

Introduction
Alterations in cell metabolism are a characteristic of many cancers (Cairns et al., 2011), and the metabolic program of rapidly proliferating cancer cells supports the biomass production needed to produce daughter cells (Vander Heiden et al., 2009). The M2 isoform of pyruvate kinase (PKM2) is preferentially expressed in cancer, where complex regulation of its activity is important for control of cell metabolism (Chaneton and Gottlieb, 2012; Mazurek, 2011). While most studies addressing the role of PKM2 in cancer metabolism have relied on analysis of cultured cells, cell culture does not recapitulate the metabolic tumor microenvironment (Vaupel et al., 1989) or the tumor cell heterogeneity that exists in vivo (Salk et al., 2010). Knockdown of PKM2 in xenograft tumors has yielded contradictory results regarding the requirement for PKM2 in tumor growth (Cortes-Cros et al., 2013; Goldberg and Sharp, 2012), further highlighting the need to investigate the role of PKM2 in the context of spontaneous tumors arising in situ.
Pyruvate kinase catalyzes the final step in glycolysis by transferring the phosphate from phosphoenolpyruvate (PEP) to ADP, thereby generating pyruvate and ATP. In mammals, pyruvate kinase is encoded by two genes that can each produce two isoforms. Tissue-specific promoters drive expression of the PKL or PKR isoforms from the PKLR gene. PKR expression is exclusive to red blood cells, while PKL is expressed primarily in the liver, with low expression in the kidney (Imamura and Tanaka, 1972; Mazurek, 2011). All other tissues studied express a product of the PKM gene, which generates either the PKM1 or PKM2 isoforms by including one of two mutually-exclusive exons during mRNA splicing (Noguchi et al., 1986). The regulation of PKM splicing is dependent on multiple splicing factors that bind within the PKM1 and PKM2 exons to promote or suppress their inclusion in the mature transcript (Clower et al., 2010; David et al., 2010; Wang et al., 2012). PKM1 expression is found predominantly in differentiated adult tissues with high ATP requirements, such as the heart, brain, and muscle. PKM2 is expressed during development and in many adult tissues including the spleen, lung, and all cancers and cancer cell lines studied to date (Clower et al., 2010; Imamura and Tanaka, 1972; Mazurek, 2011).
PKM1 and PKM2 differ by 22 amino acids and have distinct regulatory properties (Mazurek, 2011). While PKM1 forms a stable, constitutively active tetramer, PKM2 activity is controlled by numerous allosteric effectors and post-translational modifications that affect its tetramer stability. Binding of fructose-1,6-bisphosphate (FBP), an upstream intermediate in glycolysis, causes PKM2 to adopt a stable, active conformation similar to that of PKM1 (Anastasiou et al., 2012; Christofk et al., 2008b). PKM2 activation by FBP can be overridden by interaction of PKM2 with tyrosine-phosphorylated proteins produced in response to growth factor signaling (Christofk et al., 2008b; Varghese et al., 2010). PKM2 activity is reduced by other post-translational modifications (Anastasiou et al., 2011; Lv et al., 2011), and metabolites other than FBP can promote PKM2 activation (Chaneton et al., 2012; Keller et al., 2012). These events illustrate the complex regulation of PKM2 activity, and although PKM2 can exist in active or inactive states as a glycolytic enzyme, the physiological significance of these states in cells or tumors is not well understood.
It is reported that PKM2 is upregulated in cancer cells and that PKM2 is the isoform expressed in all tumors. This suggests that PKM2 expression provides a selective advantage over other pyruvate kinase isoforms. Selection for PKM2 over PKM1 during xenograft tumor growth has been observed (Christofk et al., 2008a), and down-regulation of PKM2 enzymatic activity by phosphotyrosine growth signaling (Christofk et al., 2008a; Hitosugi et al., 2009; Varghese et al., 2010), cellular redox state (Anastasiou et al., 2011) and lysine acetylation (Lv et al., 2011) has been associated with tumor growth and anabolic metabolism. Conversely, high pyruvate kinase activity due to exogenous PKM1 expression or pharmacological activation of PKM2 can impair tumor growth and decrease levels of metabolites critical for biosynthesis in vivo (Anastasiou et al., 2012). Taken together, these studies support a model where the ability of PKM2 to be inactivated is important for cancer cell proliferation. However, this model creates a quandary: if low pyruvate kinase activity is favored by proliferating cancer cells, why is there selection for PKM2 expression in cancer and not inactivation of pyruvate kinase by gene mutation, deletion, or epigenetic silencing?
One possibility is that the enzymatically inactive, non-tetramer form of pyruvate kinase has an important function in cancer outside of glycolysis. Multiple non-metabolic functions unique to PKM2 have been proposed to play a vital role in cancer cell proliferation and tumor growth (Gao et al., 2012; Luo et al., 2011; Yang et al., 2012a; Yang et al., 2011; Yang et al., 2012b). In all cases, these non-metabolic functions are found only with PKM2, and not with PKM1, suggesting that one or all may be driving PKM2 selection in cancer. However, it remains unknown which, if any, are the critical functions promoting PKM2 expression in tumors.
Another possibility is that PKM2 is selected because it can exist in states of low or high enzymatic activity to allow for metabolic adaptation to different physiological situations. While reduced PKM2 activity is associated with cell proliferation, it is possible that enzyme reactivation is important for non-proliferating cancer cells that constitute a sizable fraction of tumor cells in vivo. If this hypothesis is correct, there must be a subpopulation of tumor cells where high pyruvate kinase activity is required. Studies of PKM2 in cancer to date have relied on exogenous protein overexpression or RNA interference of PKM2 expression in cell culture or xenograft tumor models (Anastasiou et al., 2011; Christofk et al., 2008a; Cortes-Cros et al., 2013; Goldberg and Sharp, 2012; Luo et al., 2011; Yang et al., 2012a; Yang et al., 2011; Yang et al., 2012b). Although these methods modulate PKM2 expression, they impair the endogenous, cell-autonomous regulation of pyruvate kinase.
In this study we report a conditional allele (PKM2fl) that allows Cre recombinase-mediated deletion of the PKM2 isoform-specific exon. Excision of PKM exon 10 selectively abrogates PKM2 protein production while still allowing PKM1 splicing and protein expression. Loss of PKM2 in a mouse model of breast cancer accelerates tumor formation. Mammary tumors from PKM2fl/fl mice show efficient deletion, suggesting that PKM2 is not absolutely required for cell proliferation or tumor growth. Only a subset of cells in PKM2Δ/Δ tumors show compensatory PKM1 expression, and high PKM1 expression is anti-correlated with cell proliferation, supporting a model where PKM2 expression in tumors facilitates low pyruvate kinase activity needed for proliferation. Nonsense and missense mutations in PKM2 occur in human tumors. These mutations include recurrent heterozygous stop mutations in PKM exon 10, consistent with the idea that tumors tolerate or favor low pyruvate kinase activity, lending further support to a model where regulation of PKM2 glycolytic activity is driving selection for this enzyme isoform in tumors.
Results
Generation and characterization of a conditional PKM2 allele
Pre-mRNA transcribed from the PKM gene is spliced to produce PKM1 or PKM2 mRNA by inclusion of exon 9 or exon 10, respectively (Figure 1A). To study the role of PKM2 in tumors in vivo, we generated a mouse model that allows conditional deletion of the PKM2-specific exon 10. LoxP sites flanking exon 10 were introduced into the PKM locus of mouse embryonic stem (ES) cells using homologous recombination (Figures 1A, S1A). These ES cells were used to generate chimeric mice, and mice generated from germline transmission of the targeted allele were crossed with mice expressing FLP recombinase to delete the neomycin resistance gene (Neor). Breeding of these mice resulted in transmission of the PKM2 conditional allele (PKM2fl) in Mendelian ratios. PKM2fl/+ and PKM2fl/fl progeny were healthy and had no overt phenotype. The presence of the PKM2fl allele in adult mice was demonstrated by Southern blot (Figures 1B, S1B, S1C). To facilitate husbandry and analysis we developed a PCR-based strategy for determination of PKM2 genotypes (Figure 1C). We next sought to verify that the PKM2fl allele allows disruption of PKM2 expression via Cre-mediated excision of PKM exon 10. Mice with PKM2fl and inducible Cre recombinase (Cre-ER) alleles were crossed to produce mouse embryonic fibroblasts (MEFs) for analysis. Treatment of these MEFs with 4-hydroxytamoxifen resulted in excision of PKM exon 10 in PKM2fl/fl cells (Figure 1D), and western blot analysis showed loss of PKM2 protein in the PKM2Δ/Δ MEFs (Figure 1E). These data confirm that the PKM2fl allele allows exon 10 deletion and disruption of PKM2 protein production.
Figure 1. Generation and validation of PKM2 conditional mice.
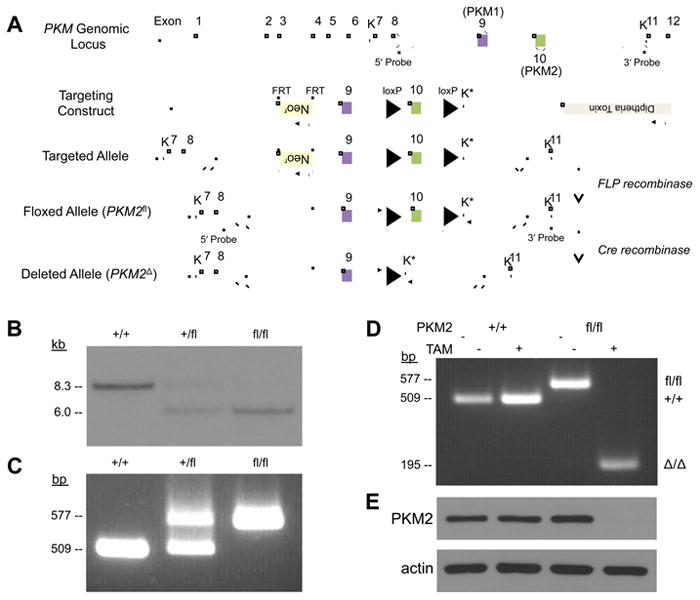
(A) The mouse PKM locus, targeting construct, and the resulting targeted, floxed and deleted alleles. The KpnI sites used for Southern blot analysis are marked with “K”, and the new KpnI site introduced by the targeting vector is marked with “K*”. The locations of the 5' and 3' Probes used for Southern blot analysis are also shown.
(B) Southern blot analysis of KpnI-digested genomic DNA using a 5' Probe. Digestion of the wild-type PKM allele yields an ∼8.3 kb fragment and the floxed allele yields a ∼6.0 kb fragment.
(C) PCR genotyping of genomic DNA from PKM2+/+, PKM2+/fl, and PKM2fl/fl mice. Genotyping primers anneal outside of the loxP sites as indicated by arrows in (A), and produce amplicons of 509 bp from the PKM2+ allele and 577 bp from the PKM2fl allele.
(D) PCR genotyping of PKM2+/+ Cre-ER and PKM2fl/fl Cre-ER MEFs that were treated with 4-hydroxytamoxifen (TAM) or mock treated. The PKM2Δ allele produces a 195 bp amplicon.
(E) Western blot analysis of PKM2 protein from MEFs as specified in (D). See also Figure S1.
PKM2 loss accelerates tumor formation and promotes liver metastasis in a mouse model of breast cancer
To investigate the role of PKM2 in tumor formation, PKM2 conditional mice were crossed to an established model of breast cancer (Xu et al., 1999). In this model, loss of the BRCA1 tumor suppressor in BRCA1fl/fl MMTV-Cre p53+/- mice results in mammary tumors by approximately one year of age. The PKM2fl allele should also undergo recombination in the Cre-expressing epithelial cells that give rise to tumors in this model. Because PKM2 has been implicated as playing an important role in cancer, we expected PKM2 deletion to result in fewer tumors or delayed tumor onset. Surprisingly, PKM2fl/fl mice showed accelerated tumor-associated mortality when compared to their PKM2+/+ counterparts (Figure 2A).
Figure 2. PKM exon 10 deletion in mammary tumors results in accelerated mortality and variable production of PKM1 mRNA.
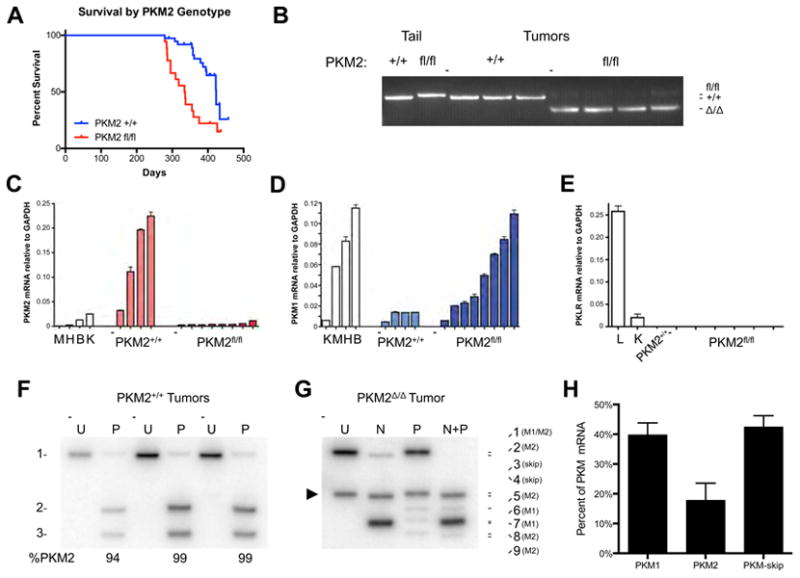
(A) Kaplan-Meier survival curve comparing PKM2+/+ and PKM2fl/fl mice with BRCA1fl/fl MMTV-Cre p53+/- alleles.
(B) PCR genotyping of the PKM2 allele in PKM2+/+ and PKM2Δ/Δ mammary tumors. Analysis of tail DNA from PKM2+/+ and PKM2fl/fl mice is shown as a control.
(C) PKM2 mRNA levels in PKM2+/+ and PKM2Δ/Δ tumors by quantitative RT-PCR, with normal mouse tissue controls: M, muscle; H, heart; B, brain; K, kidney.
(D) PKM1 mRNA levels in PKM2+/+ and PKM2Δ/Δ tumors by quantitative RT-PCR, with normal mouse tissue controls: K, kidney; M, muscle; H, heart; B, brain.
(E) PKLR mRNA levels in PKM2+/+ and PKM2Δ/Δ tumors by quantitative RT-PCR, with normal mouse tissue controls: L, liver; K, kidney.
(F) Autoradiograph of Uncut [U] and PstI [P] digested PKM cDNA amplicons. Uncut PKM1 and PKM2 amplicons are of identical length (band 1). PstI digests only the PKM2 amplicon to produce bands 2 and 3. Results from three representative PKM2+/+ tumors are shown with quantification. See Figures S2C and E for a schematic of how each band is generated.
(G) Autoradiograph of Uncut [U] and digested PKM cDNA amplicons from a representative PKM2Δ/Δ tumor. Uncut PKM1 and PKM2 amplicons are of identical length (band 1), and the amplicon corresponding to the PKM-skip mis-spliced product is marked with an arrowhead (band 3). NcoI [N] digests the PKM1 amplicon and PstI [P] digests the PKM2 amplicon. Figures S2C,D,F show how each band is generated.
(H) Quantification of PKM splicing in PKM2Δ/Δ tumors, as determined from autoradiographs as in (G). Data are displayed as means ± s.e.m, n=4. See also Figure S2.
We characterized tumors from PKM2+/+ and PKM2fl/fl mice to investigate potential factors accounting for accelerated mortality in PKM2fl/fl mice. One possibility is that PKM2 loss changes the balance of proliferation and apoptosis. Staining of tumor sections for the proliferative marker Ki-67 and for the apoptosis marker cleaved caspase-3 did not reveal overt differences between the genotypes (Figure S2A). However, macroscopic metastases were observed in livers from 3 of 5 PKM2fl/fl mice compared to 0 of 7 PKM2+/+ mice evaluated for possible metastases (Figure S2B). To our knowledge, metastasis to the liver has not been reported in this mouse model of breast cancer.
To confirm that PKM2 is deleted in tumors from PKM2fl/fl mice, we examined the efficiency of Cre-mediated exon 10 deletion by PCR. Qualitative deletion of PKM exon 10 was observed in all tumors derived from PKM2fl/fl mice (Figure 2B), and quantitative PCR (qPCR) analysis of tumor transcripts showed very low levels of PKM2 mRNA compared to wild-type tumors (Figure 2C). Because PKM2Δ/Δ tumor cells can potentially generate PKM1 protein by inclusion of exon 9 during mRNA splicing, we quantified PKM1 mRNA levels by qPCR. Some PKM2Δ/Δ tumors had low PKM1 expression at levels similar to PKM2+/+ tumors, while others had PKM1 mRNA levels approaching those found in the brain, a tissue that normally expresses PKM1 (Figure 2D). To determine whether PKM2Δ/Δ tumors had compensatory expression of the other two pyruvate kinase isoforms, PKL or PKR, we examined tumor PKLR mRNA levels by qPCR. We did not observe any mRNA expression from the PKLR gene in tumors of any genotype, suggesting that protein derived from the PKM gene was the only pyruvate kinase present (Figure 2E).
PKM2 deletion results in production of either PKM1 or a mis-spliced mRNA
We further characterized PKM mRNA splicing in tumors from PKM2+/+ and PKM2fl/fl mice. Exons 9 and 10 are of identical length but contain unique restriction enzyme sites that allow for relative quantification of PKM1 and PKM2 transcript levels (Clower et al., 2010; David et al., 2010). The alternatively spliced region between PKM exons 8 and 11 was amplified from PKM2+/+ and PKM2Δ/Δ tumor cDNA and digested for analysis (Figures S2C, S2E). Consistent with the qPCR results, PKM2 was the dominant transcript found in PKM2+/+ tumors (Figure 2F). PKM2Δ/Δ tumors displayed an overall decrease in PKM2 mRNA, with PKM1 message comprising a larger fraction of the total PKM-derived mRNAs (Figure 2G); however, this analysis also revealed an additional PKM splice variant not found in wild type tumors (compare leftmost lanes of Figures 2F and 2G). This mRNA species accounted for approximately 40% of the PKM transcript in PKM2Δ/Δ tumors (Figure 2H), was identified by sequencing to result from the splicing of exon 8 to exon 11 (Figures S2D, S2F, S2G), and is hereafter referred to as PKM-skip.
Splicing of PKM pre-mRNA to produce PKM2 transcript involves repression of exon 9 inclusion and activation of exon 10 inclusion by the binding of specific splicing factors to sequences within the alternatively spliced exons (Clower et al., 2010; David et al., 2010; Wang et al., 2012). Thus, the absence of exon 10 together with repression of exon 9 likely explains the presence of the PKM-skip mRNA species in PKM2Δ/Δ tumors (Chen et al., 2012). Splicing of PKM exon 8 to 11 causes a frameshift at the exon 8-11 junction which results in 38 missense codons and a premature stop codon more than 500 base pairs upstream of the exon 11-12 junction (Figure S2G). Aberrant splicing of PKM transcript to create a premature stop codon upstream of the exon 11-12 junction leads to message degradation by nonsense-mediated decay (Chen et al., 2012).
PKM2 deletion results in variable PKM1 protein levels but no PKM-skip protein
Because deletion of PKM2 exon 10 does not always lead to exon 9 inclusion and production of PKM1 mRNA, and because PKM1 mRNA levels varied widely across tumors, we determined the effects of PKM2 deletion on pyruvate kinase protein levels in PKM2Δ/Δ tumors. Western blotting of tumor lysates confirmed the loss of PKM2 protein in PKM2Δ/Δ tumors (Figure 3A). Variable amounts of PKM1 protein were observed in PKM2Δ/Δ tumor lysates, and overall expression of pyruvate kinase was decreased in PKM2Δ/Δ tumors relative to PKM2+/+ tumors, such that even tumors with the highest PKM1 expression had lower levels than PKM1-expressing skeletal muscle.
Figure 3. PKM exon 10 deletion in mammary tumors leads to variable expression of PKM protein.
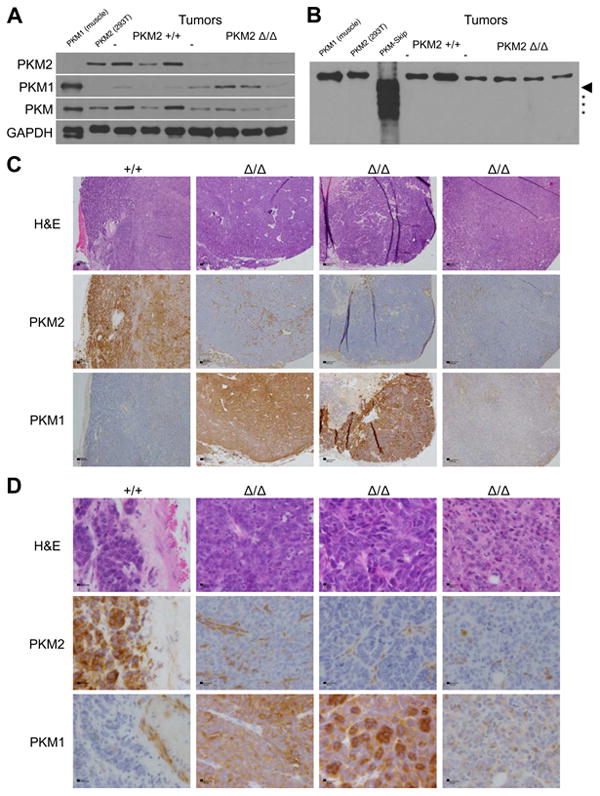
(A) PKM2, PKM1, and total PKM protein levels in PKM2+/+ and PKM2Δ/Δ tumors. Muscle and 293T cell lysates serve as PKM1 and PKM2 protein controls, respectively, with GAPDH as loading control.
(B) PKM protein in PKM2+/+ and PKM2Δ/Δ tumors visualized using a monoclonal anti-PKM antibody with an epitope common to PKM1 and PKM2. Muscle and 293T cell lysates serve as PKM1 and PKM2 protein controls, respectively. The size of full-length PKM-skip is marked with an arrowhead and PKM-skip degradation products are marked with asterisks. This blot was intentionally over-exposed.
(C) Histology and staining of one PKM2+/+ tumor and three PKM2Δ/Δ tumors for PKM2 or PKM1. Scale bars represent 200 μm.
(D) High power micrographs of the same tumors as in (C). Scale bars represent 20 μm. See also Figure S3.
We next determined whether the PKM-skip mRNA generates a protein product in PKM2Δ/Δ tumors. Wild type PKM1 and PKM2 are both 531 residues long with subunit masses of approximately 58 kD, while the predicted PKM-skip protein is 418 residues long with a predicted mass of 45 kD (Figure S3A). To verify that we could detect PKM-skip protein via western blot, PKM-skip cDNA was expressed in E. coli with an N-terminal 6x-His tag and purified using affinity chromatography. The same approach is used to produce PKM2 protein that is active both as a glycolytic enzyme (Anastasiou et al., 2012; Dombrauckas et al., 2005) and as a putative protein kinase (Gao et al., 2012; Yang et al., 2012a). Some PKM-skip product was recovered from the soluble fraction after bacterial lysis; however, the expected protein was accompanied by a series of smaller products consistent with partial degradation of an unstable protein in E. coli (Figure S3B). Both the full length and smaller polypeptides were recognized by two different anti-PKM antibodies during western blot analysis (Figure S3C). We next performed western blot analysis of tumor lysates using recombinant PKM-skip protein as a control; however, we failed to detect a band smaller than the full-length protein in any tumors (Figure 3B). These data show that PKM-skip protein is not present in appreciable quantities in PKM2Δ/Δ tumors.
Because small amounts of catalytic activity are potentially meaningful, we considered that levels of putative PKM-skip below our detection limit might retain some PKM2 activity. By comparison with published PKM2 protein structures (Christofk et al., 2008b; Dombrauckas et al., 2005), PKM-skip protein is predicted to lack the FBP binding site and the dimer-dimer interface found in the native PKM2 tetramer (Figure S3D). However, with a few amino acid changes the predicted PKM-skip monomer retains the primary structure necessary to produce the domains hosting the active site (Figure S3E). We thus evaluated whether recombinant PKM-skip retained catalytic activity and whether this protein could form dimers or tetramers. Using size exclusion chromatography, we found that recombinant PKM-skip eluted at volumes consistent with it being a population of 45 kD or smaller monomers (Figure S3F). PKM-skip protein was >230,000-fold less active per microgram than recombinant PKM2 for glycolytic pyruvate kinase activity (Figure S3G) and had no detectable protein kinase activity (Figure S3H), suggesting that PKM-skip protein would not have meaningful catalytic activity even if it were present in PKM2Δ/Δ tumors. Taken together, these data suggest that PKM-skip protein is not a confounding factor in the analysis of PKM2Δ/Δ tumors, and that PKM exon 10 deletion effectively precludes PKM2 protein production while still allowing cells to express PKM1.
Expression of PKM1 in PKM2 knockout tumors is spatially heterogeneous
To understand which cells in PKM2Δ/Δ tumors express PKM1, we performed immunohistochemistry (IHC) on fixed tumor sections. Consistent with previous reports (Christofk et al., 2008a; Mazurek, 2011), most cells in PKM2+/+ tumors stained for PKM2, but not for PKM1 (Figures 3C, 3D). PKM2Δ/Δ tumors showed PKM2 staining only in adipose or other stromal tissue associated with the tumor, with no PKM2 staining in tumor cells. PKM1 staining was variable and was not observed in every tumor cell in PKM2Δ/Δ tumors (Figures 3C, 3D). Taken together, these data suggest that PKM2 is not required by all tumor cells and that there is a differential requirement for pyruvate kinase among different tumor cell populations.
Generation of tumor cell lines selects for PKM2 expression
To further characterize PKM2Δ/Δ tumor cells, we derived cell lines from mammary tumors arising in PKM2+/+ and PKM2fl/flBRCAfl/flMMTV-Cre p53+/- mice. Generation of cell lines from PKM2Δ/Δ tumors took several weeks, and in all cases the cell lines showed high levels of PKM2 protein expression despite the deletion of PKM2 observed in the parental tumors (Figure 4A). PCR genotyping of the cell lines showed retention of at least one PKM2fl allele in all cases (Figure 4B), suggesting that the transition to tissue culture selected for rare tumor cells that had not undergone recombination at both PKM loci. In sharp contrast to the tumors from which they were derived, the tumor cell lines showed increased PKM2 mRNA levels (Figures 4C, 4D) and a reduction in PKM-skip transcript (Figure 4D).
Figure 4. Cell lines derived from PKM2Δ/Δ mammary tumors retain expression of PKM2.
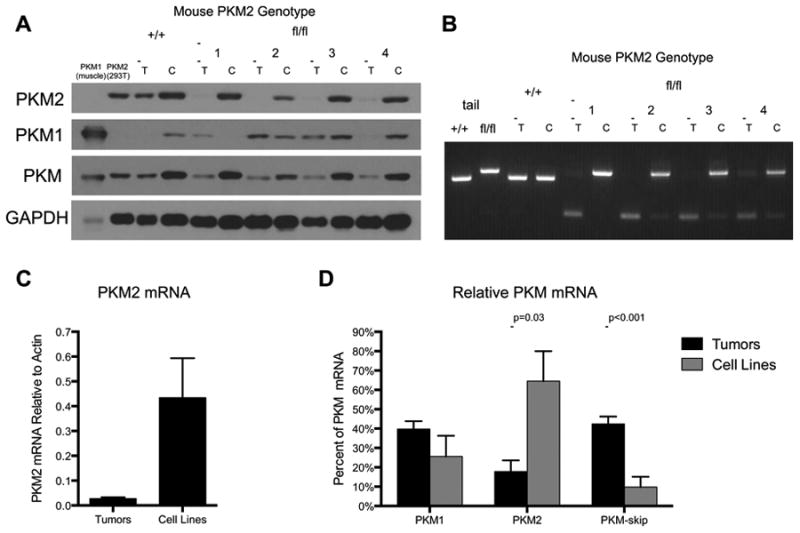
(A) PKM2, PKM1, and total PKM protein levels in tumors [T] from PKM2+/+ and PKM2fl/fl mice and their derivative cell lines [C]. Muscle and 293T cell lysates serve as PKM1 and PKM2 protein controls, respectively, with GAPDH as loading control.
(B) PCR genotyping of tumors [T] from PKM2+/+ and PKM2fl/fl mice and their derivative cell lines [C]. Analysis of tail DNA from PKM2+/+ and PKM2fl/fl mice is shown as control.
(C) PKM2 mRNA levels in PKM2Δ/Δ tumors and derivative cell lines by quantitative RT-PCR. Data are shown as means ± s.e.m, n=4 tumors, n=3 cell lines.
(D) Quantification of PKM splicing in PKM2Δ/Δ tumor-derived cell lines and parent tumors determined as shown in Figure 2G. Data are displayed as means ± s.e.m, n=4 tumors, n=4 cell lines. P-values were obtained by Student's t-test. See also Figure S4.
Introduction of inducible Cre-recombinase (Cre-ERT2) into two tumor cell lines allowed for deletion of the remaining PKM2fl allele in vitro (Figure S4A), and we found that PKM2 loss did not affect the rate of oxygen consumption in these cells or cause dependence on β-oxidation (Figure S4B). We also investigated whether non-glycolytic functions of PKM2 were active in these cells. PKM2 was not required for hypoxia- or β-catenin-induced gene expression (Figures S4C-S4J), suggesting that maintenance of those functions was not the basis for the outgrowth of rare PKM2-expressing cells during cell line establishment. Thus, despite selection for PKM2 during cell line generation, a functional PKM2 allele was not required for in vitro proliferation.
Selection against PKM1 expression, but not for PKM2 expression, in allograft tumors
Given that tissue culture conditions appear to select for PKM2-expressing cells, we focused on in vivo analysis of PKM2Δ/Δ tumor-derived cell lines to examine the consequences of constitutive PKM1 or PKM2 expression on tumor growth in the absence of endogenous PKM2. A representative cell line was stably infected with Cre-ERT2 and either no cDNA (empty vector control), FLAG-PKM1, or FLAG-PKM2. The presence of a FLAG-epitope tag does not impair the ability of exogenously expressed PKM2 to rescue any known function of the protein (Christofk et al., 2008a; Christofk et al., 2008b; Yang et al., 2011). The cells were treated to delete the remaining PKM2fl allele and injected subcutaneously into nude mice. While the control cells and the cells expressing FLAG-PKM2 were competent to form tumors, the cells expressing FLAG-PKM1 were not (Figure 5A). IHC using an anti-FLAG antibody showed that the FLAG-PKM1 expressing cells were detectable upon injection site biopsy 34 days later (Figure 5B). This suggests that constitutive PKM1 expression did not result in decreased viability, but instead promoted a state not conducive to proliferation. Interestingly, tumors derived from FLAG-PKM2-expressing cells showed heterogenous FLAG staining with only a minority of cells staining positive (Figure 5B), and most cancer cells in these tumors did not stain for PKM2 (Figure 5C). These data suggest that there was no selective pressure to retain FLAG-PKM2 expression despite deletion of the endogenous gene, and confirm that constitutive, exogenous PKM1 expression suppresses proliferation.
Figure 5. Allograft tumor growth does not select for PKM2 expression.
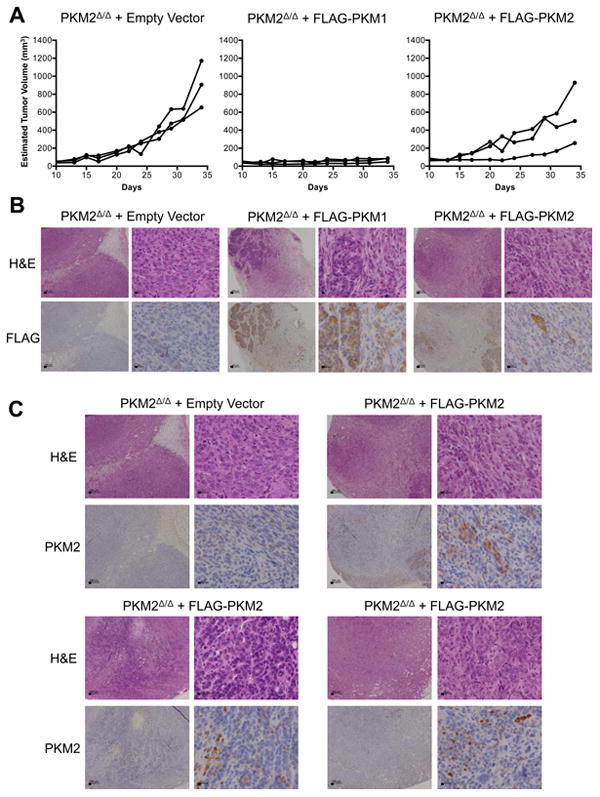
(A) Growth curves of tumors formed by PKM2Δ/Δ cells expressing empty vector, FLAG-PKM1, or FLAG-PKM2 following injection into nude mice.
(B) Representative anti-FLAG and H&E staining of tumor sections from the allograft experiment shown in (A). Scale bars represent 200 μm and 20 μm at low and high magnification, respectively.
(C) Anti-PKM2 and H&E staining of a representative PKM2Δ/Δ empty vector tumor and three PKM2Δ/Δ FLAG-PKM2 tumors. The top right PKM2Δ/Δ FLAG-PKM2 tumor is the same PKM2Δ/Δ FLAG-PKM2 tumor shown in (B). Scale bars represent 200 μm and 20 μm at low and high magnification, respectively.
PKM1 expression is found only in non-proliferating tumor cells
PKM1 staining of PKM2Δ/Δ allograft tumors showed that PKM1 is endogenously expressed in only a subpopulation of the tumor cells (Figure 6A). Because PKM1 expression suppresses tumor growth and decreased PKM2 activity is associated with pro-proliferative signaling events and anabolic metabolism (Anastasiou et al., 2012; Christofk et al., 2008a; Varghese et al., 2010), we reasoned that proliferating tumor cells might be included in the subset of cells that do not express PKM1. Sections of PKM2Δ/Δ allograft tumors were dual stained for PKM1 and the proliferative marker PCNA. Areas of high PKM1 staining had few proliferating cells, while areas of little to no PKM1 staining had many proliferating cells (Figures 6A, S5A). Quantification of this phenotype revealed that 26% of cells counted stained positive for PCNA, and of the 614 PCNA-positive cells counted, the majority (419) had low or no PKM1 staining.
Figure 6. Tumor cells proliferate in the absence of PKM2, but not in the presence of PKM1.
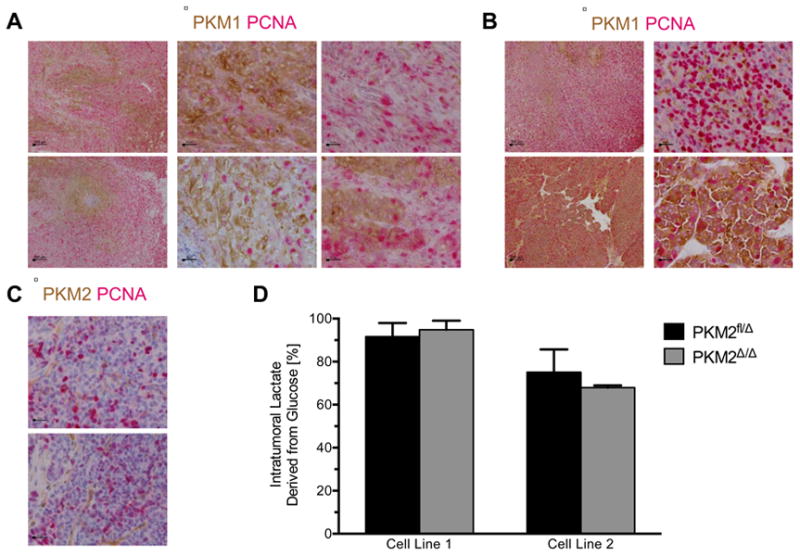
(A) PKM2Δ/Δ allograft tumors dual-stained for PKM1 (brown) and PCNA (red). Scale bars represent 200 μm and 20 μm at low and high magnification, respectively.
(B) PKM2Δ/Δ tumors from BRCAfl/flMMTV-Cre p53+/- mice dual-stained for PKM1 (brown) and PCNA (red). Scale bars represent 200 μm and 20 μm at low and high magnification, respectively.
(C) PKM2Δ/Δ tumors from BRCAfl/flMMTV-Cre p53+/- mice dual-stained for PKM2 (brown) and PCNA (red). Scale bars represent 20 μm.
(D) The percent lactate derived from glucose in PKM2fl/Δ and PKM2Δ/Δ allograft tumors was determined from 13C-lactate labeling in tumors normalized to 13C-glucose serum enrichment levels. Results from two independent tumor cell line allografts are displayed as means ± s.e.m (n=8 for Line 1 tumors of both genotypes; n=4 for Line 2 PKM2fl/Δ tumors and n = 5 for Line 2 PKM2Δ/Δ tumors). See also Figure S5.
To confirm that PKM1 expression is also associated with the non-proliferating tumor cells in endogenous tumors, we dual stained tumors from PKM2fl/flBRCAfl/flMMTV-Cre p53+/- mice for PKM1 and PCNA. We again saw a correlation between low PKM1 expression and staining with PCNA (Figures 6B, S5B). In these tumors, PCNA positive cells made up 21% of the cells counted, and 85% of PCNA positive cells had low to no PKM1 expression. Dual PKM2, PCNA staining of tumors from PKM2fl/flBRCAfl/flMMTV-Cre p53+/-mice confirmed that cells stained positive for PCNA despite a lack of PKM2 expression (Figure 6C), supporting the notion that PKM2Δ/Δ tumor cells are capable of proliferation.
Variable PKM1 expression allows normal glucose metabolism in PKM2-null tumors
Our observation of low PKM1 expression in PKM2Δ/Δ tumor cells raises the possibility that glycolysis is disrupted in PKM2Δ/Δ tumors. We therefore sought to determine if PKM2 loss resulted in gross metabolic perturbations that would be detectable as changes in glycogen or lipid storage. Spontaneous PKM2+/+ and PKM2Δ/Δ tumors exhibited similar periodic acid-Schiff staining with and without diastase pretreatment, consistent with tumors of both genotypes having similar glycogen content (Figure S5C). Oil red O staining of neutral lipid stores was also comparable between PKM2+/+ and PKM2Δ/Δ tumors, with most tumors showing very little lipid accumulation in areas devoid of fat cells (Figure S5D).
Lactate production has been associated with PKM2 expression (Christofk et al., 2008a). To investigate the effect of PKM2 deletion on tumor glucose metabolism, we determined whether PKM2Δ/Δ tumors were impaired in glucose to lactate conversion. Conscious mice harboring PKM2fl/Δ or PKM2Δ/Δ allograft tumors were intravenously infused with fully-labeled 13C-glucose to achieve stable steady-state enrichment of labeled glucose in the serum (Ayala et al., 2010; Figure S5E). Levels of labeled lactate derived from glucose, as well as the absolute concentration of lactate, were similar in both PKM2-expressing and PKM2-null tumors (Figures 6D, S5F). The relative abundance of fully-labeled lactate in tumors was greater than that found in serum regardless of PKM2 genotype, providing additional support that the tumors produce similar amounts of lactate from glucose (Figure S5G). These data are consistent with heterogeneous PKM1 expression allowing populations of PKM2Δ/Δ tumor cells to recapitulate the metabolism found in PKM2-expressing tumors.
Human cancers have PKM2 mutations and variable PKM2 expression
Given the dispensability of PKM2 for tumor growth, and the variability in pyruvate kinase expression observed in our mouse model, we sought to determine whether variable PKM2 expression might be found in primary human tumors. We performed PKM2 and PKM1 IHC on two sets of breast tumor tissue arrays containing a total of 317 cores (Figure 7A, 7B). PKM2 staining intensity scores revealed that, while 71% of the scored tumors show expression of PKM2, the majority of these had weak PKM2 staining (score 1). Moreover, a sizable fraction of the tumors were completely negative for PKM2 staining (score 0), suggesting that PKM2 expression is not consistently upregulated in human tumors (Figure 7C). An absolute requirement for high or low PKM2 expression was not found in any breast cancer subtype, although the more aggressive Her2 positive and triple negative breast tumor subtypes contained the highest percentage of PKM2 negative (score 0) tumors (Figure 7D). We found a statistically significant difference in the distribution of PKM2 IHC scores between Her2 amplified tumors and tumors that were ER/PR positive (P=0.038, Chi-square test).
Figure 7. Human breast tumors show variable expression of PKM2.
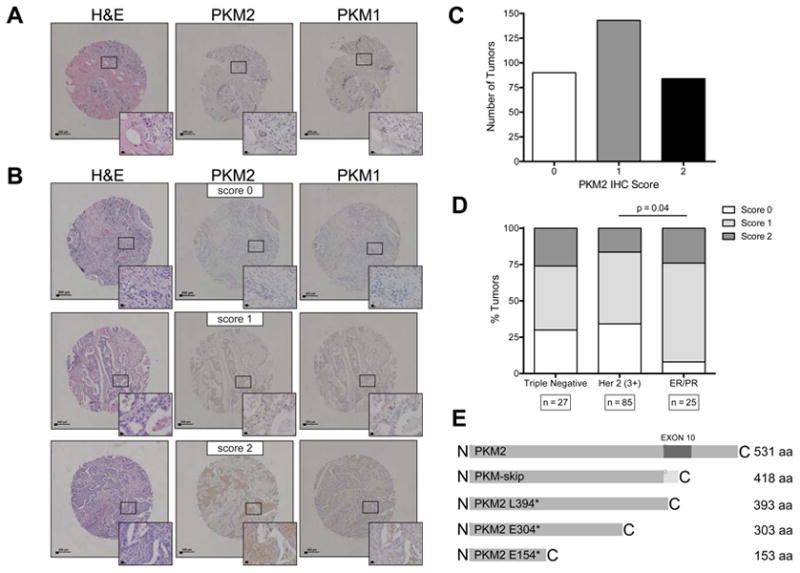
(A) Representative TMA cores containing normal human breast stained with H&E, and for PKM2 and PKM1. Insets show higher magnification. Scale bars represent 200 μm and 20 μm at low and high magnification, respectively.
(B) Representative TMA cores containing human breast tumor samples stained with H&E, and for PKM2 and PKM1. An example of each PKM2 IHC intensity score is shown (0=negative; 1=weak; 2=strong). Insets show higher magnification. Scale bars represent 200 μm and 20 μm at low and high magnification, respectively.
(C) Distribution of PKM2 IHC intensity scores of 317 tumors from two different TMAs containing breast tumor samples.
(D) Quantification of PKM2 expression in human breast tumors relative to breast tumor subtype (triple negative, Her2 amplified, or ER/PR positive). The distribution of PKM2 scores is significantly different between Her2 amplified samples and ER/PR positive samples by Chi-square test (P=0.038).
(E) Schematic showing truncations of PKM2 caused by mutations found in human cancers.
Because we observed acceleration of tumor-related mortality in our mice due to PKM2 loss, we wondered whether genetic loss or inactivation of PKM2 might also occur in some human cancers. Sequencing of human endometrial tumor samples revealed the presence of a recurrent heterozygous nonsense mutation (L394*) within exon 10 of PKM2 (Figure 7E). If translated, this message would result in a non-functional protein product similar to PKM-skip, arguing that this is a loss-of-function mutation.
Analysis of large-scale efforts to sequence human tumor tissue via The Cancer Genome Atlas (TCGA) revealed additional PKM2 mutations in 7 cancer types (Table S1). These mutations include two additional nonsense mutations (E154* and E304*) that can only generate truncated proteins smaller than PKM-skip (Figure 7E), and one frameshift mutation at V375 that results in a premature stop codon. These mutations all occur upstream of the PKM isoform-specific exons (Figure 7E, Table S1) and are expected to result in nonsense-mediated decay of the transcripts (Maquat, 2004) and render any protein product non-functional. The remaining mutations are missense mutations that are predicted to lower enzyme activity. These data are consistent with selection for loss of PKM2 activity under some situations in human cancer.
Discussion
Breast tumors in BRCAfl/flMMTV-Cre p53+/- mice progressed faster despite PKM2 deletion, and proliferation within PKM2Δ/Δ tumors was associated with low PKM1 expression. These results are in alignment with the known regulation of PKM2 activity in tumor cells, whereby PKM2 can exist in an active state like that of PKM1, or in an inactive state that is promoted by signaling events that drive cell proliferation (Christofk et al., 2008b; Lv et al., 2011; Mazurek, 2011). Following PKM exon 10 deletion, the tumor cells can create analogous states of high and low pyruvate kinase activity by titrating expression of the constitutively active PKM1 isoform. Thus, the low pyruvate kinase activity state normally induced by growth signaling can still be maintained after PKM2 deletion by repression of exon 9 during splicing to preclude PKM1 expression.
Regulation of PKM2 activity may act as an adaptive response to different cellular metabolic needs, suggesting that increased pyruvate kinase activity is important during different phases of tumor formation and progression. Tumor initiation, either at a primary or metastatic site, involves the ability to grow in a new, inappropriate tissue context. An inadequate blood supply or lack of exogenous growth and survival signals are both possible sources of nutrient stress that might limit the proliferation and survival of individual cancer cells (Vander Heiden et al., 2001; Vaupel et al., 1989). Survival during these periods of stress requires a metabolic program geared away from anabolic metabolism and toward efficient ATP production, which may explain why non-proliferating tumor cells need high pyruvate kinase activity. The ability to activate PKM2 when proliferation is not favored likely drives retention of PKM2 expression in cancer and may explain why loss of function mutations observed in human cancers are heterozygous. In this case, a reduction of pyruvate kinase activity in the cell due to mutation is beneficial for proliferative metabolism, but retention of one functional PKM allele provides metabolic flexibility to survive periods of stress experienced by cancer cells in vivo.
The PKM missense mutations found in multiple human cancer types are noteworthy because PKM2 activity is sensitive to amino acid substitution. A functional characterization of 17 mutations in PKM2 caused by rare SNPs in the human population showed that missense mutations, even in solvent exposed residues, resulted in reduced enzyme activity and stability, and in some cases altered the allosteric regulation of PKM2 (Allali-Hassani et al., 2009). Furthermore, our evaluation of a large panel of breast tumors demonstrated that PKM2 protein levels are low in many of these cancers. Because PKM2 mutations have not been found at high frequencies, the observed lower expression in some tumors may be due to decreased transcription or translation, or be the result of epigenetic silencing.
Apart from its role as a glycolytic enzyme, multiple non-metabolic functions that are specific to PKM2 have been implicated in cancer, suggesting that these non-canonical activities may drive selection for PKM2 expression in tumor cells. The fact that cells are able to form spontaneous tumors and proliferate after PKM2 deletion implies that non-metabolic functions of the PKM2 isoform are not absolutely necessary for proliferation or tumor progression. While recent reports have suggested that PKM2-dependent phosphorylation of histone H3 is required for cyclin D1 expression and progression through the cell cycle (Yang et al., 2012a), our results indicate that this mechanism is not absolutely required for cell cycle control and is dispensable for cell proliferation in at least some tumors. Other non-metabolic functions of PKM2 that promote gene expression through interactions with HIF1 (Luo et al., 2011), β-catenin (Yang et al., 2011), or by phosphorylation of the stat3 transcription factor (Gao et al., 2012), may occur in and be advantageous for populations of tumor cells, but are not required for growth of all tumors.
The ability of tumor cells to proliferate without PKM2 raises the question of whether cells with PKM2 loss and no apparent PKM1 expression require any pyruvate kinase. It is possible that very low levels of PKM1 expression provide sufficient pyruvate kinase activity to maintain glycolytic flux, which accounts for the continued conversion of glucose to lactate in PKM2Δ/Δ tumors. If so, then pyruvate kinase in cancer cells must be in excess of what is needed to allow sufficient PEP to pyruvate conversion, and pyruvate kinase activity would thus never be rate limiting at normal expression levels. However, this possibility is not consistent with the observation that both PKM1 expression and pyruvate kinase activators can suppress tumor growth by increasing pyruvate kinase activity (Anastasiou et al., 2012). If the level of pyruvate kinase activity is rate limiting in proliferating cells, then the observed loss of PKM2 without compensatory expression of other isoforms suggests that a pyruvate kinase independent mechanism for conversion of PEP to pyruvate might exist to allow glycolytic flux to continue in the absence of pyruvate kinase. The presence of such an activity has been suggested, and may allow cells to decouple glycolytic ATP generation from incorporation of glucose into central metabolic pathways (Vander Heiden et al., 2010).
The ability of PKM2Δ/Δ tumors to convert glucose to lactate despite negligible pyruvate kinase expression in many cells has another possible explanation. It may be that tumor lactate production is restricted to non-proliferating cells with high pyruvate kinase activity, while cells with low pyruvate kinase activity consume another substrate. A similar metabolic symbiosis has been suggested to support cells in hypoxic regions of tumors (Sonveaux et al., 2008).
A differential requirement for pyruvate kinase activity among tumor cell populations complicates the development of strategies to target PKM2 for cancer therapy. While PKM2 knockdown inhibits growth in vitro, the same does not hold true in vivo, as inducible knockdown of PKM2 has no effect on established xenograft tumors (Cortes-Cros et al., 2013). That finding, together with our observation of accelerated tumor mortality after PKM2 deletion, suggests that pharmacological inhibition or therapeutic knockdown of PKM2 may not be effective standalone cancer therapies. Increasing pyruvate kinase activity in the cell with small molecule activators can inhibit tumor growth (Anastasiou et al., 2012; Parnell et al., 2013); however, our finding that PKM1-expressing cancer cells persist in a non-proliferative state in vivo raises the possibility that increased pyruvate kinase activity favors survival of cancer cells under some conditions. Additionally, pharmacological activation of PKM2 activity may have no effect on the non-proliferating tumor cell population and be at best a cytostatic therapy. Finally, as at least some tumors can tolerate loss of PKM2, the deletion, mutation, or epigenetic silencing of PKM2 may provide a mechanism for resistance to PKM2 activators in a therapeutic setting. Activation or inhibition of pyruvate kinase as an adjuvant therapy may still be effective in situations where targeting PKM2 opposes the biological regulation of PKM2 activity. For example, effective PKM2 inhibition might disrupt the metabolism of the non-proliferating tumor fraction and cause cell death or sensitize the cells to cytotoxic therapy. The findings of this study highlight the importance of understanding the context-dependent metabolic needs of cancer cells to effectively target metabolism for therapeutic benefit.
Experimental Procedures
Generation of PKM2 Conditional Mice and Embryonic Fibroblasts (MEFs)
PKM2 conditional mice and embryonic fibroblasts were generated using standard protocols.
Southern Blotting, PCR Genotyping, qPCR, and Splicing Analysis
Asp718 (Roche)-digested genomic DNA was analyzed by Southern blot using standard protocols with probe binding visualized by autoradiography. Mouse genotypes were determined by PCR. qPCR reactions were performed using Fast SYBR Green Master Mix (Applied Biosystems), and PKM splicing analyzed as reported previously (Clower et al., 2010).
Western Blot and IHC
Western blots were performed using primary antibodies against PKM1 (Sigma SAB4200094), PKM2 (Cell Signaling Technology #4053), PKM (Cell Signaling Technology #3190; abcam ab6191), FLAG (Cell Signaling Technology #2368), beta-actin (abcam ab1801), or GAPDH (Cell Signaling Technology #2118). Fixed sections were stained with the following primary antibodies after antigen retrieval: PKM1 (Cell Signaling Technology #7067), PKM2 (Cell Signaling Technology #4053), PCNA (Cell Signaling Technology #2586), Ki-67 (BD Pharmingen 556003), Cleaved Caspase 3 (Cell Signaling Technology #9661), and/or FLAG (Cell Signaling Technology #2368). PKM1/PCNA dual-staining was quantified by scoring cells as PCNA-positive or -negative and PKM1-high or –low/none.
Tumor Cell Lines
Trypsin disaggregated tumor cells were propagated in DMEM with 10% fetal bovine serum, 2 mM glutamine, and penicillin/streptomycin. Cell lines were retrovirally infected with an MSCV-CreERT2-puro vector and pLHCX expression vectors and selected with puromycin and hygromycin as described previously (Christofk et al., 2008a). 1 µM 4-hydroxytamoxifen was used to induce recombination where indicated.
Allograft Tumor Experiments
5 × 106 cells were injected subcutaneously into nude mice. Tumor growth was monitored by caliper measurement in two dimensions with volumes estimated using the equation V = (π/6)(L*W2).
Tissue Microarray Analysis
PKM2 and PKM1 expression in human breast cancers was analyzed in three tissue microarrays: US BioMax BR1504, one multi-tissue TMA containing tissue from the archives of the Institute of Pathology at the University of Basel (Baumhoer et al., 2008; Schraml et al., 1999), and two breast cancer TMAs from Beth-Israel Deaconess Medical Center. Each TMA core was independently scored for PKM2 IHC intensity. PKM2 scores from US BioMax BR1504 were quantified relative to tumor subtype.
Human Tumor DNA Sequence Analysis
Whole exome sequencing of 13 endometrioid endometrial tumors demonstrated a stop codon in exon 10 of PKM (Liang et al., 2012). The PKM2 mutation was confirmed by Sequenom mass spectrometry of 234 human endometrial cancers. Additional mutations in PKM were identified through the cBIO portal (Cerami et al., 2012).
Mouse Glucose Infusion Studies
Catheters were implanted into the jugular vein of animals with allograft tumors 5-7 days before performing basal glucose turnover experiments as reported previously (Ayala et al., 2010; Jurczak et al., 2012). Conscious overnight fasted mice were infused with U-13C-glucose for 120 min to achieve steady-state enrichments without perturbing endogenous glucose homeostasis prior to tissue analysis.
Mass Spectrometry Metabolite Measurement
Metabolites were prepared for analysis with isotope-labeled internal standards. The Line 1 samples were analyzed as described previously (Jain et al., 2012). Briefly, metabolites were separated using a Shimatzu Prominince UHPLC and lactate isotopologues measured in negative ion mode using an ABSCIEX 5500 QTRAP equipped with a SelexION. GC-MS metabolite analysis employed an Agilent 7890A GC and an Agilent 5975C MS with MIDs determined as described previously (Commisso et al., 2013). The Line 2 samples were analyzed by hydrophilic interaction liquid chromatography (HILIC)/negative ion mode MS using an Open Accela 1250 U-HPLC and a Q Exactive hybrid quadrupole orbitrap mass spectrometer (Thermo Fisher Scientific; Waltham, MA).
Detailed descriptions of methods are provided in the Extended Experimental Procedures.
Supplementary Material
Highlights.
PKM2 is not required by all tumor cells
The need for pyruvate kinase is most apparent in non-proliferating tumor cells
PKM2 expression is variable in human cancers
Recurrent point mutations disrupting pyruvate kinase are found in human cancers
Acknowledgments
We thank Chu-Xia Deng for sharing the BRCA1 mouse model, Luigi Terracciano and Luigi Tornillo for providing the multi-tissue TMA, and Cynthia Clower, Sarah-Maria Fendt, Andrea J. Howell, Vivian M. Liu, Sophia Lunt, Katherine R. Mattaini, Benjamin A. Olenchock, and Kerry Pierce for technical assistance. Eric L. Bell provided advice and reagents. Denise G. Crowley, Michael Brown, Kathleen S. Cormier assisted with histology. This work was supported in part by NIH grants R01CA168653, 5P01CA117969, P30CA147882, 5P30CA14051, 5K08CA136983, DK059635, R01DK092606, R00CA131472, R01GM056203 and ADA grant 7-12-BS-09. RAD and JH acknowledge support from the Belfer Foundation. MVH acknowledges additional support from the Smith Family Foundation, the Burroughs Wellcome Fund, the Damon Runyon Cancer Research Foundation, and the Stern family. CBC is a shareholder, MVH is a shareholder and SAB member, and LCC is a shareholder, SAB member and on the Board of Directors of Agios Pharmaceuticals.
Footnotes
Supplemental information: Supplemental information includes Extended Experimental Procedures, five figures and one table.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allali-Hassani A, Wasney GA, Chau I, Hong BS, Senisterra G, Loppnau P, Shi Z, Moult J, Edwards AM, Arrowsmith CH, et al. A survey of proteins encoded by non-synonymous single nucleotide polymorphisms reveals a significant fraction with altered stability and activity. The Biochemical journal. 2009;424:15–26. doi: 10.1042/BJ20090723. [DOI] [PubMed] [Google Scholar]
- Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang JK, Shen M, Bellinger G, Sasaki AT, Locasale JW, Auld DS, et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science. 2011;334:1278–1283. doi: 10.1126/science.1211485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastasiou D, Yu Y, Israelsen WJ, Jiang JK, Boxer MB, Hong BS, Tempel W, Dimov S, Shen M, Jha A, et al. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat Chem Biol. 2012:1–9. doi: 10.1038/nchembio.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala JE, Samuel VT, Morton GJ, Obici S, Croniger CM, Shulman GI, Wasserman DH, McGuinness OP. Standard operating procedures for describing and performing metabolic tests of glucose homeostasis in mice. Dis Model Mech. 2010;3:525–534. doi: 10.1242/dmm.006239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumhoer D, Tornillo L, Stadlmann S, Roncalli M, Diamantis EK, Terracciano LM. Glypican 3 expression in human nonneoplastic, preneoplastic, and neoplastic tissues: a tissue microarray analysis of 4,387 tissue samples. American journal of clinical pathology. 2008;129:899–906. doi: 10.1309/HCQWPWD50XHD2DW6. [DOI] [PubMed] [Google Scholar]
- Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaneton B, Gottlieb E. Rocking cell metabolism: revised functions of the key glycolytic regulator PKM2 in cancer. Trends in biochemical sciences. 2012;37:309–316. doi: 10.1016/j.tibs.2012.04.003. [DOI] [PubMed] [Google Scholar]
- Chaneton B, Hillmann P, Zheng L, Martin AC, Maddocks OD, Chokkathukalam A, Coyle JE, Jankevics A, Holding FP, Vousden KH, et al. Serine is a natural ligand and allosteric activator of pyruvate kinase M2. Nature. 2012;491:458–462. doi: 10.1038/nature11540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, David CJ, Manley JL. Concentration-dependent control of pyruvate kinase M mutually exclusive splicing by hnRNP proteins. Nat Struct Mol Biol. 2012;19:346–354. doi: 10.1038/nsmb.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008a;452:230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature. 2008b;452:181–186. doi: 10.1038/nature06667. [DOI] [PubMed] [Google Scholar]
- Clower CV, Chatterjee D, Wang Z, Cantley LC, Vander Heiden MG, Krainer AR. The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and cell metabolism. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:1894–1899. doi: 10.1073/pnas.0914845107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin JA, Thompson CB, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature. 2013;497:633–637. doi: 10.1038/nature12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes-Cros M, Hemmerlin C, Ferretti S, Zhang J, Gounarides JS, Yin H, Muller A, Haberkorn A, Chene P, Sellers WR, et al. M2 isoform of pyruvate kinase is dispensable for tumor maintenance and growth. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:489–494. doi: 10.1073/pnas.1212780110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David CJ, Chen M, Assanah M, Canoll P, Manley JL. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. 2010;463:364–368. doi: 10.1038/nature08697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombrauckas JD, Santarsiero BD, Mesecar AD. Structural basis for tumor pyruvate kinase M2 allosteric regulation and catalysis. Biochemistry. 2005;44:9417–9429. doi: 10.1021/bi0474923. [DOI] [PubMed] [Google Scholar]
- Gao X, Wang H, Yang JJ, Liu X, Liu ZR. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Molecular cell. 2012;45:598–609. doi: 10.1016/j.molcel.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg MS, Sharp PA. Pyruvate kinase M2-specific siRNA induces apoptosis and tumor regression. J Exp Med. 2012;209:217–224. doi: 10.1084/jem.20111487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitosugi T, Kang S, Vander Heiden MG, Chung TW, Elf S, Lythgoe K, Dong S, Lonial S, Wang X, Chen GZ, et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Science signaling. 2009;2:ra73. doi: 10.1126/scisignal.2000431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura K, Tanaka T. Multimolecular forms of pyruvate kinase from rat and other mammalian tissues. I Electrophoretic studies J Biochem. 1972;71:1043–1051. doi: 10.1093/oxfordjournals.jbchem.a129852. [DOI] [PubMed] [Google Scholar]
- Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, Souza AL, Kafri R, Kirschner MW, Clish CB, Mootha VK. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science. 2012;336:1040–1044. doi: 10.1126/science.1218595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurczak MJ, Lee AH, Jornayvaz FR, Lee HY, Birkenfeld AL, Guigni BA, Kahn M, Samuel VT, Glimcher LH, Shulman GI. Dissociation of inositol-requiring enzyme (IRE1alpha)-mediated c-Jun N-terminal kinase activation from hepatic insulin resistance in conditional X-box-binding protein-1 (XBP1) knock-out mice. J Biol Chem. 2012;287:2558–2567. doi: 10.1074/jbc.M111.316760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller KE, Tan IS, Lee YS. SAICAR stimulates pyruvate kinase isoform M2 and promotes cancer cell survival in glucose-limited conditions. Science. 2012;338:1069–1072. doi: 10.1126/science.1224409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H, Cheung LW, Li J, Ju Z, Yu S, Stemke-Hale K, Dogruluk T, Lu Y, Liu X, Gu C, et al. Whole-exome sequencing combined with functional genomics reveals novel candidate driver cancer genes in endometrial cancer. Genome research. 2012;22:2120–2129. doi: 10.1101/gr.137596.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Hu H, Chang R, Zhong J, Knabel M, O'Meally R, Cole RN, Pandey A, Semenza GL. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 2011;145:732–744. doi: 10.1016/j.cell.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv L, Li D, Zhao D, Lin R, Chu Y, Zhang H, Zha Z, Liu Y, Li Z, Xu Y, et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Molecular cell. 2011;42:719–730. doi: 10.1016/j.molcel.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maquat LE. Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nature reviews Molecular cell biology. 2004;5:89–99. doi: 10.1038/nrm1310. [DOI] [PubMed] [Google Scholar]
- Mazurek S. Pyruvate kinase type M2: a key regulator of the metabolic budget system in tumor cells. The international journal of biochemistry & cell biology. 2011;43:969–980. doi: 10.1016/j.biocel.2010.02.005. [DOI] [PubMed] [Google Scholar]
- Noguchi T, Inoue H, Tanaka T. The M1- and M2-type isozymes of rat pyruvate kinase are produced from the same gene by alternative RNA splicing. J Biol Chem. 1986;261:13807–13812. [PubMed] [Google Scholar]
- Parnell KM, Foulks JM, Nix RN, Clifford A, Bullough J, Luo B, Senina A, Vollmer D, Liu J, McCarthy V, et al. Pharmacologic Activation of PKM2 Slows Lung Tumor Xenograft Growth. Mol Cancer Ther. 2013 doi: 10.1158/1535-7163.MCT-13-0026. [DOI] [PubMed] [Google Scholar]
- Salk JJ, Fox EJ, Loeb LA. Mutational heterogeneity in human cancers: origin and consequences. Annual review of pathology. 2010;5:51–75. doi: 10.1146/annurev-pathol-121808-102113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schraml P, Kononen J, Bubendorf L, Moch H, Bissig H, Nocito A, Mihatsch MJ, Kallioniemi OP, Sauter G. Tissue microarrays for gene amplification surveys in many different tumor types. Clinical cancer research : an official journal of the American Association for Cancer Research. 1999;5:1966–1975. [PubMed] [Google Scholar]
- Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008;118:3930–3942. doi: 10.1172/JCI36843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (New York, NY) 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Locasale JW, Swanson KD, Sharfi H, Heffron GJ, Amador-Noguez D, Christofk HR, Wagner G, Rabinowitz JD, Asara JM, et al. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science (New York, NY) 2010;329:1492–1499. doi: 10.1126/science.1188015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Plas DR, Rathmell JC, Fox CJ, Harris MH, Thompson CB. Growth factors can influence cell growth and survival through effects on glucose metabolism. Molecular and cellular biology. 2001;21:5899–5912. doi: 10.1128/MCB.21.17.5899-5912.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varghese B, Swaminathan G, Plotnikov A, Tzimas C, Yang N, Rui H, Fuchs SY. Prolactin inhibits activity of pyruvate kinase M2 to stimulate cell proliferation. Mol Endocrinol. 2010;24:2356–2365. doi: 10.1210/me.2010-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaupel P, Kallinowski F, Okunieff P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer research. 1989;49:6449–6465. [PubMed] [Google Scholar]
- Wang Z, Chatterjee D, Jeon HY, Akerman M, Vander Heiden MG, Cantley LC, Krainer AR. Exon-centric regulation of pyruvate kinase M alternative splicing via mutually exclusive exons. J Mol Cell Biol. 2012;4:79–87. doi: 10.1093/jmcb/mjr030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Wagner KU, Larson D, Weaver Z, Li C, Ried T, Hennighausen L, Wynshaw-Boris A, Deng CX. Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nat Genet. 1999;22:37–43. doi: 10.1038/8743. [DOI] [PubMed] [Google Scholar]
- Yang W, Xia Y, Hawke D, Li X, Liang J, Xing D, Aldape K, Hunter T, Alfred Yung WK, Lu Z. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell. 2012a;150:685–696. doi: 10.1016/j.cell.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Xia Y, Ji H, Zheng Y, Liang J, Huang W, Gao X, Aldape K, Lu Z. Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature. 2011;480:118–122. doi: 10.1038/nature10598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Zheng Y, Xia Y, Ji H, Chen X, Guo F, Lyssiotis CA, Aldape K, Cantley LC, Lu Z. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nature cell biology. 2012b;14:1295–1304. doi: 10.1038/ncb2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.