

Abstract
Six novel C9-methyl-5-substituted-2,4-diaminopyrrolo[2,3-d]pyrimidines 18–23 were synthesized as potential inhibitors of dihydrofolate reductase (DHFR) and as anti-opportunistic agents. These compounds represent the only examples of 9-methyl substitution in the carbon–carbon bridge of 2,4-diaminopyrrolo[2,3-d]pyrimidines. The analogs 18–23 were synthesized in a concise eight-step procedure starting from the appropriate commercially available aromatic methyl ketones. The key step involved a Michael addition reaction of 2,4,6-triaminopyrimidine to the appropriate 1-nitroalkene, followed by ring closure of the nitro adducts via a Nef reaction. The compounds were evaluated as inhibitors of DHFR from Pneumocystis carinii (pc), Toxoplasma gondii (tg), Mycobacterium avium (ma) and rat liver (rl). The biological result indicated that some of these analogs are potent inhibitors of DHFR and some have selectivity for pathogen DHFR. Compound 23 was a two digit nanomolar inhibitor of tgDHFR with 9.6-fold selectivity for tgDHFR.
1. Introduction
Opportunistic infections caused by pathogenic organisms such as Pneumocystis, Toxoplasma gondii (tg), and Mycobacterium avium (ma) are the principal cause of morbidity and mortality in AIDS patients.2 Inhibitors of dihydrofolate reductase (DHFR) from Pneumocystis and Toxoplasma are useful anti-opportunistic agents but DHFR inhibitors have yet to be clinically applied for M. avium infections. DHFR inhibitors that display potent activity and high selectivity for these pathogenic organisms versus human DHFR remain elusive for the treatment of opportunistic infection in AIDS patients. Infections caused by Pneumocystis or Toxoplasma are currently treated with weak but selective DHFR inhibitors such as trimethoprim (TMP 1) and pyrimethamine (PYR 2) (Fig. 1), and their action is augmented with sulfonamides. The combination therapy however often results in severe side effects to sulfa drugs and leads to discontinuation of treatment.3 Trimetrexate (TMQ, 3) and piretrexim (PTX, 4) (Fig. 1) are potent inhibitors of both pathogenic and human DHFR which results in high toxicity to host cells. Hence 3 and 4 are used in combination with leucovorin (N5-formyltetrahydrofolate) which is necessary to rescue the host cells.4 Thus the development of agents that display both high potency and selectivity for pathogenic DHFR remains a desirable goal for the treatment of opportunistic infections in AIDS patients.
Figure 1.

Known inhibitors of pathogenic DHFR.
Gangjee et al.5–10 reported two-atom bridged classical and non-classical, 6–5 bicyclic analogs, as potential DHFR inhibitors (Fig. 2). The structure–activity relationship (SAR) studies revealed that a N9- or C9-methyl group in the two-atom bridged side chain of the 6–5 system significantly contributes to the DHFR inhibitory activity and selectivity. Thus, in the furo[2,3-d]pyrimidine analogs (5–10) (Fig. 2), the N9- or C9-methyl compounds were more potent than the corresponding N9- or C9-desmethyl compounds. Molecular modeling using Sybyl 6.710 suggested that the 9-methyl moiety in furo[2,3-d]pyrimidines interacts with Val115 in human DHFR which may account for their increased potency against human DHFR. In addition, molecular modeling also indicated that the 9-methyl group restricts the rotation of the C8–C9 bond (compounds 7 and 8) and leads to a decreased number of accessible low energy conformations conducive for binding to human DHFR.10 Thus the classical 9-methyl substituted compound 8 was twice as potent against human DHFR and displayed 10-fold greater activity in cell culture as compared to the corresponding desmethyl analog 7.10
Figure 2.

Two atom bridged furo- and pyrrolo[2,3-d] pyrimidines.
In the two-carbon atom bridged pyrrolo[2,3-d]pyrimidine series 11–14 (Fig. 2), N9-methylation of the classical compound 11 maintained the potency but increased the selectivity for tgDHFR. Similarly, the non-classical analog 14 was more potent than the corresponding desmethyl compound 13.5,6
Classical anti-folates such as 5–8, 11, 12, 15 and 16 (Fig. 2) have a polar l-glutamate side chain, and hence utilize carrier-mediated active transport mechanisms for uptake into cells. However, those pathogenic organisms that lack active transport mechanisms are not susceptible to classical anti-folates. Non-classical anti-folates without the polar l-glutamate side chain such as TMP, TMQ and PTX, are lipophilic and have been used to target these pathogenic microorganisms.
In the two-atom bridged 6–6 bicyclic system, a methyl group in the side chain of the bridge was reported to contribute to the DHFR inhibitory activity.11 Miwa et al.12 reported the classical pyrrolo[2,3-d]pyrimidine anti-folates 15 and 16 as DHFR inhibitors. These compounds possess a three-carbon atom bridge. The C10-methylated compound 16 was similar in activity to the desmethylated compound 15. Rosowsky et al.13 reported the two-carbon atom bridged non-classical pyrrolo[2,3-d]pyrimidine analog 17 as a potent inhibitor of DHFR. It was therefore of interest to introduce a methyl group at the C9-position of the bridge in pyrrolo[2,3-d]pyrimidines to determine the effect on inhibitory activity and selectivity against pathogenic DHFR. Thus compounds 18–23 (Fig. 2), were synthesized as potential inhibitors of DHFR and as potential anti-opportunistic agents. X-ray crystal structure of similar furo[2,3-d]pyrimidine analogs with pcDHFR indicated that the side chain phenyl substituent has a hydrophobic interaction with Phe69 in pcDHFR;9 similar interactions were anticipated with tgDHFR (Phe91) and maDHFR (Val58). These hydrophobic interactions would be absent with mammalian DHFR where an Asn64 replaces a Phe or Val. Thus large hydrophobic aromatic rings, such as naphthyl, fluorenyl, biphenyl and substituted phenyls were also chosen for compounds 19–21 to enhance the hydrophobic interactions with the pathogenic DHFR.
2. Results and discussion
Gangjee et al. reported7–10 the synthesis of two-carbon atom bridged furo[2,3-d]pyrimidines 5–10 by utilizing a Wittig reaction. It was anticipated that a similar Wittig reaction (Scheme 1) of compound I and substituted (1-chloro-ethyl)-benzene should afford intermediate III, which on hydrogenation would give the desired target compounds. However the Wittig reaction of I and (1-chloro-ethyl)-benzene under a variety of different conditions of time, base, temperature and solvent failed to afford the desired product III. A second synthetic strategy was devised in which compound I was to be converted to II by first reduction to the 5-hydroxymethyl analog followed by bromination. This was to be followed by a Wittig reaction with appropriately substituted ketones. However the reduction of I to the corresponding 5-hydroxymethyl analog could not be accomplished despite several attempts with a variety of reducing agents. Thus the proposed synthetic methodology in Scheme 1 was unsuccessful.
Scheme 1.

Attempted synthesis of target compounds.
The failure of the Wittig reaction, as well as the conversion of I to its 5-hydroxymethyl analog, prompted the search for an alternate synthetic procedure. Taylor and Liu14,15 had reported the synthesis of 2,4-diamino-pyrrolo[2,3-d]pyrimidines via a Nef reaction of a nitro intermediate. The key nitro intermediates were in turn obtained by a Michael addition of 5-unsubstituted-2,4,6-triaminopyrimidine and the appropriate 1-nitroalkenes. The 1-nitroalkenes were synthesized from appropriately substituted aldehydes and nitromethane by a Henry reaction (nitroaldol condensation).16 For the synthesis of target compounds 18–23 the appropriately substituted aldehydes were not commercially available. The synthesis of these aldehydes was first attempted using a Heck coupling (Scheme 2),17 however low yields (<10%) along with problems in purification of the desired product prompted the search for alternate synthetic strategies.
Scheme 2.
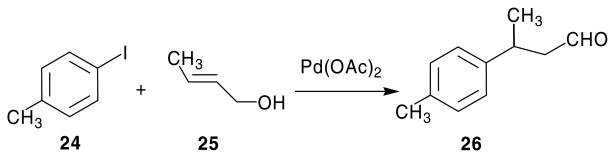
Heck coupling for the synthesis of 26.
Our alternate method was to generate the α,β-unsaturated aldehyde 30 via a Wittig reaction from 27, followed by reduction of the double bond to afford the desired aldehydes (Scheme 3). Unfortunately the initial Wittig reaction was not successful. At this stage it was decided to synthesize the esters 32–36,18 which could be reduced by LiAlH4 to the primary alcohols,19 followed by oxidation with PCC to aldehydes.20 The total yield for this two step procedure was reasonably good (40%) for 46, but purification of the compounds remained a problem, and the use of impure aldehyde for the subsequent step was not feasible. In view of the fact that the ester could be reduced to the corresponding aldehyde with DIBAL-H, an alterate synthetic approach was adopted in which the double bond of the α,β-unsaturated esters 32–36 were reduced by catalytic hydrogenation,21 followed by reduction with DIBAL-H to afford the aldehydes 42–46 in very high yield (two steps >80%, Scheme 3).22
Scheme 3.

Synthesis of aldehydes 42–46.
The Henry reaction of aldehydes 42–47 with nitromethane gave the expected nitro alcohol intermediates 48–53 (Scheme 4). This reaction was carried out under a variety of different reaction conditions using different bases (K2CO3, NaOEt, NaOMe, NaOH and KF), different solvents (MeOH, EtOH, THF, CHCl3 and i-PrOH), and different temperatures (from room temperature to heating to reflux).23–26 The optimized conditions in our hands involved using i-PrOH as the solvent, KF as a weak base and stirring at room temperature overnight to afford compounds 48–53 in >85% yield.27 Under these conditions the product was readily purified via column chromatography.
Scheme 4.

Synthesis of 18–23.
The formation of 1-nitroalkenes 54–59 was carried out under mild reaction conditions with MsCl and Et3N at 0 °C and stirring for one half hour to afford 54–59 in >90% yield.28,29 The Michael addition of 2,4,6-triaminopyrimidine to the 1-nitroalkenes 54–59 afforded the nitro adducts 60–65.30 The transformation of the nitro adduct to the carbonyl intermediate was the key step in forming the desired pyrrolo[2,3-d]pyrimidines. There are numerous Nef reaction conditions available for converting nitro adducts to carbonyl intermediates.31 However the one-pot conditions reported by Taylor and Liu.14,15 were particularly appealing. Thus the nitro adducts were first treated with aqueous NaOH solution followed by aqueous H2SO4 solution to afford the final products 18–23 (Scheme 4). The compounds were evaluated as inhibitors of pcDHFR, tgDHFR and maDHFR. Rat liver (rl) DHFR served as the mammalian standard.
Pneumocystis carinii
DHFR (Table 1): against pcDHFR the most potent analog bore a 2-naphthyl group (19). Increasing the bulk of the naphthyl to a fluorenyl group (20) or a biphenyl (21) caused a drop in activity as did the 3,4,5-triOMe substitution (23). However, the most detrimental substitution was the 2,5-diOMe (22), which was about 10-fold less potent than 19. The unsubstituted phenyl analog 18 was the least active. The compounds were not selective against pcDHFR and C9-methylation (23) did not improve activity compared to the desmethyl analog 17. In fact, the addition of a 9-CH3 to 17 caused a 3-fold decrease in activity. This result was unexpected since the 9-CH3 was expected to interact hydrophobically or via van der Walls interaction with the close Ile123 of pcDHFR.32 Perhaps the decrease in activity is due to an inability to accommodate both a bulky substitution at the C5-position along with a C9-CH3.
Table 1. Inhibitory concentration (IC50 μM) and selectivity ratios against rlDHFR, pcDHFR, tgDHFR and maDHFR versus rlDHFR by compounds 18–23a.
| Compound | pc | rl | rl/pc | tg | rl/tg | ma | rl/ma |
|---|---|---|---|---|---|---|---|
| 17b | 0.77 | 0.20 | 0.26 | 0.037 | 5.4 | 0.077 | 2.6 |
| 18 | 17.1 | 0.696 | 0.04 | 0.138 | 5 | 5.34 | 0.13 |
| 19 | 0.859 | 0.0399 | 0.05 | 0.0148 | 2.7 | 0.109 | 0.37 |
| 20 | 2.09 | 0.0567 | 0.03 | 0.0483 | 1.2 | 0.246 | 0.23 |
| 21 | 1.99 | 0.142 | 0.07 | 0.0717 | 2 | 0.0747 | 1.9 |
| 22 | 8.06 | 0.516 | 0.06 | 0.16 | 3.2 | 1.86 | 0.28 |
| 23 | 2.22 | 0.396 | 0.2 | 0.0414 | 9.6 | 0.321 | 1.23 |
| TMP | 12 | 180.0 | 14 | 2.8 | 65 | 0.30 | 600 |
| PTX | 0.013 | 0.0033 | 0.26 | 0.0043 | 0.76 | 0.00061 | 5.3 |
| TMQ | 0.042 | 0.003 | 0.07 | 0.01 | 0.3 | 0.0015 | 2.0 |
All enzymes were assayed spectroscopically in a solution containing 90 micromolar dihydrofolate, 119 μM NADPH, 41 mM Na phosphate buffer, and 8.9 mM 2-mercaptoethanol at pH 7.4 and 37 C. Rat liver and Toxoplasma DHFR are assayed in the presence of 150 mM KCl. Reactions are initiated with an amount of enzyme yielding a change in OD at 340 nM of 0.035/min.
The data for 17 was taken from Ref. 13.
Toxoplasma gondii
DHFR (Table 1): the compounds were significantly more potent and selective against tgDHFR compared to pcDHFR. The most potent compound was again the 2-naphthyl analog 19 which had an IC50 value of 15 nM. Compounds 20, 21 and 23 were almost as active, with IC50 values between 37 and 72 nM, indicating that bulk on the phenyl ring was highly conducive to tgDHFR inhibition. The unsubstituted phenyl analog 18 and the 2,5-dimethyl analog 22 were the least active compounds. Once again 9-CH3 substitution as in 23 did not significantly increase potency against tgDHFR compared to the desmethyl analog 17. Selectivity, though improved from that toward pcDHFR, was not significant, with the best compound 23 being 9-fold more selective compared to rlDHFR. This was not very different from the desmethyl analog.
Mycobacterium avium
DHFR (Table 1): against maDHFR the most potent analog was 23. All analogs were more potent against the bacterial maDHFR than against pcDHFR. Comparing 17 with 23 indicates that 9-CH3 substitution has a detrimental effect on maDHFR inhibition. There was no selectivity for maDHFR for this series of analogs. The most potent analogs were 17 and 21 indicating that neither N9-methylation nor bulk at C5 contributes significantly to activity against maDHFR.
Rat liver DHFR (Table 1): compounds 19 and 20 were potent nanomolar inhibitors of mammalian DHFR. These two analogs have bulky aromatic substitutions and were not expected to interact with rlDHFR which contain an Asn in place of Phe69 of pcDHFR. It is possible that the compounds bind to tgDHFR and to rlDHFR in different orientations that perhaps allow potent inhibitory activity for both analogs against both enzymes. Again there is no advantage of the 9-CH3 substitution over the 9-desmethyl compound.
In summary, the phenyl unsubstituted analog 18 and the 2,5-diOMe substituted analog 22 were the least potent DHFR inhibitors tested against the four enzymes. The most potent compound against rat liver DHFR and tgDHFR was the 2-naphthyl analog 19; compounds 17 and 19 were similar in potency against pcDHFR. The other analogs did not show any particular trend with respect to all DHFR, however differences in activities of the compounds against specific DHFR were noted. Direct comparison of the N9-CH3 analog 23 with the corresponding N9-desmethyl analog 17 indicates that the N9-CH3 group is detrimental to activity. Bulky substitutions on the C5-position did not afford significant increase in potency (except against maDHFR) or selectivity. Thus the SAR is different against different DHFR and the SAR of each enzyme target needs to be compared separately with the mammalian standard. In this limited series N9-methylation and C5-bulky substitutions does not afford both potent and selective inhibitors for any of the pathogenic DHFR evaluated. However, the 2 digit nanomolar potency of compounds 19 and 20 against tgDHFR and 21 against tgDHFR and maDHFR, indicates that potency can be maintained or improved with some bulky substitutions at C5 compared to the phenyl analog 18.
3. Experimental
Melting points were determined on a Mel-Temp II melting point apparatus with FLUKE 51 K/J electronic thermometer and were uncorrected. Nuclear magnetic resonance spectra for proton (1H) were recorded on a Bruker WH-300 (300 MHz) spectrometer. The chemical shift values were expressed in ppm (parts per million) relative to tetramethylsilane as internal standard; s = singlet, d = double, t = triplet, q = quartet, m = multiplet, br s = broad singlet. The relative integrals of peak areas agreed with those expected for the assigned structures. High-resolution mass spectra (HRMS), using electron impact (EI), were recorded on a VG AUTOSPEC (Fisons Instruments) micromass (EBE Geometry) double focusing mass spectrometer. Thin-layer chromatography (TLC) was performed on POLYGRAM Sil G/UV254 silica gel plates with fluorescent indicator, and the spots were visualized under 254 and 366 nm illumination. Proportions of solvents used for TLC were by volume. Column chromatography was performed on 230–400 mesh silica gel purchased from Aldrich Chemical Co., Milwaukee, WI. All evaporations were carried out in vacuo with a rotary evaporator. Analytical samples were dried in vacuo (0.2 mmHg) in an Abderhalden drying apparatus over P2O5 at 75–110 °C. Elemental analysis was performed by Altlantic Microlabs, Norcross, GA. Element compositions are within ±0.4% of calculated values. Fractional moles of water or organic solvents frequently found in some analytical samples could not be prevented despite 24–48 h of drying in vacuo and were confirmed where possible by their presence in the 1H NMR spectra. All solvents and chemicals were purchased from Aldrich Chemical Co. and Fisher Scientific and were used as received.
3.1. General procedure for the synthesis of E-ethyl 3-(substituted-phenyl)but-2-enoate (32–36)
To a suspension of NaH (290 mg, 11.9 mmol) in 30 mL of dry THF was added triethyl phosphonoacetate (2.67 g, 11.9 mmol). The mixture was stirred for 1 h at room temperature and then the appropriate commercially available arylmethylketone (11.9 mmol) in 30 mL THF was added dropwise. After stirring at room temperature for 3 days, the reaction mixture was concentrated by evaporation of the solvent and then diluted with 300 mL of water. The resulting aqueous solution was extracted with Et2O (100 mL × 3) and the organic layers combined and dried over Na2SO4. The solvent was removed under reduced pressure to give 2–3 mL of a liquid residue, which was loaded on a silica gel column (15 × 150 mm) and eluted with hexane, 5% EtOAc in hexane and then 10% EtOAc in hexane. The fractions containing the product (TLC) were pooled and evaporated to afford 32–36. Here only the E-isomer of the product was isolated and characterized. The Z-isomer was difficult to isolate from the mixture (E/Z), and was not characterized.
3.2. E-Ethyl 3-(2-naphthyl)but-2-enoate (32)
Using the general procedure described above, compound 32 was obtained from 2-acetonaphthone as a colorless oil in 75% yield. Rf 0.53 (hexane/EtOAc/3:1). 1H NMR (CDCl3): δ 1.34 (t, 3H, CH3), 2.69 (s, 3H, CH3), 4.23 (q, 2H, CH2), 6.29 (s, 1H, CH), 7.47–7.48 (m, 7H, Ar-H × 7).
3.3. E-Ethyl 3-(2-fluorenyl)but-2-enoate (33)
Using the general procedure described above, compound 33 was obtained from 2-acetylfluorene as an off-yellow solid in 61% yield. Mp: 87–89 °C; Rf 0.49 (hexane/EtOAc/3:1). 1H NMR (CDCl3): δ 1.33 (t, 3H, CH3), 2.64 (s, 3H, CH3), 3.91 (s, 2H, CH2), 4.23 (q, 2H, CH2), 6.21 (s, 1H, CH), 7.30–7.81 (m, 7H, Ar-H × 7).
3.4. E-Ethyl 3-(4-biphenyl)but-2-enoate (34)
Using the general procedure described above, compound 34 was obtained from 4-acetyl-biphenyl as an off-white solid in 80% yield. Mp: 63–65 °C (lit. 63–64 °C); (lit.33 63–64° C); Rf 0.55 (hexane/EtOAc/3:1). 1H NMR (CDCl3): δ 1.33 (t, 3H, CH3), 2.64 (s, 3H, CH3), 4.26 (q, 2H, CH2), 6.21 (s, 1H, CH), 7.34–7.71 (m, 9H, Ar-H × 9).
3.5. E-Ethyl 3-(2,5-dimethoxyphenyl)but-2-enoate (35)
Using the general procedure described above, compound 35 was obtained from 2,5-dimethoxyacetophenone as a colorless oil in 74% yield. Rf 0.49 (hexane/EtOAc/3:1). 1H NMR (CDCl3): δ 1.28 (t, 3H, CH3), 2.56 (s, 3H, CH3), 3.76 (s, 3H, OCH3), 3.78 (s, 3H, OCH3), 4.18 (q, 2H, CH2), 5.90 (s, 1H, CH), 6.72–6.89 (m, 3H, Ar-H × 3).
3.6. E-Ethyl 3-(3,4,5-trimethoxyphenyl)but-2-enoate (36)
Using the general procedure described above, compound 36 was obtained from 3,4,5-trimethoxyacetophenone as an off-white solid in 63% yield. Mp: 57–59 °C; (lit.34 55–57 °C); Rf 0.50 (hexane/EtOAc/3:1). 1H NMR (CDCl3): δ 1.30 (t, 3H, CH3), 2.55 (s, 3H, CH3), 3.86 (s, 3H, OCH3), 3.89 (s, 6H, 2 × OCH3), 4.20 (q, 2H, CH2), 6.09 (s, 1H, CH), 6.75 (s, 2H, Ar-H × 2).
3.7. General procedure for synthesis of (±)-ethyl 3-(substituted-phenyl)butanoate (37-41)
Ethyl E-3-(substituted-phenyl)but-2-enoate (32–36) (11.7 mmol) was dissolved in 150 mL of EtOAc in a 500 mL flask, and 5% Pd/C (2.5 g) was added. The suspension was stirred under 1 atm H2 until the disappearance of the starting material (TLC). The catalyst was filtered and the solvent evaporated under reduced pressure to afford 37–41, which was used directly for the next step without further purification.
3.8. (±)-Ethyl 3-(2-naphthyl)butanoate (37)
Using the general procedure described above, compound 37 was obtained from E-ethyl 3-(2-naphthyl)but-2-enoate 32 as a colorless oil in 95% yield. Rf 0.65 (hexane/EtOAc/3:1). 1H NMR (CDCl3): δ 1.17 (t, 3H, CH3), 1.39 (d, 3H, CH3), 2.65–2.72 (m, 2H, CH2), 3.46–3.48 (m, 1H, CH), 4.07 (q, 2H, CH2), 7.37–7.45 (m, 3H, Ar-H × 3), 7.79–7.81 (m, 4H, Ar-H × 4).
3.9. (±)-Ethyl 3-(2-fluorenyl)butanoate (38)
Using the general procedure described above, compound 38 was obtained from E-ethyl 3-(2-fluorenyl)but-2-enoate 33 as a colorless oil in 97% yield. Rf 0.71 (hexane/EtOAc/3:1). 1H NMR (CDCl3): δ 1.18 (t, 3H, CH3), 1.34 (d, 3H, CH3), 2.55–2.70 (m, 2H, CH2), 3.32–3.40 (m, 1H, CH), 3.80 (s, 2H, CH2), 4.07 (q, 2H, CH2), 7.22–7.65 (m, 4H, Ar-H × 4), 7.70–7.76 (m, 3H, Ar-H × 3).
3.10. (±)-Ethyl 3-(4-biphenyl)butanoate (39)
Using the general procedure described above, compound 39 was obtained from E-ethyl 3-(4-biphenyl)but-2-enoate 34 as a colorless oil in 98% yield. Rf 0.70 (hexane/EtOAc/3:1). 1H NMR (CDCl3): δ 1.19 (t, 3H, CH3), 1.35 (d, 3H, CH3), 2.65–2.70 (m, 2H, CH2), 3.34–3.41 (m, 1H, CH), 4.10 (q, 2H, CH2), 7.27–7.72 (m, 9H, Ar-H × 9).
3.11. (±)-Ethyl 3-(2,5-dimethoxyphenyl)butanoate (40)
Using the general procedure described above, compound 40 was obtained from E-ethyl 3-(2,5-dimethoxyphenyl)but-2-enoate 35 as a colorless oil in 98% yield. Rf 0.60 (hexane/EtOAc/3:1). 1H NMR (CDCl3): δ 1.18 (t, 3H, CH3), 1.25 (d, 3H, CH3), 2.43–2.70 (m, 2H, CH2), 3.57–3.71 (m, 1H, CH), 3.72 (s, 3H, OCH3), 3.75 (s, 3H, OCH3), 4.08 (q, 2H, CH2), 6.67–6.79 (m, 3H, Ar-H × 3).
3.12. (±)-Ethyl 3-(3,4,5-trimethoxyphenyl)butanoate (41)
Using the general procedure described above, compound 41 was obtained from E-ethyl 3-(3,4,5-trimethoxyphenyl)but-2-enoate 36 as a colorless oil in 100% yield. Rf 0.60 (hexane/EtOAc/3:1). 1H NMR (CDCl3): δ 1.18 (t, 3H, CH3), 1.28 (d, 3H, CH3), 2.47–2.63 (m, 2H, CH2), 3.18–3.23 (m, 1H, CH), 3.81 (s, 3H, OCH3), 3.85 (s, 6H, 2 × OCH3), 4.08 (q, 2H, CH2), 6.43 (s, 2H, Ar-H × 2).
3.13. General procedure for synthesis of (±)-3-(substituted-phenyl)butylaldehyde (42–46)
To a cooled (−78 °C) solution of (±)-ethyl 3-(substituted-phenyl)butanoate (37-41) (10 mmol) in 15 mL of dry CH2Cl2 was added dropwise a solution of DIBAL-H (10 mL, 10 mmol, 1.0 M in hexane solution). After the reaction mixture was stirred at −78 °C for 30 min, methanol (6 mL) was added dropwise and then the reaction mixture was allowed to warm to room temperature. To this was added a solution of saturated NH4Cl (10 mL) and the mixture was extracted with CHCl3 (25 mL × 3). The organic layers were combined and washed with 1 N HCl (5 mL) and brine (5 mL) and dried (MgSO4). The solvent was removed under reduced pressure to give 2–3 mL of an oil, which was transferred to a silica gel column (15 × 150 mm), and eluted with 5% EtOAc in hexane and 10% EtOAc in hexane. Fractions containing the product (TLC) were pooled and evaporated to afford 42–46.
3.14. (±)-3-(2-Naphthyl)butylaldehyde (42)
Using the general procedure described above, compound 42 was obtained from (±)-ethyl 3-(2-naphthyl)butanoate 37 as a colorless oil in 85% yield. Rf 0.60 (hexane/EtOAc/3:1). 1H NMR (CDCl3): δ 1.42 (d, 3H, CH3), 2.65–2.95 (m, 2H, CH2), 3.45–3.70 (m, 1H, CH), 7.35–7.82 (m, 7H, Ar-H × 7), 9.75 (s, 1H, CHO).
3.15. (±)-3-(2-Fluorenyl)butylaldehyde (43)
Using the general procedure described above, compound 43 was obtained from (±)-ethyl 3-(2-fluorenyl)butanoate 38 as a colorless oil in 80% yield. Rf 0.62 (hexane/EtOAc/3:1).1H NMR (CDCl3): δ 1.39 (d, 3H, CH3), 2.65–2.95 (m, 2H, CH2), 3.30–3.60 (m, 1H, CH), 3.88 (s, 2H, CH2), 7.23–7.76 (m, 7H, Ar-H × 7), 9.74 (s, 1H, CHO).
3.16. (±)-3-(4-Biphenyl)butylaldehyde (44)
Using the general procedure described above, compound 44 was obtained from (±)-ethyl 3-(4-biphenyl)butanoate 39 as a colorless oil in 75% yield. Rf 0.61 (hexane/EtOAc/3:1). 1H NMR (CDCl3): δ 1.35 (d, 3H, CH3), 2.55–2.90 (m, 2H, CH2), 3.37–3.50 (m, 1H, CH), 7.23–7.58 (m, 9H, Ar-H × 9), 9.23 (s, 1H, CHO).
3.17. (±)-3-(2,5-Dimethoxyphenyl)butrylaldehyde (45)
Using the general procedure described above, compound 45 was obtained from (±)-ethyl 3-(2,5-dimethoxyphenyl)butanoate 40 as a colorless oil in 85% yield. Rf 0.55 (hexane/EtOAc/3:1). 1H NMR (CDCl3): δ 1.29 (d, 3H, CH3), 2.54–2.74 (m, 2H, CH2), 3.60–3.65 (m, 1H, CH), 3.83 (s, 3H, OCH3), 3.86 (s, 3H, OCH3), 6.69–6.80 (m, 3H, Ar-H × 3).
3.18. (±)-3-(3,4,5-Trimethoxyphenyl)butrylaldehyde (46)
Using the general procedure described above, compound 46 was obtained from (±)-ethyl 3-(3,4,5-trimethoxyphenyl)butanoate 41 as a colorless oil in 81% yield. Rf 0.38 (hexane/EtOAc/3:1). 1H NMR (CDCl3): δ 1.28 (d, 3H, CH3), 2.65–2.72 (m, 2H, CH2), 3.28–3.33 (m, 1H, CH), 3.82 (s, 3H, OCH3), 3.85 (s, 6H, 2 × OCH3), 6.42 (s, 2H, Ar-H × 2), 9.71 (s, 1H, CHO).
3.19. General procedure for the synthesis of 1-nitro-4-(substituted-phenyl)-2-pentanol (48–53)
To a solution of (±)-3-(substituted-phenyl)-butrylaldehyde (42–47) (10 mmol) in 10 mL of iso-propanol was added KF (29 mg, 0.5 mmol) and nitromethane (2.5 mL, 40 mmol). The reaction mixture was stirred at room temperature overnight, and the solid KF was filtered and the solvent and excess nitromethane were evaporated under reduced pressure. The residue was dissolved in ethyl acetate (50 mL) and washed with water (2×10 mL), and dried (MgSO4). After evaporating the solvent, the residue was dissolved in 2 mL of CHCl3 and was loaded on a 15 × 150 mm silica gel column and eluted with hexane, hexane/EtOAc/10:1 and hexane/EtOAc/7:1. Fractions containing the product (TLC) were pooled and evaporated to afford 48–53.
3.20. 1-Nitro-4-phenyl-2-pentanol (48)
Using the general procedure described above, compound 48 was obtained from (±)-3-phenyl-butrylaldehyde 47 as a colorless oil in 70% yield. Rf 0.43 (CHCl3). 1H NMR (DMSO-d6): δ 1.22 (d, 3H, CH3), 1.50–1.90 (m, 2H, CH2), 2.75–3.10 (m, 1H, CH), 3.65–4.15 (m, 1H, CH), 4.25–4.80 (m, 2H, CH2), 5.36–5.43 (m, 1H, OH), 7.19–7.32 (m, 5H, Ar-H × 5).
3.21. 1-Nitro-4-(2-naphthyl)-2-pentanol (49)
Using the general procedure described above, compound 49 was obtained from (±)-3-(2-naphthyl)-butrylaldehyde 42 as a colorless oil in 80% yield. Rf 0.40 (hexane/EtOAc/3:1). 1H NMR (CDCl3): δ 1.38 (d, 3H, CH3), 1.65–2.20 (m, 2H, CH2), 2.54 (d, 1H, OH), 3.05–3.25 (m, 1H, CH), 3.90–4.15 (m, 1H, CH), 4.20–4.60 (m, 2H, CH2), 7.34–7.83 (m, 7H, Ar-H × 7).
3.22. 1-Nitro-4-(2-fluorenyl)-2-pentanol (50)
Using the general procedure described above, compound 50 was obtained from (±)-3-(2-fluorenyl)-butrylaldehyde 43 as a colorless oil in 67% yield. Rf 0.45 (hexane/EtOAc/3:1). 1H NMR (CDCl3): δ 1.34 (d, 3H, CH3), 1.60–1.95 (m, 2H, CH2), 2.52 (d, 1H, OH), 3.00–3.30 (m, 1H, CH), 3.89 (s, 2H, CH2), 3.90–4.10 (m, 1H, CH), 4.15–4.50 (m, 2 H, CH2), 7.20–7.95 (m, 7H, Ar-H × 7).
3.23. 1-Nitro-4-(4-biphenyl)-2-pentanol (51)
Using the general procedure described above, compound 51 was obtained from (±)-3-(4-biphenyl)-butrylaldehyde 44 as a colorless oil in 72% yield. Rf 0.51 (hexane/EtOAc/3:1). 1H NMR (CDCl3): δ 1.35 (d, 3H, CH3), 1.60–2.00 (m, 2H, CH2), 2.53 (d, 1H, OH), 2.95–3.20 (m, 1H, CH), 4.00–4.20 (m, 1H, CH), 4.20–4.50 (m, 2H, CH2), 7.25–7.74 (m, 9H, Ar-H × 9).
3.24. 1-Nitro-4-(2,5-dimethoxyphenyl)-2-pentanol (52)
Using the general procedure described above, compound 52 was obtained from (±)-3-(2,5-dimethoxyphenyl)-butrylaldehyde 45 as a colorless oil in 75% yield. Rf 0.37 (hexane/EtOAc/3:1). 1H NMR (CDCl3): δ 1.29 (d, 3H, CH3), 1.40–1.90 (m, 2H, CH2), 2.54 (d, 1H, OH), 3.32–3.51 (m, 1H, CH), 3.78 (s, 3H, OCH3), 3.82 (s, 3H, OCH3), 3.85–4.05 (m, 1H, CH), 4.15–4.40 (m, 2H, CH2), 6.72–6.86 (m, 3H, Ar-H × 3).
3.25. 1-Nitro-4-(3,4,5-trimethoxyphenyl)-2-pentanol (53)
Using the general procedure described above, compound 53 was obtained from (±)-3-(3,4,5-trimethoxyphenyl)-butrylaldehyde 46 as a colorless oil in 78% yield. Rf 0.45 (CHCl3). 1H NMR (CDCl3): δ 1.43 (d, 3H, CH3), 1.60–1.88 (m, 2H, CH2), 2.56 (d, 1H, OH), 2.89–3.02 (m, 1H, CH), 3.83 (s, 3H, OCH3), 3.86 (s, 6H, 2 × OCH3), 3.94–4.04 (m, 1H, CH), 4.29–4.43 (m, 2H, CH2), 6.41 (s, 2H, Ar-H × 2).
3.26. General procedure for the synthesis of (±)-1-nitro-4-(substituted-phenyl)-1-pentene (54–59)
To a solution of 1-nitro-4-(substituted-phenyl)-2-pentanol (48–53) (10mmol) in 15mL of dry CH2Cl2 at 0 °C, was added methanesulfonyl chloride (1.18 g, 0.8 mL, 10 mmol), followed by Et3N (2.2 g, 2.8 mL, 20 mmol). The mixture was stirred at 0 °C for 20 min, and then poured into 30 mL of water and the aqueous mixture extracted with CH2Cl2 (50 mL × 3). The organic layers were combined and dried (MgSO4). After evaporating the solvent, the residue was dissolved in 1 mL of CH2Cl2 and was placed on a 15 × 150 mm silica gel column and eluted with hexane, hexane/EtOAc/50:1. Fractions containing the product (TLC) were pooled and evaporated to afford 54–59.
3.27. (±)-1-Nitro-4-phenyl-l-pentene (54)
Using the general procedure described above, compound 54 was obtained from (±)-1-nitro-4-phenyl-2-pentanol 48 as a light yellow oil in 90% yield. Rf 0.63 (hexane/EtOAc/3:1). 1H NMR (CDCl3): δ 1.35 (d, 3H, CH3), 2.48–2.62 (m, 2H, CH2), 2.91–3.00 (m, 1H, CH), 6.88 (d, 1H, CH), 7.11–7.35 (m, 6H, CH and Ar-H × 5).
3.28. (±)-1-Nitro-4-(2-naphthyl)-l-pentene (55)
Using the general procedure described above, compound 55 was obtained from (±)-1-nitro-4-(2-naphthyl)-2-pentanol 49 as a light yellow oil in 91% yield. Rf 0.73 (hexane/EtOAc/3:1). 1H NMR (CDCl3): δ 1.41 (d, 3H, CH3), 2.50–2.80 (m, 2H, CH2), 3.00–3.30 (m, 1H, CH), 6.87 (d, 1H, CH), 7.15–7.90 (m, 8H, CH and Ar-H × 7).
3.29. (±)-1-Nitro-4-(2-fluorenyl)-l-pentene (56)
Using the general procedure described above, compound 56 was obtained from (±)-1-nitro-4-(2-fluorenyl)-2-pentanol 50 as a light yellow oil in 90% yield. Rf 0.69 (hexane/EtOAc/3:1). 1H NMR (CDCl3): δ 1.30 (d, 3H, CH3), 2.33–2.60 (m, 2H, CH2), 2.75–3.00 (m, 1H, CH), 3.79 (s, 2H, CH2), 6.78 (d, 1H, CH), 7.00–7.90 (m, 8H, CH and Ar-H × 7).
3.30. (±)-1-Nitro-4-(4-biphenyl)-l-pentene (57)
Using the general procedure described above, compound 57 was obtained from (±)-1-nitro-4-(4-biphenyl)-2-pentanol 51 as a light yellow oil in 92% yield. Rf 0.69 (hexane/EtOAc/3:1). 1H NMR (CDCl3): δ 1.38 (d, 3H, CH3), 2.49–2.65 (m, 2H, CH2), 2.98–3.15 (m, 1H, CH), 6.91 (d, 1H, CH), 7.14–7.70 (m, 10H, CH and Ar-H × 9).
3.31. (±)-1-Nitro-4-(2,5-dimethoxyphenyl)-l-pentene (58)
Using the general procedure described above, compound 58 was obtained from (±)-1-nitro-4-(2,5-dimethoxyphenyl)-2-pentanol 52 as a light yellow oil in 90% yield. Rf 0.60 (hexane/EtOAc/3:1). 1H NMR (CDCl3): δ 1.26 (d, 3H, CH3), 2.46–2.25 (m, 2H, CH2), 3.30–3.50 (m, 1H, CH), 3.76 (s, 3H, OCH3), 3.77 (s, 3H, OCH3), 6.70–6.80 (m, 3H, Ar-H × 3), 6.87 (d, 1H, CH), 7.16–7.25 (m, 1H, CH).
3.32. (±)-1-Nitro-4-(3,4,5-trimethoxyphenyl)-l-pentene (59)
Using the general procedure described above, compound 59 was obtained from (±)-1-nitro-4-(3,4,5-trimethoxyphenyl)-2-pentanol 53 as a light yellow oil in 91% yield. Rf 0.40 (hexane/EtOAc/3:1). 1H NMR (CDCl3): δ 1.32 (d, 3H, CH3), 2.46–2.52 (m, 2H, CH2), 3.30–3.14 (m, 1H, CH), 3.84 (s, 3H, OCH3), 3.86 (s, 3H, OCH3), 6.41 (s, 2H, Ar-H × 2), 6.91 (d, 1H, CH), 7.10–7.19 (m, 1H, CH).
3.33. General procedure for the synthesis of 5-[1-(nitromethyl)-3-substituted arylbutyl]pyrimidine-2,4,6-triamine (60–65)
A mixture of (±)-1-nitro-4-(substituted-aryl)-pentene (54–59) (5 mmol) and (5 mmol) of 2,4,6-triaminopyrimidine in 20 mL water and 20 mL ethyl acetate was stirred at 50–55 °C for 24 h. The solid dissolved on heating. The reaction mixture was poured into 200 mL ethyl acetate, washed with water (2×40 mL), dried (Na2SO4) and purified by silica gel column chromatography with 5% CH3OH in CHCl3. Fractions containing the product (TLC) were pooled and evaporated to afford 60–65.
3.34. 5-[(1-Nitromethyl)-3-phenylbutyl]pyrimidine-2,4,6-triamine (60)
Using the general procedure described above, compound 60 was obtained from (±)-1-nitro-4-phenyl-1-pentene 54 as a yellow solid in 82% yield. Mp: 70–73 °C; Rf 0.30 (CHCl3:CH3OH/5:1). 1H NMR (DMSO-d6): δ 1.13–1.20 (m, 3H, CH3), 1.60–2.30 (m, 2H, CH2), 2.50–2.65 (m, 1H, CH), 2.97–3.54 (m, 1H, CH), 4.65–4.80 (m, 2H, CH2), 5.05–5.35 (m, 4H, 2 × NH2), 5.40–5.65 (m, 2H, NH2), 7.10–7.90 (m, 5H, Ar-H × 5). Anal. Calcd for C15H20N6O2·0.2CH3OH: C, 56.56; H, 6.50; N, 26.04. Found: C, 56.73; H, 6.40; N, 25.87.
3.35. 5-[3-(2-Naphthyl)-1-(nitromethyl)butyl]pyrimidine-2,4,6-triamine (61)
Using the general procedure described above, compound 61 was obtained from (±)-1-nitro-4-(2-naphthyl)-1-pentene 55 as a yellow solid in 81% yield. Mp: 75–77 °C; Rf 0.50 (CHCl3:CH3OH/5:1). 1H NMR (DMSO-d6): δ 1.23–1.30 (m, 3H, CH3), 1.65–2.30 (m, 2H, CH2), 2.60–2.90 (m, 1H, CH), 3.55–3.75 (m, 1H, CH), 4.65-4.95 (m, 2H, CH2), 5.09–5.53 (m, 6H, 3 × NH2), 7.36–7.83 (m, 7H, Ar-H × 7). Anal. Calcd for C19H22N6O2: C, 62.28; H, 6.05; N, 22.94. Found: C, 62.44; H, 6.03; N, 22.60.
3.36. 5-[3-(2-Fluorenyl)-1-((nitromethyl)butyl]pyrimidine-2,4,6-triamine (62)
Using the general procedure described above, compound 62 was obtained from (±)-1-nitro-4-(2-fluorenyl)-1-pentene 56 as a yellow solid in 75% yield. Mp: 78–80 °C; Rf 0.53 (CHCl3:CH3OH/5:1). 1H NMR (DMSO-d6): δ 1.19–1.26 (m, 3H, CH3), 1.15–2.25 (m, 2H, CH2), 2.50–2.80 (m, 1H, CH), 3.90–3.65 (m, 1H, CH), 3.86 (s, 2H, CH2), 4.65–4.90 (m, 2H, CH2), 5.03–5.60 (m, 6H, 3 × NH2), 7.18–7.86 (m, 7H, Ar-H × 7). Anal. Calcd for C22H24N6O2: C, 65.33; H, 5.98; N, 20.78. Found: C, 65.14; H, 5.99; N, 20.66.
3.37. 5-[3-(4-Biphenyl)-1-(nitromethyl)butyl]pyrimidine-2,4,6-triamine (63)
Using the general procedure described above, compound 63 was obtained from (±)-1-nitro-4-(4-biphenyl)-1-pentene 57 as a yellow solid in 78% yield. Mp: 75–76 °C; Rf 0.45 (CHCl3:CH3OH/5:1). 1H NMR (DMSO-d6): δ 1.22 (d, 3H, CH3), 1.65-2.25 (m, 2H, CH2), 2.30–2.55 (m, 1H, CH), 3.70–3.95 (m, 1H, CH), 4.65–4.90 (m, 2H, CH2), 5.30–5.51 (m, 6H, 3 × NH2), 7.20–7.70 (m, 9H, Ar-H × 9). Anal. Calcd for C21H24N6O2: C, 64.27; H, 6.16; N, 21.41. Found: C, 64.22; H, 6.17; N, 21.48.
3.38. 5-[3-(2,5-Dimethoxyphenyl)-1-(nitromethyl)butyl]pyrimidine-2,4,6-triamine (64)
Using the general procedure described above, compound 64 was obtained from (±)-1-nitro-4-(2,5-dimethoxyphenyl)-1-pentene 58 as a yellow solid in 80% yield. Mp: 80–83 °C; Rf 0.41 (CHCl3:CH3OH/5:1). 1H NMR (DMSO-d6): δ 1.05–1.16 (m, 3H, CH3), 1.65–2.15 (m, 2H, CH2), 2.85–3.10 (m, 1H, CH), 3.40–3.60 (m, 1 H, CH), 3.66 (s, 3H, OCH3), 3.68 (s, 3H, OCH3). 4.65–4.90 (m, 2H, CH2), 5.20–5.70 (m, 6H, 3 × NH2), 6.72–6.85 (m, 3H, Ar-H × 3). Anal. Calcd for C17H24N6O4·0.4H2O: C, 53.23; H, 6.52; N, 21.91. Found: C, 52.89; H, 6.38; N, 21.70.
3.39. 5-[3-(3,4,5-Trimethoxyphenyl)1-(nitromethyl)butyl]pyrimidine-2,4,6-triamine (65)
Using the general procedure described above, compound 65 was obtained from (±)-1-nitro-4-(3,4,5-trimethoxyphenyl)-1-pentene 59 as a yellow solid in 79% yield. Mp: 84–87 °C; Rf 0.41 (CHCl3:CH3OH/5:1). 1H NMR (DMSO-d6): δ 1.19 (d, 3H, CH3), 1.62–2.20 (m, 2H, CH2), 2.85–3.10 (m, 1H, CH), 3.40–3.60 (m, 1H, CH), 3.74 (s, 3H, OCH3), 3.76 (s, 6H, 2 × OCH3), 4.69–4.78 (m, 2H, CH2), 5.10–5.60 (m, 6H, 3 × NH2), 6.44 (s, 2H, Ar-H × 2). Anal. Calcd for C15H20N6O2·0.6-H2O: C, 51.81; H, 6.57; N, 20.14. Found: C, 52.06; H, 6.43; N, 19.75.
3.40. General procedure for the synthesis of (±)-2,4-diamino-5-[2-methyl-2-(substituted-pheny)]ethyl-pyrrolo[2,3-d]pyrimidine (18–23)
To a solution of NaOH (0.24 g, 6.0 mmol) in 3.0 mL of water was added (1.0 mmol) of (±)-1-nitro-2-(2,4,6-triaminopyrimidin-5-yl)-4-(substituted-phenyl)-pentane (60–65) and the mixture was stirred at room temperature for 2 h. This mixture was added dropwise to a solution of 98% H2SO4 (0.55mL, 0.98 g, 10.0 mmol) in 4.0 mL of water at 0 °C. After stirring for an additional 3 h, the pH of the mixture was adjusted to 7 with 2 N NaOH at 0 °C. The mixture was stirred at room temperature for another 1 h and then acidified with acetic acid (0.5 mL). The precipitated solid was collected by filtration, washed with water, followed by ethyl acetate, and dried in vacuum to afford an off-white solid. This solid was dissolved in 5 mL of methanol and 100 mg of silica gel was added and the solvent was removed to form a plug, which was loaded on a silica gel column (15 × 150 mm), and eluted with 4% methanol in chloroform. Fractions containing the product (TLC) were pooled and evaporated to afford 18–23.
3.41. (±)-2,4-Diamino-5-(2-methyl-2-phenyl)ethyl-pyrrolo[2,3-d]pyrimidine (18)
Using the general procedure described above, compound 18 was obtained from (±)-1-nitro-2-(2,4,6-triaminopyrimidin-5-yl)-4-phenyl-pentane 60 as an off-white solid in 49% yield. Mp: >250 °C; Rf 0.65 (CHCl3:CH3OH/5:1). 1H NMR (DMSO-d6): δ 1.18 (d, 3H, CH3), 2.89–2.98 (m, 3H, CH and CH2), 5.42 (s, 2H, NH2), 5.98 (s, 2H, NH2), 6.23 (s, 1H, CH), 7.14–7.23 (m, 5H, Ar-H × 5), 10.35 (s, 1H, NH). Anal. Calcd for C15H17N5: C, 67.39; H, 6.41; N, 26.20. Found: C, 67.25; H, 6.41; N, 26.08.
3.42. (±)-2,4-Diamino-5-[2-methyl-2-(2-naphthyl)]ethyl-pyrrolo[2,3-d]pyrimidine (19)
Using the general procedure described above, compound 19 was obtained from (±)-1-nitro-2-(2,4,6-triaminopyrimidin-5-yl)-4-(2-naphthyl)-pentane 61 as an off-white solid in 53% yield. Mp: >200 °C; Rf 0.64 (CHCl3:CH3OH/5:1). 1H NMR (DMSO-d6): δ 1.31 (d, 3H, CH3), 2.95–3.20 (m, 3H, CH and CH2), 5.35 (s, 2H, NH2), 5.96 (s, 2H, NH2), 6.23 (s, 1H, CH), 7.46–7.92 (m, 7H, Ar-H × 7), 10.27 (s, 1H, NH). Anal. Calcd for C15H17N5·0.6H2O: C, 69.53; H, 6.20; N, 21.34. Found: C, 69.61; H, 6.25; N, 21.02.
3.43. (±)-2,4-Diamino-5-[2-methyl-2-(2-fluorenyl)]ethyl-pyrrolo[2,3-d]pyrimidine (20)
Using the general procedure described above, compound 20 was obtained from (±)-1-nitro-2-(2,4,6-triaminopyrimidin-5-yl)-4-(2-fluorenyl)-pentane 62 as an off-white solid in 48% yield. Mp: >200 °C; Rf 0.68 (CHCl3:CH3OH/5:1). 1H NMR (DMSO-d6): δ 1.27 (d, 3H, CH3), 2.85–3.15 (m, 3H, CH and CH2), 3.87 (s, 2H, CH2), 5.37 (s, 2H, NH2), 5.96 (s, 2H, NH2), 6.28 (s, 1H, CH), 7.15–7.95 (m, 7H, Ar-H × 7), 10.32 (s, 1H, NH). Anal. Calcd for C22H21N5·0.8H2O: C, 71.44; H, 6.16; N, 18.94. Found: C, 71.28; H, 6.10; N, 18.63.
3.44. (±)-2,4-Diamino-5-[2-methyl-2-(4-biphenyl)]ethyl-pyrrolo[2,3-d]pyrimidine (21)
Using the general procedure described above, compound 21 was obtained from (±)-1-nitro-2-(2,4,6-triaminopyrimidin-5-yl)-4-(4-biphenyl)-pentane 63 as an off-white solid in 45% yield. Mp: >230 °C; Rf 0.66 (CHCl3:CH3OH/5:1). 1H NMR (DMSO-d6): δ 1.22 (d, 3H, CH3), 2.85–3.15 (m, 3H, CH and CH2), 5.42 (s, 2H, NH2), 5.99 (s, 2H, NH2), 6.29 (s, 1H, CH), 7.33–7.64 (m, 9H, Ar-H × 9), 10.36 (s, 1H, NH). Anal. Calcd for C21H21N5·1.2H2O: C, 69.09; H, 6.46; N, 19.18. Found: C, 69.33; H, 6.07; N, 18.83.
3.45. (±)-2,4-Diamino-5-[2-methyl-2-(2,5-dimethoxyphenyl)]ethyl-pyrrolo[2,3-d]pyrimidine (22)
Using the general procedure described above, compound 22 was obtained from (±)-1-nitro-2-(2,4,6-triaminopyrimidin-5-yl)-4-(2,5-dimethoxyphenyl)-pentane 64 as an off-white solid in 50% yield. Mp: >250 °C; Rf 0.57 (CHCl3:CH3OH/5:1). 1H NMR (DMSO-d6): δ 1.08 (d, 3H, CH3), 2.36–3.30 (m, 3H, CH and CH2), 3.70 (s, 3H, OCH3), 3.78 (s, 3H, OCH3), 5.38 (s, 2H, NH2), 6.20 (s, 2H, NH2), 6.39 (s, 1H, CH), 6.72–6.90 (m, 3H, Ar-H × 3), 10.39 (s, 1H, NH). Anal. Calcd for C17H21N5O2: C, 62.37; H, 6.47; N, 21.39. Found: C, 62.21; H, 6.59; N, 21.30.
3.46. (±)-2,4-Diamino-5-[2-methyl-2-(3,4,5-triOMepheny)]ethyl-pyrrolo[2,3-d]pyrimidine(23)
Using the general procedure described above, compound 23 was obtained from (±)-1-nitro-2-(2,4,6-triaminopyrimidin-5-yl)-4-(3,4,5-trimethoxyphenyl)-pentane 65 as an off-white solid in 49% yield. Mp: >250 °C; Rf 0.53 (CHCl3:CH3OH/5:1). 1H NMR (DMSO-d6): δ 1.18 (d, 3H, CH3), 2.84–2.94 (m, 3H, CH and CH2), 3.60 (s, 3H, OCH3), 3.65 (s, 6H, 2 × OCH3), 5.36 (s, 2H, NH2), 5.92 (s, 2H, NH2), 6.31 (s, 1H, CH), 6.51 (s, 2H, Ar-H × 2), 10.34 (s, 1H, NH). Anal. Calcd C18H23N5O3·0.1H2O: C, 60.19; H, 6.51; N, 19.50. Found: C, 59.86; H, 6.62; N, 19.24.
3.47. Sources of DHFR enzymes
Dihydrofolate reductase from P. carinii was produced as the recombinant enzyme expressed in Escherichia coli.35 The sequence of the protein was identical to that predicted for the previously reported gene sequence.36
Dihydrofolate reductase from T. gondii was isolated directly from the RH strain of T. gondii grown in culture on mutant Chinese hamster ovary cells lacking dihydrofolate reductase (CHO/dhfr−, American Type Culture Collection 3952 CL).36 The organisms are introduced into a confluent monolayer of the cells and harvested when they have lysed the mammalian cells. The 100,000g supernate is stored in liquid nitrogen.
Mycobacterium avium used in these studies was a clinical isolate from Indiana University School of Medicine, Department of Pathology; it was identified as a serovar 4. The strain was maintained on Lowenstein-Jensen slants (Baxter Scientific) grown at room temperature. To produce enzyme, the organism was grown in Middle-brook 7H-9 liquid medium at 37 °C to an OD660 of 0.5–0.7, which took several weeks. At harvest, the bacteria were sedimented by centrifugation, sonicated, and the 100,000g supernate was stored under liquid nitrogen until assay. These supernates contained both dihydrofolate reductase and dihydroopteroate synthetase activity.
Rat liver dihydrofolate reductase was prepared from livers of adult female Sprague–Dawley rats. The 100,000g supernate prepared from crude homogenates was partially purified by ammonium sulfate precipitation; the 50–90% precipitate was redissolved and stored in liquid nitrogen.
3.48. DHFR assay
The spectrophotometric assay for dihydrofolate reductase was modified to optimize for temperature, substrate concentration, and cofactor concentration for each enzyme form assayed. The standard assay contained sodium phosphate-buffer, pH 7.4 (40.7 mM), 2-mercaptoethanol (8.9 mM), NADPH (0.117 mM), dihydrofolic acid (0.092 mM), and sufficient enzyme to produce a change in OD340 of 0.035/min. KCL (150 mM) was added to the reaction for the T. gondii, M. avium, and rat liver enzymes. All assays were performed at 37 °C. The reaction was followed for 3 min with continuous recording. Activity under these conditions was linear with enzyme concentration over at least a 4-fold range. Background activity in the absence of dihydrofolic acid was near zero and was subtracted from all rates.
3.49. Determination of IC50 values
Dihydrofolate reductase was assayed in the presence of a series of concentrations of inhibitor to produce a range of rates from 1% to 90% of the uninhibited rate. At least three concentrations were required for calculation; most curves contained five or more concentrations. Semilogarithmic plots of the data yielded normal sigmoidal curves for most inhibitors. The concentration yielding 50% inhibition (IC50) was calculated using Prism 4.0 (GraphPad).
Acknowledgments
This work was supported, in part, by a grant from the NIH, National Institutes of Allergy and Infections Disease AI 47759 (AG).
Footnotes
See Ref. 1.
References
- 1.Presented in part at 225th American Chemical Society National Meeting. New Orleans, LA. March 2003; Amercian Chemical Society; Washington, DC. 2003. MEDI 134. [Google Scholar]
- 2.Gangjee A, Elzein E, Kothare M, Vasudevan A. Curr Pharm Des. 1996;2:263. [Google Scholar]
- 3.Klepser ME, Klepser TB. Drugs. 1997;53:40. doi: 10.2165/00003495-199753010-00004. [DOI] [PubMed] [Google Scholar]
- 4.Masur H, Polis MA, Tuazon CU, Ogata-Arakaki D, Kovacs JA, Katz D, Hilt D, Simmons T, Feuerstein I, Lundgren B, Lane HC, Chabner BA, Allegra CJ. J Infect Dis. 1992;167:1422. doi: 10.1093/infdis/167.6.1422. [DOI] [PubMed] [Google Scholar]
- 5.Gangjee A, Mavandadi F, Queener SF, McGuire JJ. J Med Chem. 1995;38:2158. doi: 10.1021/jm00012a016. [DOI] [PubMed] [Google Scholar]
- 6.Gangjee A, Mavandadi F, Queener SF. J Med Chem. 1997;40:1173. doi: 10.1021/jm960717q. [DOI] [PubMed] [Google Scholar]
- 7.Gangjee A, Devraj R, McGuire JJ, Kisliuk RL, Queener S, Barrows LR. J Med Chem. 1994;37:1169. doi: 10.1021/jm00034a015. [DOI] [PubMed] [Google Scholar]
- 8.Gangjee A, Devraj R, McGuire JJ, Kisliuk RL. J Med Chem. 1995;38:3798. doi: 10.1021/jm00019a009. [DOI] [PubMed] [Google Scholar]
- 9.Gangjee A, Guo X, Queener SF, Cody V, Galitsky N, Luft JR, Pangborn W. J Med Chem. 1998;41:1263. doi: 10.1021/jm970537w. [DOI] [PubMed] [Google Scholar]
- 10.Gangjee A, Zeng Y, McGuire JJ, Kisliuk RL. J Med Chem. 2000;43:3125. doi: 10.1021/jm000130i. [DOI] [PubMed] [Google Scholar]
- 11.Gangjee A, Adair O, Queener SF. J Med Chem. 1999;42:2447. doi: 10.1021/jm990079m. [DOI] [PubMed] [Google Scholar]
- 12.Miwa T, Hitaka T, Akimoto H, Nomura H. J Med Chem. 1991;34:555. doi: 10.1021/jm00106a012. [DOI] [PubMed] [Google Scholar]
- 13.Rosowsky A, Chen H, Fu H, Queener SF. Bioorg Med Chem. 2003;11:59. doi: 10.1016/s0968-0896(02)00325-5. [DOI] [PubMed] [Google Scholar]
- 14.Taylor EC, Liu B. Tetrahedron Lett. 1999;40:4023. [Google Scholar]
- 15.Taylor EC, Liu B. Tetrahedron Lett. 1999;40:4027. [Google Scholar]
- 16.Luzzio FA. Tetrahedron. 2001;57:915. [Google Scholar]
- 17.Melpolder JB, Heck RF. J Org Chem. 1976;41:265. [Google Scholar]
- 18.Galemmo RA, Jr, Johnson WH, Jr, Learn KS, Lee TDY, Huang FC, Campbell HF, Youssefyeh R, O'Rourke SV, Schuessler G. J Med Chem. 1990;33:2828. doi: 10.1021/jm00172a024. [DOI] [PubMed] [Google Scholar]
- 19.Albrecht M, Riether C. Synthesis. 1997:957. [Google Scholar]
- 20.Crombie L, Mistry J. Tetrahedron Lett. 1975;31:2647. [Google Scholar]
- 21.Durman J, Elliott J, McElroy AB, Warren S. J Chem Soc Perkin Trans. 1985;1:1237. [Google Scholar]
- 22.Asaoka M, Shima K, Fujii N, Takei H. Tetrahedron. 1988;44:4757. [Google Scholar]
- 23.Ambros R, Schneider MR, Von Angerer S. J Med Chem. 1990;33:153. doi: 10.1021/jm00163a026. [DOI] [PubMed] [Google Scholar]
- 24.Chen Q, Huggins MT, Lightner DA, Norona W, McDonagh AF. J Am Chem Soc. 1999;121:9253. [Google Scholar]
- 25.Sinhababu AK, Borchardt RT. Tetrahedron Lett. 1983;24:227. [Google Scholar]
- 26.Gairaud CB, Lappin GR. J Org Chem. 1953;15:1. [Google Scholar]
- 27.Kambe S. Chem Soc Bull JP. 1968;41:1444. [Google Scholar]
- 28.Urban FJ, Breitenbach R, Murtiashaw CW, Vanderplas BC. Tetrahedron: Asymmetry. 1995;6:321. [Google Scholar]
- 29.Melton J, McMurry JE. J Org Chem. 1975;40:2138. [Google Scholar]
- 30.Taylor EC, Liu B. J Org Chem. 2003;68:9938. doi: 10.1021/jo030248h. [DOI] [PubMed] [Google Scholar]
- 31.Pinnick HW. Org React. 1990;38:655. [Google Scholar]
- 32.Cody V, Galitsky N, Luft JR, Pangborn W, Queener SF, Gangjee A. Acta Cryst D: Biol Cryst. 2002;D58:1393. doi: 10.1107/S0907444902010442. [DOI] [PubMed] [Google Scholar]
- 33.Anderson PL, Brittian DA. Chem Abstr. 1975;82:170341. Ger. Offen. DE 2439294, 1975. [Google Scholar]
- 34.Moffett RB. J Med Chem. 1964;7:319. doi: 10.1021/jm00333a015. [DOI] [PubMed] [Google Scholar]
- 35.Bartlett MS, Shaw M, Navaran P, Smith JW, Queener SF. Antimicrob Agents Chemother. 1995;39:2441. doi: 10.1128/aac.39.11.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Edman JC, Edman U, Cao M, Lundgren B, Kovacs JA, Santi DV. Proc Natl Acad Sci USA. 1989;85:8625. doi: 10.1073/pnas.86.22.8625. [DOI] [PMC free article] [PubMed] [Google Scholar]