Res Rev. 1995;13:301–314. doi: 10.1016/0165-0173(96)00004-5. [DOI] [PubMed] [Google Scholar]
- Heilig M, Koob GF, Ekman R, Britton KT. Corticotropin-releasing factor and neuropeptide Y: role in emotional integration. Trends Neurosci. 1994;13:80–85. doi: 10.1016/0166-2236(94)90079-5. [DOI] [PubMed] [Google Scholar]
- Fendt M, Koch M, Schnitzler HU. NMDA receptors in the pontine brainstem are necessary for fear potentiation of the startle response. Eur J Pharmacol. 1996;13:1–6. doi: 10.1016/S0014-2999(96)00749-2. [DOI] [PubMed] [Google Scholar]
- Alon T, Zhou L, Perez CA, Garfield AS, Friedman JM, Heisler LK. Transgenic mice expressing green fluorescent protein under the control of the corticotropin-releasing hormone promoter. Endocrinology. 2009;13:5626–5632. doi: 10.1210/en.2009-0881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meloni EG, Gerety LP, Knoll AT, Cohen BM, Carlezon WA Jr. Behavioral and anatomical interactions between dopamine and corticotropin-releasing factor in the rat. J Neurosci. 2006;13:3855–3863. doi: 10.1523/JNEUROSCI.4957-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan AM, Kaminski KL, Sanchez-Watts G, Ponzio TA, Kuzmiski JB, Bains JS, Watts AG. MAP kinases couple hindbrain-derived catecholamine signals to hypothalamic adrenocortical control mechanisms during glycemia-related challenges. J Neurosci. 2011;13:18479–18491. doi: 10.1523/JNEUROSCI.4785-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel HR, Qi Y, Hawkins EJ, Hileman SM, Elmquist JK, Imai Y, Ahima RS. Neuropeptide Y deficiency attenuates responses to fasting and high-fat diet in obesity-prone mice. Diabetes. 2006;13:3091–3098. doi: 10.2337/db05-0624. [DOI] [PubMed] [Google Scholar]
- Segal-Lieberman G, Trombly DJ, Juthani V, Wang X, Maratos-Flier E. NPY ablation in C57BL/6 mice leads to mild obesity and to an impaired refeeding response to fasting. Am J Physiol Endocrinol Metab. 2003;13:E1131–E1139. doi: 10.1152/ajpendo.00491.2002. [DOI] [PubMed] [Google Scholar]
- Wortley KE, Anderson KD, Yasenchak J, Murphy A, Valenzuela D, Diano S, Yancopoulos GD, Wiegand SJ, Sleeman MW. Agouti-related protein-deficient mice display an age-related lean phenotype. Cell Metab. 2005;13:421–427. doi: 10.1016/j.cmet.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Sainsbury A, Cusin I, Doyle P, Rohner-Jeanrenaud F, Jeanrenaud B. Intracerebroventricular administration of neuropeptide Y to normal rats increases obese gene expression in white adipose tissue. Diabetologia. 1996;13:353–356. doi: 10.1007/BF00418353. [DOI] [PubMed] [Google Scholar]
- Schwartz MW, Baskin DG, Bukowski TR, Kuijper JL, Foster D, Lasser G, Prunkard DE, Porte D Jr, Woods SC, Seeley RJ. et al. Specificity of leptin action on elevated blood glucose levels and hypothalamic neuropeptide Y gene expression in ob/ob mice. Diabetes. 1996;13:531–535. doi: 10.2337/diab.45.4.531. [DOI] [PubMed] [Google Scholar]
- Schwartz MW, Seeley RJ, Campfield LA, Burn P, Baskin DG. Identification of targets of leptin action in rat hypothalamus. J Clin Invest. 1996;13:1101–1106. doi: 10.1172/JCI118891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian M, White R, Parker MG. Metabolic regulation by the nuclear receptor corepressor RIP140. Trends Endocrinol Metab. 2006;13:243–250. doi: 10.1016/j.tem.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Rosell M, Jones MC, Parker MG. Role of nuclear receptor corepressor RIP140 in metabolic syndrome. Biochim Biophys Acta. 2011;13:919–928. doi: 10.1016/j.bbadis.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonardsson G, Steel JH, Christian M, Pocock V, Milligan S, Bell J, So PW, Medina-Gomez G, Vidal-Puig A, White R. et al. Nuclear receptor corepressor RIP140 regulates fat accumulation. Proc Natl Acad Sci USA. 2004;13:8437–8442. doi: 10.1073/pnas.0401013101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castet A, Herledan A, Bonnet S, Jalaguier S, Vanacker JM, Cavailles V. Receptor-interacting protein 140 differentially regulates estrogen receptor-related receptor transactivation depending on target genes. Mol Endocrinol. 2006;13:1035–1047. doi: 10.1210/me.2005-0227. [DOI] [PubMed] [Google Scholar]
- Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J. et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;13:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- Meloni EG, Reedy CL, Cohen BM, Carlezon WA Jr. Activation of raphe efferents to the medial prefrontal cortex by corticotropin-releasing factor: correlation with anxiety-like behavior. Biol Psychiatry. 2008;13:832–839. doi: 10.1016/j.biopsych.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons AM, Thiele TE. Neuropeptide Y conjugated to saporin alters anxiety-like behavior when injected into the central nucleus of the amygdala or basomedial hypothalamus in BALB/cJ mice. Peptides. 2010;13:2193–2199. doi: 10.1016/j.peptides.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deo GS, Dandekar MP, Upadhya MA, Kokare DM, Subhedar NK. Neuropeptide Y Y1 receptors in the central nucleus of amygdala mediate the anxiolytic-like effect of allopregnanolone in mice: Behavioral and immunocytochemical evidences. Brain Res. 2010;13:77–86. doi: 10.1016/j.brainres.2009.12.088. [DOI] [PubMed] [Google Scholar]
- Chalmers DT, Lovenberg TW, De Souza EB. Localization of novel corticotropin-releasing factor receptor (CRF2) mRNA expression to specific subcortical nuclei in rat brain: comparison with CRF1 receptor mRNA expression. J Neurosci. 1995;13:6340–6350. doi: 10.1523/JNEUROSCI.15-10-06340.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grill HJ, Markison S, Ginsberg A, Kaplan JM. Long-term effects on feeding and body weight after stimulation of forebrain or hindbrain CRH receptors with urocortin. Brain Res. 2000;13:19–28. doi: 10.1016/S0006-8993(00)02193-4. [DOI] [PubMed] [Google Scholar]
- Bledsoe AC, Oliver KM, Scholl JL, Forster GL. Anxiety states induced by post-weaning social isolation are mediated by CRF receptors in the dorsal raphe nucleus. Brain Res Bull. 2011;13:117–122. doi: 10.1016/j.brainresbull.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammack SE, Pepin JL, DesMarteau JS, Watkins LR, Maier SF. Low doses of corticotropin-releasing hormone injected into the dorsal raphe nucleus block the behavioral consequences of uncontrollable stress. Behav Brain Res. 2003;13:55–64. doi: 10.1016/S0166-4328(03)00133-5. [DOI] [PubMed] [Google Scholar]
- Grill HJ, Kaplan JM. The neuroanatomical axis for control of energy balance. Front Neuroendocrinol. 2002;13:2–40. doi: 10.1006/frne.2001.0224. [DOI] [PubMed] [Google Scholar]
- Grill HJ. Distributed neural control of energy balance: contributions from hindbrain and hypothalamus. Obesity (Silver Spring) 2006;13(Suppl 5):216S–221S. doi: 10.1038/oby.2006.312. [DOI] [PubMed] [Google Scholar]
- Sawchenko PE, Swanson LW. Central noradrenergic pathways for the integration of hypothalamic neuroendocrine and autonomic responses. Science. 1981;13:685–687. doi: 10.1126/science.7292008. [DOI] [PubMed] [Google Scholar]
- Potter E, Behan DP, Linton EA, Lowry PJ, Sawchenko PE, Vale WW. The central distribution of a corticotropin-releasing factor (CRF)-binding protein predicts multiple sites and modes of interaction with CRF. Proc Natl Acad Sci U S A. 1992;13:4192–4196. doi: 10.1073/pnas.89.9.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behan DP, Potter E, Lewis KA, Jenkins NA, Copeland N, Lowry PJ, Vale WW. Cloning and structure of the human corticotrophin releasing factor-binding protein gene (CRHBP) Genomics. 1993;13:63–68. doi: 10.1006/geno.1993.1141. [DOI] [PubMed] [Google Scholar]
- Burrows HL, Nakajima M, Lesh JS, Goosens KA, Samuelson LC, Inui A, Camper SA, Seasholtz AF. Excess corticotropin releasing hormone-binding protein in the hypothalamic-pituitary-adrenal axis in transgenic mice. J Clin Invest. 1998;13:1439–1447. doi: 10.1172/JCI1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerc P, Coll Constans MG, Lulka H, Broussaud S, Guigne C, Leung-Theung-Long S, Perrin C, Knauf C, Carpene C, Penicaud L. et al. Involvement of cholecystokinin 2 receptor in food intake regulation: hyperphagia and increased fat deposition in cholecystokinin 2 receptor-deficient mice. Endocrinology. 2007;13:1039–1049. doi: 10.1210/en.2006-1064. [DOI] [PubMed] [Google Scholar]
- Samuelson LC, Isakoff MS, Lacourse KA. Localization of the murine cholecystokinin A and B receptor genes. Mamm Genome. 1995;13:242–246. doi: 10.1007/BF00352408. [DOI] [PubMed] [Google Scholar]
- Miyasaka K, Ichikawa M, Ohta M, Kanai S, Yoshida Y, Masuda M, Nagata A, Matsui T, Noda T, Takiguchi S. et al. Energy metabolism and turnover are increased in mice lacking the cholecystokinin-B receptor. J Nutr. 2002;13:739–741. doi: 10.1093/jn/132.4.739. [DOI] [PubMed] [Google Scholar]
- Horinouchi Y, Akiyoshi J, Nagata A, Matsushita H, Tsutsumi T, Isogawa K, Noda T, Nagayama H. Reduced anxious behavior in mice lacking the CCK2 receptor gene. Eur Neuropsychopharmacol. 2004;13:157–161. doi: 10.1016/S0924-977X(03)00103-2. [DOI] [PubMed] [Google Scholar]
- Chen H, Kent S, Morris MJ. Is the CCK2 receptor essential for normal regulation of body weight and adiposity? Eur J Neurosci. 2006;13:1427–1433. doi: 10.1111/j.1460-9568.2006.05016.x. [DOI] [PubMed] [Google Scholar]
- Leibowitz SF, Sladek C, Spencer L, Tempel D. Neuropeptide Y, epinephrine and norepinephrine in the paraventricular nucleus: stimulation of feeding and the release of corticosterone, vasopressin and glucose. Brain Res Bull. 1988;13:905–912. doi: 10.1016/0361-9230(88)90025-1. [DOI] [PubMed] [Google Scholar]
- Gutman AR, Yang Y, Ressler KJ, Davis M. The role of neuropeptide Y in the expression and extinction of fear-potentiated startle. J Neurosci. 2008;13:12682–12690. doi: 10.1523/JNEUROSCI.2305-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CaH Landis WA. The Startle Pattern. New York: Farrar and Rinehart; 1939. [Google Scholar]
- Blackshaw S, Snyder SH. Developmental expression pattern of phototransduction components in mammalian pineal implies a light-sensing function. J Neurosci. 1997;13:8074–8082. doi: 10.1523/JNEUROSCI.17-21-08074.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byerly MS, Simon J, Lebihan-Duval E, Duclos MJ, Cogburn LA, Porter TE. Effects of BDNF, T3, and corticosterone on expression of the hypothalamic obesity gene network in vivo and in vitro. Am J Physiol Regul Integr Comp Physiol. 2009;13:R1180–R1189. doi: 10.1152/ajpregu.90813.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;13:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]