

Abstract
The classical antifolate N-{4-[(2,4-diamino-5-ethyl-7H-pyrrolo[2,3-d]pyrimidin-6-yl)sulfanyl]benzoyl}-l-glutamic acid (2) and 15 nonclassical analogues (3–17) were synthesized as potential dihydrofolate reductase (DHFR) inhibitors and as antitumor agents. 5-Ethyl-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamine (20) served as the key intermediate to which various aryl thiols and a heteroaryl thiol were appended at the 6-position via an oxidative addition reaction. The classical analogue 2 was synthesized by coupling the benzoic acid derivative 18 with diethyl l-glutamate followed by saponification. The classical compound 2 was an excellent inhibitor of human DHFR (IC50 = 66 nM) as well as a two digit nanomolar (<100 nM) inhibitor of the growth of several tumor cells in culture. Some of the nonclassical analogues were potent and selective inhibitors of DHFR from two pathogens (Toxoplasma gondii and Mycobacterium avium) that cause opportunistic infections in patients with compromised immune systems.
Introduction
Folate metabolism is an attractive target for chemotherapy as a result of its crucial role in the biosynthesis of nucleic acid precursors.1 Inhibition of folate-dependent enzymes in cancer cells, in microbial cells, and protozoan cells provides compounds that have found clinical utility as antitumor, antimicrobial, and antiprotozoal agents.2,3
As part of a continuing effort to develop novel classical antifolates as antitumor agents, Gangjee et al.4 reported the synthesis of N-{4-[(2,4-diamino-5-methyl-7H-pyrrolo[2,3-d]-pyrimidin-6-yl)sulfanyl]benzoyl}-l-glutamic acid (1a; Figure 1) as a potent dual inhibitor of dihydrofolate reductase (DHFRa) and thymidylate synthase (TS) with IC50 values in the 10−7 M range. Compound 1a also demonstrated potent inhibitory activity against the growth of several human tumor cell lines in the National Cancer Institute (NCI)5 preclinical in vitro screen, with GI50 values of 10−7 to 10−8 M or lower. The DHFR inhibitory potency and tumor cell inhibitory activities of 1a were in part attributed to its C5-methyl group. Molecular modeling (Figure 2) using Sybyl 7.06 indicated that the C5-substituent of 6–5 ring-fused bicyclic pyrrolo[2,3-d]pyrimidines (red) could mimic the C5-substituent of 6–6 ring-fused bicyclic pteridines or pyrido[2,3-d]pyrimidines (green) such as 5-methyl-5-deaza methotrexate or 5-methyl-5-deaza aminopterin in which the 5-methyl group was reported to have a conducive hydrophobic interaction with human DHFR.7,8 Molecular modeling of 1a also suggested that homologation of the C5-methyl to an ethyl could further enhance the hydrophobic interaction with human DHFR at Val115 and perhaps the antitumor activity as well. Figure 3 shows the superimposition of the homologated 5-ethyl classical compound 2 (Figure 1) onto MTX (not shown) in the X-ray crystal structure of MTX in human DHFR (PDB 1U72). The van der Waals distance of the methyl moiety of the 5-ethyl group from Val115 is about 3.5 Å, which is much closer than that for the 5-methyl group of 1a. Thus, compound 2, the C5-ethyl homologue of 1a, was designed and synthesized to explore this possibility.
Figure 1.

Figure 2.

Superimposition of a 6–5 pyrrolo[2,3-d]pyrimidine ring system (in red) on to a 6–6 pyrido[2,3-d]pyrimidine ring system (in green) illustrating that the 5-Me moiety of the 6–5 system mimics the 5-Me of the 6–6 system.
Figure 3.
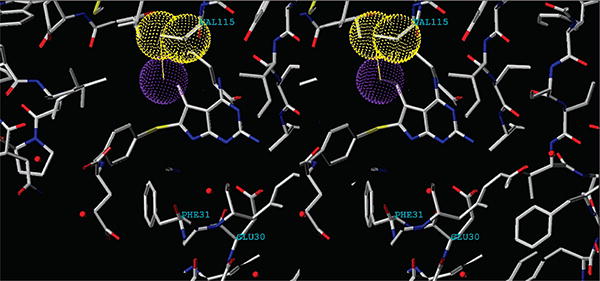
Stereoview. Compound 2 superimposed on to methotrexate (not shown) in the X-ray crystal structure of MTX and human DHFR. The van der Waals surface of the methyl moiety (in purple) of the 5-ethyl group interacts with the van der Waals surface of the methyl and methylene carbon of Val115 (in yellow) in hDHFR. This interaction affords better hDHFR inhibitory activity for 2 than the 5-methyl moiety in 1a. The hDHFR Phe31 and Glu30 are also labeled.
A disadvantage of classical antifolates as antitumor agents is that they require an active transport mechanism to enter cells, which, when impaired, causes tumor resistance.9 In addition, cells that lack these transport mechanisms, including many bacterial and protozoan cells, are not susceptible to the action of classical antifolates. In an attempt to overcome these potential drawbacks, nonclassical lipophilic antifolates have been developed as antitumor agents that do not require the folate transport system(s) but enter cells via diffusion.10
An additional aspect of nonclassical antifolates involves their utility in opportunistic infections. The principal causes of death in patients with acquired immunodeficiency syndrome (AIDS) are opportunistic infections caused by Pneumocystis jirovecii (previously Pneumocystis carinii, pc) and Toxoplasma gondii (tg).11 Current therapeutic regimens include the use of selective but weak inhibitors of protozoal DHFR such as trimethoprim (TMP) and pyrimethamine (Figure 1), in combination with sulfonamides to enhance potency.11 Toxicity of the sulfa drug component of these combinations is often a serious problem. In addition, the potent but toxic nonclassical antifolates trimetrexate (TMQ) and piritrexim (PTX; Figure 1), coadministered with leucovorin for host rescue, are also used.11 Serious toxicities associated with the use of TMQ and PTX often force the cessation of treatment.11 Mycobacterium avium (M. avium) complex or MAC infection also adversely affects the quality of life of AIDS patients.11 Thus, it is of considerable interest to incorporate selectivity and potency into a single nonclassical antifolate that can be used alone to treat these infections.
Gangjee et al.12 recently described the design and synthesis of several nonclassical 5-methyl-6-[(substituted-phenyl)sulfanyl]-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamines (1b–i) as DHFR inhibitors against opportunistic pathogens that infect AIDS patients. Some of these compounds were potent and selective against DHFR from both tg and M. avium compared to mammalian DHFR. Compound 1b with a 1-naphthyl substituent is 16-fold more potent and equally selective against tgDHFR as the clinically used TMP. The enhanced selectivity and potency of these analogues were in part attributed to the hydrophobic interaction between the 6-hydrophobic phenyl ring with Phe69 in pcDHFR, Phe91 in tgDHFR, and Val58 in maDHFR (vs the lack of interaction with hydrophilic Asn64 in hDHFR in the same region). Hydrophobic interactions at this region of the molecule with DHFR is complemented with the hydrophobic interaction of the 5-methyl moiety with Ile123 (pcDHFR), Val151 (tgDHFR), and Ile102 (maDHFR). The 5-methyl group also affects the conformations of the 6-side chain in these molecules and could contribute to the potency and/or selectivity. Molecular modeling (Sybyl 7.0)6 suggested that an extension of the 5-methyl group to an ethyl group might enhance the potency and selectivity against some pathogenic DHFR. Thus, a series of nonclassical 5-ethyl-6-[(substituted-phenyl)-sulfanyl]-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamines (3–17) were also synthesized.
Chemistry
Syntheses of 3–18 (Scheme 1) were achieved by oxidative thiolation of intermediate 20 with the appropriate thiophenol as previously reported.13 Intermediate 20 was in turn obtained from the reaction of malononitrile with 1-hydroxy-2-butanone (21) via a two-step procedure reported by Taylor et al.14 Coupling of 18 with diethyl-l-glutamate using isobutyl chloroformate as the activating agent15 afforded the diester 19. Final saponification of the diester gave the desired diacid 2 in 70% over two steps.
Scheme 1.

Biological Evaluation and Discussion
DHFR Inhibition
Compounds 2–17 were evaluated as inhibitors of Escherichia coli (ec) and recombinant human (rh) DHFR.4 The inhibitory potency (IC50) values are listed in Table 1 and compared with MTX, TMP, and the previously reported values for 1a.4 The most potent inhibitor against both ecDHFR and hDHFR in the C5-ethyl series was the classical analogue 2. Against human DHFR, 2 was 3-fold more potent than the 5-methyl substituted parent compound 1a, thus supporting our hypothesis that homologation of the C5-methyl of 1a to an ethyl would enhance its interaction with DHFR.
Table 1. Inhibitory Concentrations (IC50 in μM) and Selectivity Ratios for 2–17 Against Isolated Human and ecDHFRa.
| cmpd | ecDHFRb | human DHFRc | selectivity ratios human DHFR/ecDHFR |
|---|---|---|---|
| 1ad | 0.016 | 0.21 | 13 |
| 2 | 0.002 | 0.066 | 33 |
| 3 | 0.15 | 2.9 | 20 |
| 4 | 0.15 | 6.0 | 40 |
| 5 | 0.09 | 5.8 | 64.4 |
| 6 | 0.14 | 2.7 | 19.3 |
| 7 | 0.16 | >31 (30%) | >194 |
| 8 | nde | >31 (36%) | nde |
| 9 | 0.28 | 2.8 | 10 |
| 10 | 0.14 | 1.4 | 10 |
| 11 | nde | >28 (30%) | nde |
| 12 | 0.32 | 2.9 | 9.1 |
| 13 | 0.87 | 5.8 | 6.7 |
| 14 | 0.32 | >32 (35%) | >100 |
| 15 | 0.17 | 18 | 106 |
| 16 | 0.16 | 1.4 | 8.75 |
| 17 | 3.2 | 32 | 10 |
| methotrexate | 0.0066 | 0.022 | 3.33 |
| TMP | 0.02 | 680 | 34 000 |
The percent inhibition was determined at a minimum of four inhibitor concentrations within 20% of the 50% point. The standard deviations for determination of 50% points were within ± 10% of the value given.
Kindly provided by Dr. R. L. Blakely, St. Jude Children's Hospital, Memphis TN.
Kindly provided by Dr. J. H. Friesheim, Medical College of Ohio, Toledo, OH.
Data derived from ref 4.
nd = not determined.
In the nonclassical analogues, compound 5 with a 4-nitrophenyl substitution was the most potent compound in this series against ecDHFR, but it was 45-fold less potent than the classical 2. A number of the nonclassical compounds in this series showed good selectivity for ecDHFR as compared to human DHFR. Compounds 5, 7, 14, and 15 were 64- to 194-fold more selective for ecDHFR than human DHFR. Thus the 6-substituted phenyl analogs were reasonably selective for bacterial DHFR. The structure–activity relationship (SAR) among these analogues suggests that selectivity for ecDHFR over hDHFR is independent of the electronic nature of the substituent but that mono meta-substitution is most favorable to ecDHFR selectivity.
Compounds 3–17 were also evaluated as inhibitors against pcDHFR, tgDHFR, maDHFR, and rlDHFR (which served as the mammalian standard) and compared with TMP, TMQ, and the previously reported C5-methyl analogues 1b–i where applicable. The results (IC50) are reported in Table 2.
Table 2. Inhibitory Concentrations (IC50) in μM and Selectivity Ratios for 3–17 against P. carinii (pc), T. gondii (tg), M. avium (ma), and Rat Liver (rl) DHFRa.
| inhibition concn (IC50 mM) | selectivity ratio (IC50/IC50) | ||||||
|---|---|---|---|---|---|---|---|
|
|
|
||||||
| cmpd | pcDHFR | tgDHFR | maDHFR | rlDHFR | rl/pc | rl/tg | rl/ma |
| 3 | 18.4(20%)b | 3.15 | 19.8 | 40.1 | nd | 12.7 | 2.0 |
| 1b | 37.3 | 0.16 | 0.70 | 4.57 | 0.1 | 28.5 | 6.5 |
| 4 | 13.88 | 5.12 | 6.52 | 8.62 | 0.6 | 1.7 | 1.3 |
| 5 | 8.68 | 0.32 | 0.37 | 3.2 | 0.4 | 10.0 | 8.6 |
| 6 | 26.6 | 11.6 | 7.40 | 10.1 | 0.4 | 0.9 | 1.4 |
| 1c | 42.9 | 6.94 | 4.40 | 47.7 | 1.1 | 6.9 | 10.8 |
| 7 | 4.02 | 0.50 | 0.42 | 6.33 | 1.6 | 12.6 | 15.1 |
| 1d | 16.7 | 8.93 | 4.00 | 19 | 1.1 | 2.1 | 4.8 |
| 8 | 11.9 | 1.54 | 0.70 | 5.48 | 0.5 | 3.6 | 7.8 |
| 1h | 20(14%)b | 20.0 | 19.0 | 110 | 9.1 | 5.5 | 5.8 |
| 9 | 20.7 | 27.6 | 16.9 | 81.6 | 3.9 | 3.0 | 4.8 |
| 10 | 7.19 | 0.22 | 0.35 | 1.17 | 0.2 | 5.3 | 3.3 |
| 11 | 20.6 | 3.80 | 12.0 | 4.0 | 0.2 | 1.1 | 0.33 |
| 1g | 112 | 2.69 | 0.69 | 31.3 | 0.28 | 11.6 | 45.4 |
| 12 | 53 | 4.62 | 3.07 | 31.8 | 0.6 | 6.9 | 10.4 |
| 1i | 14.6 | 0.17 | 0.1 | 7.8 | 0.5 | 45.9 | 78 |
| 13 | 6.04 | 0.12 | 0.10 | 4.05 | 0.7 | 33.8 | 40.5 |
| 14 | 28.3 | 1.09 | 0.77 | 31 | 1.1 | 28.4 | 40.5 |
| 15 | 21.8 | 0.18 | 0.88 | 5.6 | 0.3 | 31.1 | 6.36 |
| 16 | 9.58 | 0.16 | 0.11 | 3.55 | 0.4 | 22.2 | 32.3 |
| 17 | 30.2 | 7.10 | 18.4 | 10.1 | 0.3 | 1.4 | 0.55 |
| TMQ | 0.042 | 0.010 | 0.0015 | 0.003 | 0.07 | 0.3 | 2 |
| TMP | 12 | 2.8 | 0.3 | 180 | 15 | 64 | 600 |
These assays were carried out at 37 °C under conditions of saturating substrate (90 μM dihydrofolic acid) and cofactor (119 μM NADPH) in the presence of 150 mM KCl.
Value in parentheses is the percent inhibition at the concentration indicated.
None of the analogues were potent or selective inhibitors of pcDHFR.
For tgDHFR in the 6-phenyl substituted analogues 5–15 and 17, compounds 13 and 15 were most potent followed by 5, 7, and 10. Compounds with a single 4-halogen (6 and 8), 3,4-diCl (9), or 2,6-disubstituted compounds (11 and 17) were comparatively poor inhibitors. Among the 6-bicyclic compounds 3, 4, and 16, only the quinoline 16 has activity comparable to 13 and 15. The naphthyl analogues were 10-fold less potent. Selectivity for tgDHFR was the highest for 13 with 14, 15, and 16 in the same range. Thus, 13 with a 2,5-diOMePh was the most potent and selective. For maDHFR, the most potent analogues were again 13 and 16, with 10 a close third. These were followed by 5, 7, 8, 14, and 15.
With the exception of 7, the trends for potency against maDHFR was quite similar to tgDHFR. Selectivity for maDHFR was dependent on the substitution with 13 and 14 being the most selective, followed by 16. Once again, as with tgDHFR, 13 was the most potent and selective analogue of the series.
The 5-ethyl compounds were also compared with the corresponding 5-methyl analogues from the previous study.12 This comparison afforded variable results. Compound 13, the most potent and selective analogue for both tgDHFR and maDHFR, was not much different in both potency and selectivity from its 5-methyl analogue 1i. Similarly, compounds 9 and 1h were not very different. However, 4 and 1b, 7 and 1c, 8 and 1d, and 12 and 1g (for maDHFR) were quite different with respect to potency and selectivity for one or both pathogen DHFR. For the 2,5-diOMePh analogues and the 3,4-diCl analogues, the 5-substitution of methyl or ethyl is immaterial; however, for the 2-naphthyl analogue, there is a significant difference in the 5-methyl and 5-ethyl analogues. Clearly these results reflect the ease (or difficulty) with which the particular enzyme simultaneously accommodates both the 5- and the 6-substitutions.
Compound 13 emerges as the best analogue being 35-fold and 39-fold more selective for tgDHFR and maDHFR, respectively. In addition, it was 23-fold more potent against tgDHFR than TMP and 3-fold more potent as TMP against maDHFR.
In Vitro Human Tumor Cell Growth Inhibition
The classical analogue 2 was also evaluated as inhibitors of the in vitro growth of CCRF-CEM human leukemia cells and of three CCRF-CEM sublines (Table 3) with single defined mechanisms of resistance to MTX during continuous exposure. The results (Table 3) indicate that the classical 6-ethyl analogue 2 was highly cytotoxic against the growth of CCRF-CEM cells in culture and had an EC50 about 3-fold more potent than the parent 5-methyl compound 1a. This indicates that homologation of the C5-methyl to an ethyl for the classical 5-substituted 2,4-diamino pyrolo[2,3-d]pyrimindine is highly conducive to tumor growth inhibition in culture and may indicate better transport and more potent hDHFR inhibition.
Table 3.
Growth Inhibition of Parental CCRF-CEM Human Leukemia Cells and Sub-lines with Single, Defined Mechanisms of MTX Resistance during Continuous (0–120 h) Exposure to MTX, 1a, or 2. Values Presented Are Average ± Range for n = 2 and Average ± S.D. for n > 2.
| EC50, nM | ||||
|---|---|---|---|---|
|
|
||||
| drug | CCRF-CEM | R1a (↑ DHFR) |
R2b (↓ uptake) |
R30dmc (↓ Glun) |
| MTX | 13.3 ± 1.5 (n = 4) |
410 ± 130 (n = 4) |
1950 ± 150 (n = 2) |
15.7 ± 0.9 (n = 3) |
| 1ad | 190 ± 10 (n = 2) |
8300 ± 800 (n = 2) |
7700 ± 300 (n = 2) |
515 ± 15 (n = 2) |
| 2 | 68 ± 11 (n = 2) |
5700 ± 200 (n = 2) |
6700 ± 300 (n = 2) |
200 ± 25 (n = 2) |
CCRF-CEM subline resistant to MTX solely as a result of a 20-fold increase in wild-type DHFR protein and activity.16
CCRF-CEM subline resistant as a result of decreased uptake of MTX.17
CCRF-CEM subline resistant to MTX solely as a result of decreased polyglutamylation; this cell line has 1% of the FPGS specific activity (measured with MTX as the folate substrate) of parental CCRF-CEM.18
Data from ref 4.
DHFR overexpressing line R116 was 84-fold cross-resistant to 2, which is slightly higher than the cross-resistance of R1 to MTX (31-fold), indicating that DHFR is likely the primary target of this 2,4-diamino-pyrrolo[2,3-d]pyrimidine. The MTX-transport resistant subline R2,17 which does not express functional reduced folate carrier (RFC), is 99-fold cross-resistant to 2, while it is 147-fold cross-resistant to MTX. The data suggest that 2 utilizes the RFC as its primary means of transport. The R30dm subline18 expressing low levels of folylpolyglutamate synthetase (FPGS) is 3-fold cross-resistant to 2 under continuous exposure conditions, suggesting that polyglutamates of 2 must be considered as part of its mechanism of action.
Metabolite protection studies were performed to further elucidate the mechanism of action of 2. At concentrations of drug that inhibited the growth of CCRF-CEM cells by ≥90%, leucovorin at ≥0.1 μM was able to fully protect against the effects of MTX and 2 (data not shown). This is consistent with an antifolate mechanism of action for 2. Further studies in CCRF-CEM examined the ability of thymidine (TdR) and hypoxanthine (Hx) to protect against growth inhibition of 2 (Table 4). These metabolites can be salvaged to produce dTTP and the purine dNTPs required for DNA synthesis and, thus, bypass the MTX blockade.19 As described in Experimental Section, in T-lymphoblast cell lines like CCRF-CEM, TdR can only be tested in the presence of dCyd, which reverses its toxic effects; however, dCyd has no protective effect on MTX either alone or in paired combination with either Hx or TdR (Table 4; footnote). The data (Table 4) show that for 2, neither TdR nor Hx alone protected to any significant extent; both metabolites were required to achieve significant protection. This indicates that both purine and thymidylate syntheses are inhibited and is consistent with DHFR being the primary target of 2.
Table 4. Protection of CCRF-CEM Human Leukemia Cells against the Growth Inhibitory Effects of MTX and 2 by 10 μM Hx, 5 μM TdR, and Their Combinationa.
| relative growthb (%) | ||||
|---|---|---|---|---|
|
|
||||
| drug | no addition | 5 μM TdR | 10 μM Hx | Hx+TdR |
| MTX (40 nM) | 11 ± 0 | 13 ± 1 | 14 ± 1 | 74 ± 1 |
| 2 (5000 nM) | 12 ± 0 | 15 ± 0 | 12 ± 0 | 74 ± 2 |
Growth is expressed relative to quadruplicate cultures not treated with either drug or metabolite and is the average ± range for duplicate-treated samples. The experiment was repeated with similar results.
Deoxycytidine (dCyd; 10 μM) was present in all the above cultures to prevent the inhibition of growth caused in T-cell leukemias like CCRF-CEM by TdR (see Experimental Section). In typical results, dCyd alone had no effect on CCRF-CEM growth (99 ± 3% of control) and did not protect against growth inhibition by either drug (data not shown). Hx alone (97 ± 1% of control) or in the presence of dCyd (98 ± 1% of control) did not affect CCRF-CEM growth and did not protect against MTX-induced growth inhibition (table above). TdR alone inhibited growth of CCRF-CEM (31 ± 1% of control), but TdR+dCyd was essentially not growth inhibitory (95 ± 2% of control) and neither protected against MTX-induced growth inhibition (table above). Similarly, Hx+TdR+dCyd did not appreciably inhibit growth of CCRF-CEM (96 ± 4% of control).
Compound 2 was selected by the NCI for evaluation in its in vitro preclinical antitumor screening program.5 The ability of compound 2 to inhibit the growth of tumor cell lines was measured as GI50 values, the concentration required to inhibit the growth of tumor cells in culture by 50% as compared to a control. In 25 of the 59 cell lines, compound 2 showed GI50 values of ≤1 × 10−7 M (Table 5). In three of the leukemia cell lines, four of the colon cancer cell lines, and one melanoma, the GI50 values for 2 were two digit nanomolar (<100 nM) and paralleled the hDHFR IC50 value. In addition, the spectrum of tumor inhibition (GI50 ≤ 1 × 10−7 M) was increased from 11 for 1a to 25 for 2, indicating that homologation of the 5-methyl to the 5-ethyl was instrumental in increasing the spectrum of tumor inhibition in culture. It was also interesting to note that compound 2 was not a general cell poison but showed selectivity both within a type of tumor cell line as well as across different tumor cell lines with inhibitory values that in some instances differed by 10 000-fold (data not shown).
Table 5. Cytotoxic Evaluation (GI50, M) of Compounds 1a4 and 2 against Selected Tumor Cell Lines5.
| cell line | GI50 of 1a (M) | GI50 of 2 (M) |
|---|---|---|
| Leukemia | ||
| CCRF-CEM | 2.2 × 10−7 | 1.7 × 10−8 |
| K-562 | 2.7 × 10−8 | 1.2 × 10−8 |
| MOLT-4 | <1.0 × 10−8 | 4.8 × 10−7 |
| RPMI-8226 | <1.0 × 10−8 | 5.9 × 10−7 |
| SR | –a | <1.0 × 10−8 |
| Non-Small Cell Lung Cancer | ||
| NCI-H460 | 6.3 × 10−7 | 1.8 × 10−7 |
| Colon Cancer | ||
| HCT-116 | – | 2.3 × 10−8 |
| HT29 | 1.6 × 10−7 | 9.8 × 10−8 |
| – | KM12 | 5.6 × 10−8 |
| SW-620 | 1.2 × 10−7 | 2.6 × 10−8 |
| Central Nervous System Cancer | ||
| SF-268 | – | 4.3 × 10−7 |
| SF-295 | – | 6.6 × 10−7 |
| SF-539 | – | 2.4 × 10−7 |
| U-251 | 8.2 × 10–7 | 5.3 × 10−7 |
| Melanoma | ||
| LOX IMVI | 1.7 × 10−7 | 4.4 × 10−8 |
| UACC-62 | – | 8.8 × 10−7 |
| Ovarian Cancer | ||
| OVCAR-8 | – | 7.4 × 10−7 |
| Renal Cancer | ||
| ACHN | – | 4.4 × 10−7 |
| CAKI-1 | – | 1.7 × 10−7 |
| SN12C | – | 6.0 × 10−7 |
| UO-31 | – | 3.0 × 10−7 |
| Prostate Cancer | ||
| PC-3 | 4.4 × 10−7 | 4.0 × 10−7 |
| DU-145 | – | 3.7 × 10−7 |
| Breast Cancer | ||
| MCF7 | – | 1.7 × 10−7 |
| MDA-MB-435 | – | 6.6 × 10−7 |
– = concn > 1.0 × 10−7 M.
In summary, diethyl N-{4-[(2,4-diamino-5-ethyl-7H-pyrrolo-[2,3-d]pyrimidin-6-yl)sulfanyl]benzoyl}-l-glutamic acid 2, the C5-ethyl homologue of compound 1a, and its nonclassical analogues 3–17 were designed and synthesized to investigate the effect of homologation of the 5-methyl substituent on DHFR inhibitory potency, selectivity, and antitumor activity. The biological results indicate that elongation of the C5-methyl classical, 5,6-disubstituted pyrrolo[2,3-d]pyrimidine (1a) to an ethyl group (2) increases the inhibitory activity against both DHFR and the growth of tumor cells in culture and significantly increases the spectrum of tumor inhibition in culture compared to the C5-methyl parent compound 1a. The DHFR inhibitory activity of the 15 nonclassical analogues indicates that these compounds are potent inhibitors of ecDHFR, tgDHFR, and maDHFR, with moderate to good selectivity compared to mammalian DHFR. SAR for the nonclassical analogues indicates that the 2,5-dimethoxy substitution on the phenyl ring provides the most potent and selective inhibitor for tgDHFR and maDHFR. This 2,5-dimethoxy phenyl substitution occurs in several other potent and selective DHFR inhibitors, such as PTX. Potency and selectivity was also found with the unsubstituted phenyl analogue and to some extent with electron withdrawing chloro and nitro substitutions. Homologation of the 5-methyl group to an ethyl had variable effects on the inhibition of pathogenic DHFR and mammalian DHFR and are dependent on the nature of the 6-position substituent.
Experimental Section
All evaporations were carried out in vacuo with a rotary evaporator. Analytical samples were dried in vacuo (0.2 mmHg) in a CHEM-DRY drying apparatus over P2O5 at 80 °C. Melting points were determined on a MEL-TEMP II melting point apparatus with FLUKE 51 K/J electronic thermometer and are uncorrected. Nuclear magnetic resonance spectra for proton (1H NMR) were recorded on a Bruker WH-300 (300 MHz) spectrometer. Chemical shift values are expressed in ppm (parts per million) relative to tetramethylsilane as internal standard; s = singlet, d = doublet, t = triplet, q = quartet, m = mutiplet, br = broad singlet. The relative integrals of peak areas agreed with those expected for the assigned structures. Thin-layer chromatography (TLC) was performed on Polygram Sil G/UV254 silica gel plates with fluorescent indicator, and the spots were visualized under 254 and 366 nm illumination. Proportions of solvents used for TLC are by volume. Column chromatography was performed on 230–400 mesh silica gel purchased from Aldrich, Milwaukee, WI. LCMS analysis was performed with an HP 1100 series LC/MSD equipped with a Waters Nova-Pak C18 column and with a Finnigan LCQ equipped with a Phenomenox Luna C18 column. Elemental analyses were performed by Atlantic Microlab, Inc., Norcross, GA. Element compositions are within ±0.4% of the calculated values. Fractional moles of water or organic solvents frequently found in some analytical samples of antifolates could not be prevented in spite of 24–48 h of drying in vacuo and were confirmed where possible by their presence in the 1H NMR spectra. All solvents and chemicals were purchased from Aldrich Chemical Co. or Fisher Scientific and were used as received.
General Procedure for the Synthesis of Compounds 3–18
To a solution of 5-ethyl-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamine (20;14 2–3 mmol) in EtOH/H2O (1:1) was added two equivalents of the appropriately substituted aromatic thiol, and the reaction mixture heated to reflux. At reflux, two equivalents of I2 was added and the reaction continued for 2 h. when TLC showed the disappearance of starting material 20. At this point, the reaction was stopped and the reaction mixture was allowed to cool. The precipitated disulfide was filtered out and the filtrate was evaporated to dryness under reduced pressure. To the resulting residue was added methanol and silica gel, and this suspension was evaporated to dryness. The silica gel plug was loaded on a dry silica gel column and flushed with CHCl3 (500 mL) and eluted with a gradient of 1% to 5% MeOH in CHCl3. Fractions containing the product were pooled and evaporated to afford pure 5-ethyl-6-[(substituted-phenyl)sulfanyl]-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamines 3–19.
5-Ethyl-6-(1-naphthylsulfanyl)-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamine (3)
Compound 3 was synthesized from 20 (0.35 g, 2 mmol), 1-naphthalene thiol (0.66 g, 4 mmol), and iodine (1.02 g, 4 mmol) using the general procedure described above to afford 159 mg (24%) of 3 as a white solid: mp 291.7 °C (dec); TLC Rf 0.57 (CHCl3/MeOH, 5:1, silica gel); 1H NMR (DMSO-d6) 0.99–1.07 (t, 3 H, 5-CH2CH3), 2.76–2.78 (q, 2 H, CH2CH3), 5.60 (s, 2 H, 2- or 4-NH2 exch with D2O), 6.21 (s, 2 H, 2- or 4-NH2 exch with D2O), 6.81–8.25 (m, 7H, C10H7), 11.01 (s, 1 H, 7-H). Anal. (C18H17N5S) C, H, N, S.
5-Ethyl-6-(2-naphthylsulfanyl)-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamine (4)
Compound 4 was synthesized from 20 (0.35 g, 2 mmol), 2-naphthalene thiol (0.66 g, 4 mmol), and iodine (1.02 g, 4 mmol) using the general procedure described above to afford 298 mg (45%) of 4 as a white solid: mp 231 °C (dec); TLC Rf 0.57 (CHCl3/MeOH, 5:1, silica gel); 1H NMR (DMSO-d6) 0.99–1.07 (t, 3 H, 5-CH2CH3), 2.76–2.78 (q, 2 H, CH2CH3), 5.60 (s, 2 H, 2- or 4-NH2 exch with D2O), 6.21 (s, 2 H, 2- or 4-NH2 exch with D2O), 6.81–8.25 (m, 7H, C10H7), 11.01 (s, 1 H, 7-H). Anal. (C18H17N5S.0.20H2O) C, H, N, S.
5-Ethyl-6-[(4-nitrophenyl)sulfanyl]-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamine (5)
Compound 5 was synthesized from 20 (0.35 g, 2 mmol), 4-nitrobenzenethiol (0.62 g, 4 mmol), and iodine (1.02 g, 4 mmol) using the general procedure described above to afford 228 mg (35%) of 5 as an orange solid: mp 286.4 °C (dec); TLC Rf 0.55 (CHCl3/MeOH, 5:1, silica gel); 1H NMR (DMSO-d6) 0.99–1.04 (t, 3 H, 5-CH2CH3), 2.72–2.75 (q, 2 H, CH2CH3), 5.67 (s, 2 H, 2- or 4-NH2 exch with D2O), 6.29 (s, 2 H, 2- or 4-NH2 exch with D2O), 7.16–7.18 (d, 2 H, 3′,5′-CH), 8.11–8.14 (d, 2 H, 2′, 6′-CH) 11.09 (s, 1 H, 7-H). Anal. (C14H14N6O2S•0.40H2O) C, H, N, S.
6-[(4-Bromophenyl)sulfanyl]-5-ethyl-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamine (6)
Compound 6 was synthesized from 20 (0.35 g, 2 mmol), 4-bromobenzenethiol (0.75 g, 4 mmol), and iodine (1.02 g, 4 mmol) using the general procedure described above to afford 273 mg (38%) of 6 as a white solid: mp 272.5 °C (dec); TLC Rf 0.55 (CHCl3/MeOH, 5:1, silica gel); 1H NMR (DMSO-d6) 0.98–1.03 (t, 3 H, 5-CH2CH3), 2.72–2.74 (q, 2 H, CH2CH3), 5.58 (s, 2 H, 2- or 4-NH2 exch with D2O), 6.19 (s, 2 H, 2- or 4-NH2 exch with D2O), 6.91–6.94 (d, 2 H, 3′,5′-CH), 7.43–7.45 (d, 2 H, 2′, 6′-CH) 10.97 (s, 1 H, 7-H). Anal. (C14H14N5SBr•0.30H2O) C, H, N, S, Br.
6-[(3-Chlorophenyl)sulfanyl]-5-ethyl-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamine (7)
Compound 7 was synthesized from 20 (0.35 g, 2 mmol), 3-chlorobenzenethiol (0.57 g, 4 mmol), and iodine (1.02 g, 4 mmol) using the general procedure described above to afford 88 mg (14%) of 7 as a white solid: mp 244.5 °C TLC Rf 0.56 (CHCl3/MeOH, 5:1, silica gel); 1H NMR (DMSO-d6) 0.99–1.03 (t, 3 H, 5-CH2CH3), 2.73–2.76 (q, 2 H, CH2CH3), 5.62 (s, 2 H, 2- or 4-NH2 exch with D2O), 6.23 (s, 2 H, 2- or 4-NH2 exch with D2O), 6.95–7.29 (m, 4 H, C6H4), 11.01 (s, 1 H, 7-H). Anal. (C14H14N5SCl) C, H, N, S, Cl.
6-[(4-Chlorophenyl)sulfanyl]-5-ethyl-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamine (8)
Compound 8 was synthesized from 20 (0.35 g, 2 mmol), 4-chlorobenzenethiol (0.57 g, 4 mmol), and iodine (1.02 g, 4 mmol) using the general procedure described above to afford 95 mg (15%) of 8 as an off-white solid: mp 245.1 °C (dec); TLC Rf 0.56 (CHCl3/MeOH, 5:1, silica gel); 1H NMR (DMSO-d6) 0.99–1.03 (t, 3 H, 5-CH2CH3), 2.73–2.75 (q, 2 H, CH2CH3), 5.66 (s, 2 H, 2- or 4-NH2 exch with D2O), 6.27 (s, 2 H, 2- or 4-NH2 exch with D2O), 6.98–7.01 (d, 2 H, 3′,5′-CH), 7.31–7.34 (d, 2 H, 2′, 6′-CH) 11.02 (s, 1 H, 7-H). Anal. (C14H14N5SCl•0.40H2O) C, H, N, S, Cl.
6-[(3,4-Dichlorophenyl)sulfanyl]-5-ethyl-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamine (9)
Compound 9 was synthesized from 20 (0.35 g, 2 mmol), 3,4-dichlorobenzenethiol (0.71 g, 4 mmol), and iodine (1.02 g, 4 mmol) using the general procedure described above to afford 168 mg (24%) of 9 as a white solid: mp 300.7 °C (dec); TLC Rf 0.56 (CHCl3/MeOH, 5:1, silica gel); 1H NMR (DMSO-d6) 0.98–1.02 (t, 3 H, 5-CH2CH3), 2.74–2.76 (q, 2 H, CH2CH3), 5.53 (s, 2 H, 2- or 4-NH2 exch with D2O), 6.13 (s, 2 H, 2- or 4-NH2 exch with D2O), 6.80–7.03 (m, 3 H, C6H3), 11.02 (s, 1 H, 7-H). Anal. (C14H13N5SCl2) C, H, N, S, Cl.
6-[(2,4-Dichlorophenyl)sulfanyl]-5-ethyl-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamine (10)
Compound 10 was synthesized from 20 (0.35 g, 2 mmol), 2,4-dichlorobenzenethiol (0.71 g, 4 mmol), and iodine (1.02 g, 4 mmol) using the general procedure described above to afford 91 mg (13%) of 10 as a white solid: mp 291.6 °C (dec); TLC Rf 0.56 (CHCl3/MeOH, 5:1, silica gel); 1H NMR (DMSO-d6) 0.98–1.03 (t, 3 H, 5-CH2CH3), 2.68–2.71 (q, 2 H, CH2CH3), 5.65 (s, 2 H, 2- or 4-NH2 exch with D2O), 6.26 (s, 2 H, 2- or 4-NH2 exch with D2O), 7.30–7.62 (m, 3 H, C6H3), 11.01 (s, 1 H, 7-H). Anal. (C14H13N5SCl2•0.02CHCl3) C, H, N, S, Cl.
6-[(2,6-Dichlorophenyl)sulfanyl]-5-ethyl-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamine (11)
Compound 11 was synthesized from 20 (0.35 g, 2 mmol), 2,6-dichlorobenzenethiol (0.71 g, 4 mmol), and iodine (1.02 g, 4 mmol) using the general procedure described above to afford 126 mg (18%) of 11 as a white solid: mp 295.2 °C (dec); TLC Rf 0.56 (CHCl3/MeOH, 5:1, silica gel); 1H NMR (DMSO-d6) 0.75–0.80 (t, 3 H, 5-CH2CH3), 2.69–2.71 (q, 2 H, CH2CH3), 5.50 (s, 2 H, 2- or 4-NH2 exch with D2O), 6.07 (s, 2 H, 2- or 4-NH2 exch with D2O), 7.29–7.50 (m, 3 H, C6H3), 10.87 (s, 1 H, 7-H). Anal. (C14H13N5SCl2) C, H, N, S, Cl.
5-Ethyl-6-[(4-methoxyphenyl)sulfanyl]-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamine (12)
Compound 12 was synthesized from 20 (0.35 g, 2 mmol), 4-methoxybenzenethiol (0.56 g, 4 mmol), and iodine (1.02 g, 4 mmol) using the general procedure described above to afford 143 mg (23%) of 12 as an off-white solid: mp 276.7 °C (dec); TLC Rf 0.55 (CHCl3/MeOH, 5:1, silica gel); 1H NMR (DMSO-d6) 1.06–1.11 (t, 3 H, 5-CH2CH3), 2.89–2.91 (q, 2 H, CH2CH3), 3.79 (s, 3 H, 4-OCH3), 5.58 (s, 2 H, 2- or 4-NH2 exch with D2O), 6.19 (s, 2 H, 2- or 4-NH2 exch with D2O), 6.91–6.94 (d, 2 H, 3′,5′-CH), 7.43–7.45 (d, 2 H, 2′, 6′-CH) 10.97 (s, 1 H, 7-H). Anal. (C15H17N5SO) C, H, N, S.
6-[(2,5-Dimethoxyphenyl)sulfanyl]-5-ethyl-7H-pyrrolo[2,3-d]-pyrimidine-2,4-diamine (13)
Compound 13 was synthesized from 20 (0.35 g, 2 mmol), 2,5-dimethoxybenzenethiol (0.68 g, 4 mmol), and iodine (1.02 g, 4 mmol) using the general procedure described above to afford 204 mg (30%) of 13 as a white solid: mp 239.5 °C, TLC Rf 0.55 (CHCl3/MeOH, 5:1, silica gel); 1H NMR (DMSO-d6) 0.98–1.03 (t, 3 H, 5-CH2CH3), 2.55–2.69 (q, 2 H, CH2CH3), 3.52 (s, 3 H, OCH3), 3.85 (s, 3 H, OCH3), 5.59 (s, 2 H, 2- or 4-NH2 exch with D2O), 6.21 (s, 2 H, 2- or 4-NH2 exch with D2O), 6.62–6.91 (m, 3 H, C6H3), 10.89 (s, 1 H, 7-H). Anal. (C16H19N5SO2) C, H, N, S.
5-Ethyl-6-[(3-methoxyphenyl)sulfanyl]-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamine (14)
Compound 14 was synthesized from 20 (0.35 g, 2 mmol), 3-methoxybenzenethiol (0.56 g, 4 mmol), and iodine (1.02 g, 4 mmol) using the general procedure described above to afford 155 mg (25%) of 14 as an off-white solid: mp 182.2 °C, TLC Rf 0.55 (CHCl3/MeOH, 5:1, silica gel); 1H NMR (DMSO-d6) 0.98–1.03 (t, 3 H, 5-CH2CH3), 2.72–2.74 (q, 2 H, CH2CH3), 3.66 (s, 3 H, 3-OCH3), 5.58 (s, 2 H, 2- or 4-NH2 exch with D2O), 6.19 (s, 2 H, 2- or 4-NH2 exch with D2O), 6.91–6.94 (d, 2 H, 3′,5′-CH), 7.43–7.45 (d, 2 H, 2′, 6′-CH) 10.97 (s, 1 H, 7-H). Anal. (C15H17N5SO) C, H, N, S.
5-Ethyl-6-(phenylsulfanyl)-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamine (15)
Compound 15 was synthesized from 20 (0.35 g, 2 mmol), thiophenol (0.44 g, 4 mmol), and iodine (1.02 g, 4 mmol) using the general procedure described above to afford 180 mg (32%) of 15 as an off-white solid: mp 275 °C (dec); TLC Rf 0.55 (CHCl3/MeOH, 5:1, silica gel); 1H NMR (DMSO-d6) 0.99–1.03 (t, 3 H, 5-CH2CH3), 2.74–2.76 (q, 2 H, CH2CH3), 5.63 (s, 2 H, 2- or 4-NH2 exch with D2O), 6.24 (s, 2 H, 2- or 4-NH2 exch with D2O), 6.98–7.28 (m, 5 H, C6H5), 10.99 (s, 1 H, 7-H). Anal. (C14H15N5S•0.20H2O) C, H, N, S.
5-Ethyl-6-(8-quinolinylsulfanyl)-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamine (16)
Compound 16 was synthesized from 20 (0.35 g, 2 mmol), 2-quinolinethiol (0.64 g, 4 mmol), and iodine (1.02 g, 4 mmol) using the general procedure described above to afford 292 mg (44%) of 16 as a light brown solid: mp 260.5 °C (dec); TLC Rf 0.57 (CHCl3/MeOH, 5:1, silica gel); 1H NMR (DMSO-d6) 0.96–1.08 (t, 3 H, 5-CH2CH3), 2.76–2.79 (q, 2 H, CH2CH3), 5.65 (s, 2 H, 2- or 4-NH2 exch with D2O), 6.28 (s, 2 H, 2- or 4-NH2 exch with D2O), 7.47–8.21 (m, 6 H, C9H6) 11.10 (s, 1 H, 7-H). Anal. (C17H16N6S•1.10H2O) C, H, N, S.
6-[(2,6-Dimethylphenyl)sulfanyl]-5-ethyl-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamine (17)
Compound 17 was synthesized from 20 (0.35 g, 2 mmol), 2,6-dimethylbenzenethiol (0.55 g, 4 mmol), and iodine (1.02 g, 4 mmol) using the general procedure described above to afford 346 mg (56%) of 17 as a white solid: mp 290.5 °C (dec); TLC Rf 0.55 (CHCl3/MeOH, 5:1, silica gel); 1H NMR (DMSO-d6) 1.01 (t, 3 H, 5-CH2CH3), 2.78 (q, 2 H, CH2CH3), 2.08 (s, 6 H, 2- and 6-CH3), 5.58 (s, 2 H, 2- or 4-NH2 exch with D2O), 6.19 (s, 2 H, 2- or 4-NH2 exch with D2O), 6.91–6.94 (d, 2 H, 3′,5′-CH), 7.43–7.45 (d, 2 H, 2′, 6′-CH) 10.97 (s, 1 H, 7-H). Anal. (C16H19N5S) C, H, N, S.
4-[(2,4-Diamino-5-ethyl-7H-pyrrolo[2,3-d]pyrimidin-6-yl)sulfanyl]benzoic acid (18)
To a mixture of 20 (500 mg, 2.8 mmol) and 4-mercaptobenzoic acid (900 mg, 5.8 mmol) in EtOH/H2O (3:2) at reflux was added 2 equiv of iodine. The resulting mixture turned into a suspension within 1 min, and the precipitate formed dissolved to form a dark solution within half an hour. The reflux was continued for another 1.5 to 2 h, until TLC showed the disappearance of the starting materials and the formation of desired product. The reaction mixture was filtered to remove the excess disulfide, and the filtrate was evaporated to dryness under reduced pressure. EtOAc was added to the resulting residue, the mixture was refluxed for 15 min, and the precipitate formed was collected through filtration and recrystallized from MeOH to afforded 0.93 g (quantitative) of 18 as white crystals: mp >250 °C (dec); Rf = 0.45 (CHCl3/MeOH, 5:1); 1H NMR (DMSO-d6) δ 0.98–1.03 (t, 3 H, CH2CH3), 2.78–2.90 (q, 2 H, CH2CH3), 7.08–7.11 (d, 2 H, C6H4), 7.29 (s, 2 H, 2- or 4-NH2), 7.82–7.85 (d, 2 H, C6H4), 8.02 (s, 2 H, 4- or 2-NH2), 11.51 (br, 1 H, 7-NH), 12.20 (s, 1 H, COOH). HRMS (m/z) calcd for C15H15 N5O2S, 329.0946; found, 329.0959.
Diethyl N-{4-[(5-Ethyl-2-amino-3,4-dihydro-4-oxo-7H-pyrrolo[2,3-d]pyrimidin-6-yl)thio]benzoyl}-l-glutamate (19)
To a solution of 18 (125 mg, 0.38 mmol) in anhydrous DMF (9 mL) was added triethylamine (130 μL), and the mixture was stirred under nitrogen at room temperature for 5 min. The resulting solution was cooled to 0 °C, isobutyl chloroformate (130 μL 1.0 mmol) was added, and the mixture was stirred at 0 °C for 30 min. At this time. TLC (MeOH/CHCl3, 1:5) indicated the formation of the activated intermediate at Rf 0.55 and the disappearance of the starting acid at Rf 0.18. Diethyl-l-glutamate hydrochloride (240 mg, 1.0 mmol) was added to the reaction mixture followed immediately by triethylamine (130 μL, 1.0 mmol). The reaction mixture was slowly allowed to warm to room temperature and stirred under nitrogen for 24 h. TLC showed the formation of one major spot at Rf = 0.61 (MeOH/CHCl3, 1:5). The reaction mixture was evaporated to dryness under reduced pressure. The residue was dissolved in a minimum amount of CH3Cl/MeOH, 4:1, and chromatographed on a silica gel column (3 × 10 cm) with 4% MeOH in CHCl3 as the eluent. Fractions that showed the desired single spot at Rf 0.61 were pooled and evaporated to dryness and crystallized from ethyl ether to afford 165 mg (85%) of 19 as white crystals: mp 197.6–200 °C; 1H NMR (DMSO-d6) δ 0.98–1.03 (t, 3 H, C6-CH2CH3), 1.08–1.25 (m, 6 H, OEt), 1.90–2.08 (m, 2 H, Glu β-CH2), 2.32–2.35 (t, 2 H, Glu γ-CH2), 2.78–2.90 (q, 2 H, C6-CH2CH3), 4.00–4.13 (m, 4 H, OEt), 4.33–4.40 (m, 1 H, Glu α-CH), 6.25 (br, 2 H, 2- or 4-NH2), 6.90 (s, 2 H, 2- or 4-NH2), 7.06–7.08 (d, 2 H, Ph–CH), 7.76–7.78 (d, 2 H, Ph–CH), 8.69 (d, 1 H, -CONH), 11.45 (s, 1 H, 7-NH). HRMS (m/z) calcd for C24H30N6O5S, 514.1998; found, 514.2010.
N-{4-[(5-Ethyl-2-amino-3,4-dihydro-4-oxo-7H-pyrrolo[2,3-d]-pyrimidin-6-yl)thio]-benzoyl}-l-glutamic Acid (2)
To a solution of the diester 19 (150 mg, 0.3 mmol) in MeOH (10 mL) was added 1 N NaOH (6 mL), and the mixture was stirred under nitrogen at room temperature for 16 h. TLC showed the disappearance of the starting material (Rf = 0.55) and formation of one major spot at the origin (MeOH/CHCl3, 1:5). The reaction mixture was evaporated to dryness under reduced pressure. The residue was dissolved in water (10 mL), the resulting solution was cooled in an ice bath, and the pH was adjusted to 3–4 with dropwise addition of 1 N HCl. The resulting suspension was frozen in a dry ice–acetone bath and thawed in the refrigerator to 4–5 °C and filtered. The residue was washed with a small amount of cold water and ethyl acetate and dried in vacuo using P2O5 to afford 120 mg (88%) of 2 as a yellow powder: mp 213–215 °C (dec); 1H NMR (DMSO-d6) δ 0.98–1.03 (t, 3 H, C6-CH2CH3), 1.91–2.08 (m, 2 H, Glu β-CH2), 2.30–2.35 (t, 2 H, Glu γ-CH2), 2.71–2.77 (q, 2 H, C6-CH2CH3), 4.35 (m, 1 H, Glu α-CH), 5.72 (s, 2 H, 2-NH2), 6.58 (s, 2 H, 2-NH2), 7.05–7.07 (d, 2 H, Ph–CH), 7.76–7.78 (d, 2 H, Ph–CH), 8.53–8.55 (d, 1 H, -CONH), 11.24 (s, 1 H, 7-NH). Anal. (C20H22N6O5S•1.8H2O) C, H, N, S.
Cell Lines and Methods for Measuring Growth Inhibitory Potency (Table 3)
Solutions used in cell culture studies were standardized using extinction coefficients. Extinction coefficients were determined for 2 [pH 1, λmax 236 nm (29 300) and λmax 277 nm (23 400); pH 7, λmax 274 nm (22 700) and λmax 297 nm (20 200); pH 13, λmax 222 nm (30 200) and λmax 274 nm (22 400)]. Extinction coefficients for methotrexate (MTX), a gift from Immunex (Seattle, WA), were from the literature.20 Aminopterin, Hx, TdR, and deoxycytidine (dCyd) were purchased from Sigma Chemical Co. (St. Louis, MO). Other chemicals and reagents were reagent grade or higher.
Cell Lines and Methods for Measuring Growth Inhibitory Potency
Cell lines were verified to be negative for Mycoplasma contamination (Mycoplasma Plus PCR primers, Stratagene, La Jolla, CA). The human % lymphoblastic leukemia cell line CCRF-CEM21 and its MTX-resistant sublines R1,16 R2,17 and R30dm18 were cultured as described.18 R1 expresses 20-fold elevated levels of DHFR, the target enzyme of MTX. R2 has dramatically reduced MTX uptake but normal levels of MTX-sensitive DHFR. R30dm expresses 1% of the FPGS activity of CCRF-CEM and is resistant to short-term, but not continuous, MTX exposure; however, R30dm is cross-resistant in continuous exposure to antifolates that require polyglutamylation to form potent inhibitors. Growth inhibition of all cell lines by continuous drug exposure was assayed as described.18 EC50 values (drug concentration effective at inhibiting cell growth by 50%) were determined visually from plots of percent growth relative to a solvent-treated control culture versus the logarithm of drug concentration.
Protection against growth inhibition of CCRF-CEM cells was assayed by including leucovorin ((6R,S)-5-formyltetrahydrofolate) at 0.1–10 μM with a concentration of drug previously determined to inhibit growth by 90–95%; the reminder of the assay was as described. Growth inhibition was measured relative to the appropriate leucovorin-treated control; leucovorin, even at 10 μM, caused no growth inhibition in the absence of drug, however. Protection against growth inhibition of CCRF-CEM cells was assayed by including Hx (10 μM), TdR (5 μM), or dCyd (10 μM) individually, in pairs (Hx+dCyd, TdR+dCyd), or all together (Hx+TdR+dCyd), with concentrations of MTX or 2 that would inhibit growth by ≈90–95% over a growth period of ≈72 h. The growth period was limited because beyond 72 h, CCRF-CEM cells deplete TdR in the growth media and drug effects are no longer protected. dCyd is added only to alleviate the growth inhibitory effects of 5 μM TdR against CCRF-CEM cells.20 Controls with metabolites alone (no drug) in the combinations described above (in duplicate), controls with drug alone with no metabolites (in duplicate), and untreated controls with neither drugs nor metabolites (in quadruplicate) were performed. Growth inhibition was measured as percent growth relative to untreated control cells (absence of drugs and metabolites).
Supplementary Material
Acknowledgments
This work was supported in part by grants from the National Institutes of Health CA89300 (A.G.), AI44661 (A.G.), and CA43500 (J.J.M.) and the Roswell Park Cancer Institute CCSG CA16065 and CA10914 (R.L.K.). The authors thank Mr. William H. Haite for performing growth inhibition and metabolic protection studies.
Footnotes
Abbreviations: DHFR, dihydrofolate reductase; TS, thymidylate synthase; NCI, National Cancer Institute; AIDS, acquired immunodeficiency syndrome; pc, Pneumocystis carinii; tg, Toxoplasma gondii; TMP, trimethoprim; TMQ, trimetrexate; PTX, piritrexim; M. avium, Mycobacterium avium; ec, Escherichia coli; rh, recombinant human; SAR, structure–activity relationship; FPGS, folylpolyglutamate synthetase; TdR, thymidine; Hx, hypoxanthine.
Supporting Information Available: Results from elemental analyses. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Berman EM, Werbel LM. The Renewed Potential for Folate Antagonists in Contemporary Cancer Chemotherapy. J Med Chem. 1991;34:479–485. doi: 10.1021/jm00106a001. [DOI] [PubMed] [Google Scholar]
- 2.Rosowsky A. Chemistry and Biological Activity of Antifolates. In: Ellis GP, West GB, editors. Progress in Medicinal Chemistry. Elsevier Science Publishers; Amsterdam: 1989. pp. 1–252. [DOI] [PubMed] [Google Scholar]
- 3.Gangjee A, Elzein E, Kothare M, Vasudevan A. Classical and Nonclassical Antifolates as Potential Antitumor, Antipneumocystis and Antitoxoplasma Agents. Curr Pharm Des. 1996;2:263–280. [Google Scholar]
- 4.Gangjee A, Lin X, Kisliuk RL, McGuire JJ. Synthesis of N-{4-[(2,4-Diamino-5-methyl-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)thio]benzoyl}-l-glutamic Acid and N-{4-[(2-Amino-4-oxo-5-methyl-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)thio]benzoyl}-l-glutamic Acid as Dual Inhibitors of Dihydrofolate Reductase and Thymidylate Synthase and as Potential Antitumor Agents. J Med Chem. 2005;48:7215–7222. doi: 10.1021/jm058234m. [DOI] [PubMed] [Google Scholar]
- 5.We thank the Developmental Therapeutics Program of the National Cancer Institute for performing the in vitro anticancer evaluation.
- 6.Tripos Inc., 1699 South Handley Rd, Suite 303, St. Louis, MO 63144.
- 7.Piper JR, McCaleb GS, Montgomery JA, Kisliuk RL, Gaumont Y, Sirotnak FM. Synthesis and Antifolate Activity of 5-Methyl-5-deaza Analogs of Aminopterin, MTX, Folic Acid, and N10-Methyl Folic Acid. J Med Chem. 1986;29:1080–1087. doi: 10.1021/jm00156a029. [DOI] [PubMed] [Google Scholar]
- 8.Hynes JB, Harmon SJ, Floyd GG, Farrington M, Hart LD, Gale GR, Washtein WL, Sustern SS, Freisheim JH. Chemistry and Antitumor Evaluation of Selected Classical 2,4-Diaminoquinazoline Analogues of Folic Acid. J Med Chem. 1985;28:209–215. doi: 10.1021/jm00380a011. [DOI] [PubMed] [Google Scholar]
- 9.Zhao R, Goldman ID. Resistance to Antifolates. Oncogene. 2003;22:7431–7457. doi: 10.1038/sj.onc.1206946. [DOI] [PubMed] [Google Scholar]
- 10.Werbel LM. In: Folate Antagonists as Therapeutic Agents. Sirotnak FM, Burchall JJ, Ensminger W, Montgomery JA, editors. Vol. 1. Academic Press; Orlando, FL: 1984. p. 261. [Google Scholar]
- 11.Klepser ME, Klepser TB. Drug Treatment of HIV-Related Opportunistic Infections. Drugs. 1997;53:40–73. doi: 10.2165/00003495-199753010-00004. [DOI] [PubMed] [Google Scholar]
- 12.Gangjee A, Lin X, Queener SF. Design, Synthesis, and Biological Evaluation of 2,4-Diamino-5-methyl-6-substituted-pyrrolo[2,3-d]pyrimidines as Dihydrofolate Reductase Inhibitors. J Med Chem. 2004;47:3689–3692. doi: 10.1021/jm0306327. [DOI] [PubMed] [Google Scholar]
- 13.Gangjee A, Mavandadi F, Kisliuk RL, McGuire JJ, Queener SF. 2-Amino-4-oxo-5-substituted-pyrrolo[2,3-d]pyrimidines as Non-classical Antifolate Inhibitors of Thymidylate Synthase. J Med Chem. 1996;39:4563–4568. doi: 10.1021/jm960097t. [DOI] [PubMed] [Google Scholar]
- 14.Taylor EC, Patel HH, Jun JG. A One-Step Ring Transformation/Ring Annulation Approach to Pyrrolo[2,3-d]pyrimidines. A New Synthesis of the Potent Dihydrofolate Reductase Inhibitor TNP-351. J Org Chem. 1995;60:6684–6687. [Google Scholar]
- 15.Gangjee A, Zeng Y, McGuire JJ, Kisliuk RL. Synthesis of N-[4-[1-Ethyl-2-(2,4-diaminofuro[2,3-d]pyrimidin-5-yl)ethyl]benzoyl]-l-glutamic Acid as an Antifolate. J Med Chem. 2002;45:1942–1948. doi: 10.1021/jm010575m. [DOI] [PubMed] [Google Scholar]
- 16.Mini E, Srimatkandata S, Medina WD, Moroson BA, Carman MD, Bertino JR. Molecular and Karyological Analysis of Methotrexate-Resistant and –Sensitive Human Leukemic CCRF-CEM Cells. Cancer Res. 1985;45:317–325. [PubMed] [Google Scholar]
- 17.Rosowsky A, Lazarus H, Yuan GC, Beltz WR, Mangini L, Abelson HT, Modest EJ, Frei E., III Effects of Methotrexate Esters and Other Lipophilic Antifolates on Methotrexate-Resistant Human Leukemic Lymphoblasts. Biochem Pharmacol. 1980;29:648–652. doi: 10.1016/0006-2952(80)90391-3. [DOI] [PubMed] [Google Scholar]
- 18.McCloskey DE, McGuire JJ, Russell CA, Rowan BG, Bertino JR, Pizzorno G, Mini E. Decreased Folylpolyglutamate Synthetase Activity as a Mechanism of Methotrexate Resistance in CCRF-CEM Human Leukemia Sublines. J Biol Chem. 1991;266:6181–6187. [PubMed] [Google Scholar]
- 19.Hakala MT, Taylor E. The Ability of Purine and Thymidine Derivatives and of Glycine to Support the Growth of Mammalian Cells in Culture. J Biol Chem. 1959;234:126–128. [PubMed] [Google Scholar]
- 20.Grindey GB, Wang MC, Kinahan JJ. Thymidine Induced Perturbations in Ribonucleoside and Deoxyribonucleoside Triphosphate Pools in Human Leukemic CCRF-CEM Cells. Mol Pharmacol. 1979;16:601–606. [PubMed] [Google Scholar]
- 21.Foley GF, Lazarus H, Farber S, Uzman BG, Boone BA, McCarthy RE. Continuous Culture of Lymphoblasts from Peripheral Blood of a Child with Acute Leukemia. Cancer. 1965;18:522–529. doi: 10.1002/1097-0142(196504)18:4<522::aid-cncr2820180418>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.