

Abstract
Rationale
The Ca2+ sensitivity of the myofilaments is increased in hypertrophic cardiomyopathy (HCM) and other heart diseases and may contribute to a higher risk for sudden cardiac death (SCD). Ca2+ sensitization increases susceptibility to reentrant ventricular tachycardia in animal models, but the underlying mechanism is unknown.
Objective
To investigate how myofilament Ca2+ sensitization creates reentrant arrhythmia susceptibility.
Methods and Results
Using HCM mouse models (troponinT (TNT)-I79N) and a Ca2+ sensitizing drug (EMD57033), here we identify focal energy deprivation as a direct consequence of myofilament Ca2+ sensitization. To detect ATP depletion and thus energy deprivation we measured accumulation of dephosphorylated Connexin 43 (Cx43) isoform P0 as well as AMP kinase activation by Western blotting and immunostaining. No differences were detected between groups at baseline, but regional accumulation of Cx43-P0 occurred within minutes in all Ca2+ sensitized hearts, in vivo after Isoproterenol challenge and in isolated hearts after rapid pacing. Lucifer yellow dye spread demonstrated reduced gap junctional coupling in areas with Cx43-P0 accumulation. Optical mapping revealed that selectively the transverse conduction velocity (CVT) was slowed and anisotropy increased. Myofilament Ca2+ de-sensitization with blebbistatin prevented focal energy deprivation, CVT slowing and the reentrant ventricular arrhythmias.
Conclusions
Myofilament Ca2+ sensitization rapidly leads to focal energy deprivation and reduced intercellular coupling during conditions that raise arrhythmia susceptibility. This is a novel pro-arrhythmic mechanism that can increase arrhythmia susceptibility in structurally normal hearts within minutes and may therefore contribute to SCD in diseases with increased myofilament Ca2+ sensitivity.
Keywords: Myofilament Ca2+ sensitivity, ventricular arrhythmias, sudden cardiac death, Connexin43 (Cx43), familial hypertrophic cardiomyopathy, conduction velocity dispersion
INTRODUCTION
Hypertrophic cardiomyopathy (HCM) is the most common inherited cardiac disease with a prevalence of about one in 500.1 In most cases the underlying molecular defect is an autosomal dominant mutation in a sarcomeric protein (“disease of the sarcomere”).2 Mutation carriers have an increased risk for ventricular arrhythmia and sudden cardiac death (SCD) 3–6, but how sarcomeric mutations predispose to ventricular arrhythmias and SCD remains unclear.
With few exceptions, proteins with HCM mutations integrate together with wild-type protein into the sarcomere thereby altering the force producing process.7 Through an as yet unidentified pathway this initiates progressive structural changes that typically involve hypertrophy, focal and interstitial fibrosis and myofibrillar disarray, but with very large individual variability.8, 9 The structural remodeling likely creates an anatomical substrate and contributes to the increase in arrhythmia susceptibility in HCM.10, 11 However, SCD risk assessment on the basis of structural features alone is inaccurate and current algorithms to classify individual patients are unreliable.12, 13 Hence, additional factors likely contribute to arrhythmia risk and there is need to investigate the molecular events that cause SCD.
Considerable effort has been invested into identifying a common sarcomere property conferred by HCM mutations which would explain the similar downstream effects of the many different HCM mutations. A frequent finding among HCM mutations is increased myofilament Ca2+ sensitivity, which is also observed in acquired conditions associated with arrhythmias.14, 15 Myofilament Ca2+ sensitization has several immediate downstream effects and impacts Ca2+ homeostasis (by affecting cytosolic Ca2+ buffering 16), mechanics (positive inotropic, negative lusitropic) and energy demand (proportional increase in myosin ATPase activity), as reviewed in14. We have previously demonstrated that myofilament Ca2+ sensitization is proarrhythmic, in part by using transgenic mouse models that express human TroponinT (TnT) HCM mutations.17 The degree of Ca2+ sensitization correlated with the risk for ventricular reentrant-type arrhythmias, e.g. the highest arrhythmia risk was found in mice with the most sensitized myofilaments (TnT-I79N). Ca2+ sensitized hearts exhibited an increased dispersion of conduction velocity (CV, i.e. regional CV slowing), which is a highly proarrhythmic state 17–19, but was not the result of structural remodeling in the TnT mutant hearts.20, 21 Accordingly, treating control mice with the Ca2+ sensitizer drug EMD57033 (EMD) increased CV dispersion and induced arrhythmias, whereas a compound with Ca2+ desensitizing properties, blebbistatin (BLEB), prevented CV dispersion and arrhythmia induction during rapid pacing. Thus, increasing myofilament Ca2+ sensitivity raises arrhythmia risk and lowering Ca2+ sensitivity to control level prevents the occurrence of arrhythmias. How myofilament Ca2+ sensitization causes increased CV dispersion and arrhythmias is not understood and the objective of this study was to investigate the underlying pathway.
Here, we utilized the TnT-I79N mouse model, EMD and BLEB to determine the link between myofilament Ca2+ sensitivity and arrhythmias. We identify regional energy deprivation as a direct consequence of myofilament Ca2+ sensitization. Subsequent focal gap junctional uncoupling likely contributes to regional CV slowing forming the substrate that can support reentry. This mechanism generates arrhythmia susceptibility in structurally normal hearts within a short period of time and can cause lethal arrhythmias.
METHODS
A detailed description of all procedures and information about mouse models and drugs can be found in the online data supplement (supplemental material at http://circres.ahajournals.org).
RESULTS
Arrhythmias in TnT-I79N mice in vivo are associated with altered cardiac gap junctions
To investigate the mechanism responsible for the ventricular arrhythmias in the TnT-I79N HCM mouse model, we utilized a previously established catecholamine challenge protocol (Isoproterenol, (Iso), 1.5 mg/kg).17, 22 TnT-I79N mice frequently exhibited ventricular ectopy (56%) and/or ventricular tachycardia (VT, 25%), while only one mouse expressing wild-type TnT (TnT-WT) had one single premature ventricular contraction (PVC, Fig. 1B, p<0.006 by Fisher exact test). The heart rate increase was the same in both groups (from 464±11 to 620±6 in TnT-WT compared to 457±12 to 624±16 beats/min in TnT-I79N). ECG analysis revealed a progressive prolongation of QRS duration in TnT-I79N mice following Iso injection (Fig. 1C). A longer QRS duration reflects a delay in whole heart activation, which may be a consequence of the slower regional conduction previously reported.17
Fig. 1. Isoproterenol (Iso) injection in vivo induces ventricular arrhythmias, slowed ventricular depolarization and is associated with reduced expression of the gap junction protein Cx43 in TnT-I79N mice.
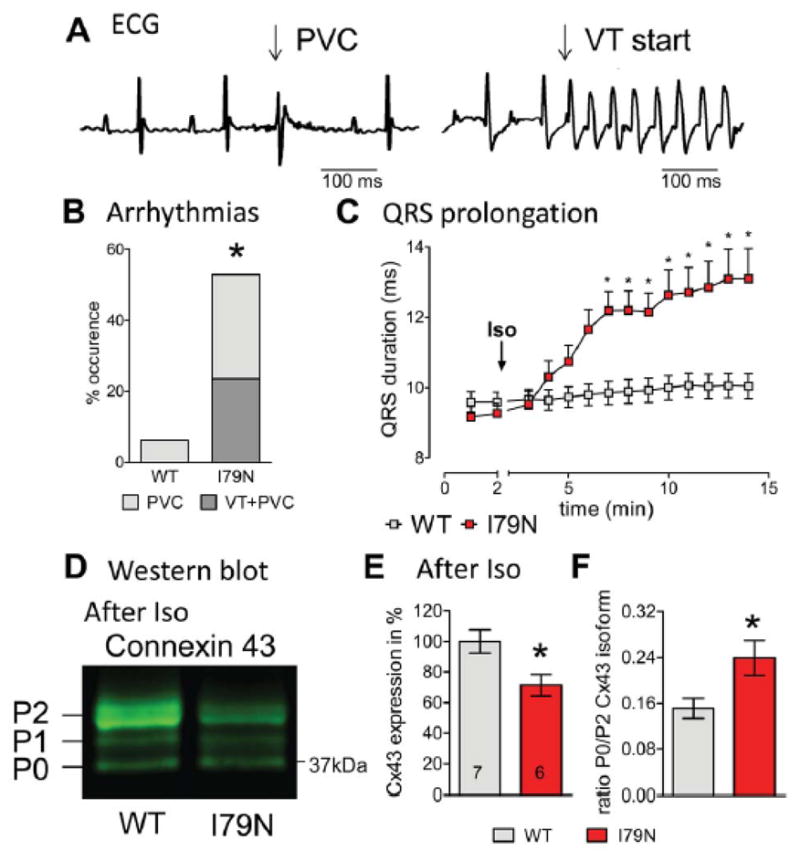
(A) Representative examples for a premature ventricular contraction (PVC) and ventricular tachycardia (VT). Summary data are presented in (B), n=16 each. (C) QRS duration was prolonged within a few minutes after Iso injection in TnT-I79N, but not TnT-WT mice. (D) Typical Western blot pattern for anti-Cx43 (isoforms P2, P1, P0 indicated) after Iso treatment, (E) Total expression of Cx43 and (F) P0/P2 isoform ratio analyzed by Western blot. N numbers are indicated in columns and are the same for (E) and (F). * p≤0.05 vs WT
Gap junction channels are important for conduction properties, they lower membrane resistance by directly connecting the cytoplasm of neighboring cells and thus facilitate electrical propagation.23 Thus, we investigated the gap junction forming protein expressed in ventricle, Connexin 43 (Cx43), in hearts collected after Iso injection and found a lower expression (72±6%; Fig. 1E; for baseline expression, see Fig. 3C,D). Cx43 has several phosphorylation sites and at least three different phosphorylation states of Cx43 are separated by SDS Polyacrylamid-gelelectrophoresis (marked as P2, P1, P0, in Fig. 1D).24, 25 P0 is less phosphorylated than P2 and often referred to as “dephosphorylated” Cx43. The ratio between the Cx43 isoforms P0/P2 was increased following Iso treatment (Fig. 1F), indicating that in addition to the overall Cx43 decrease, altered phosphorylation may be associated with changes in gap junctional function.26
Fig. 3. Rapid pacing induces changes in connexin43 (Cx43) protein expression, solubility and isoform distribution in TnT-I79N isolated hearts (pacing protocol see Supplemental Fig. IA).
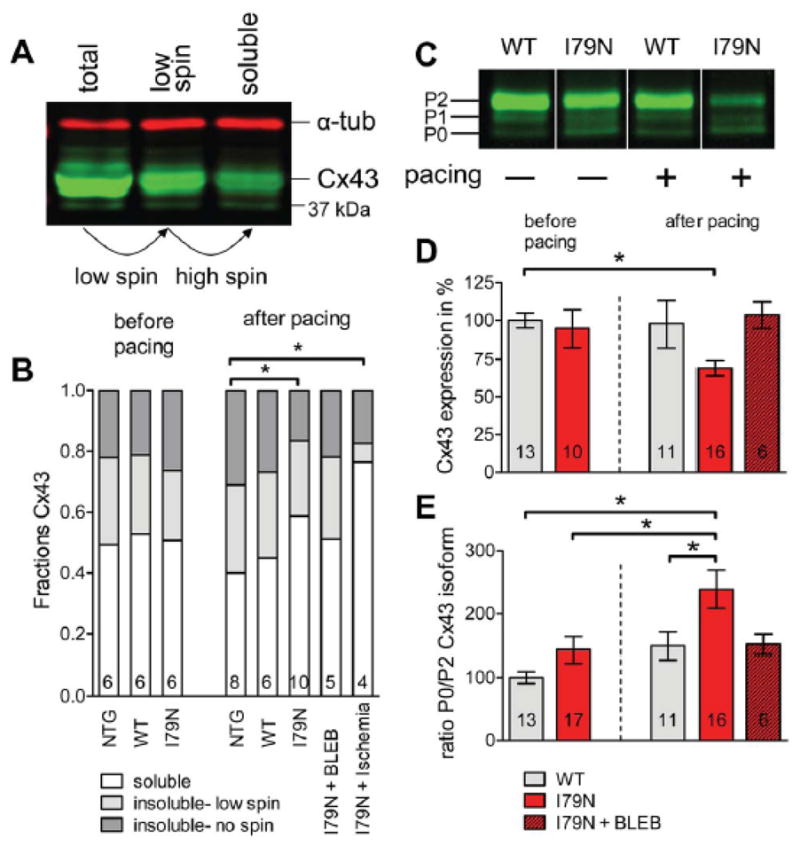
(B) Fractions of soluble (non-junctional) and insoluble (junctional) Cx43 in NTG, WT and I79N. N numbers are indicated in columns. A representative Western blot is shown (A, α-tubulin (α-tub)). The same sample volume was loaded before centrifugation (total), after 500×g centrifugation (low spin) and after 14000×g centrifugation (soluble). (C) A representative Western blot (all samples from same blot, but intermediate lanes were deleted) of WT and I79N samples before and after pacing. The different Cx43 isoforms are indicated (P2, P1, P0). (D) Summary data for Cx43 expression and (E) Summary data for the ratio of P0/P2 isoform for WT and I79N before and after pacing and I79N after pacing in the presence of BLEB (3 μM). * p≤0.05
Rapid pacing induces VT, prolongs QRS duration and increases anisotropy ratio in TnT-I79N hearts
To further dissect the mechanism responsible for the QRS prolongation and ventricular arrhythmias, we used a rapid pacing protocol in isolated hearts that led to VT in over 50% of TnT-I79N, but not in TnT-WT hearts (Supplemental Fig. IA+B). Fig. IC shows an example for VT induced during rapid pacing in an isolated TnT-I79N heart. Similar to our findings in vivo after Iso, rapid pacing of isolated hearts caused progressive QRS prolongation in TnT-I79N hearts but not in TnT-WT (Fig. 2A). QRS prolongation was prevented by the Ca2+ desensitizer BLEB, which at 3 μM reduces myofilament Ca2+ sensitivity of TnT-I79N hearts to that of non-transgenic control hearts.17 We next performed optical mapping and measured the CV along the fiber (longitudinal (CVL), fast) and across the fiber (transverse (CVT), slow) as illustrated in Fig. 2B.17 CVL in TnT-I79N was similar to control (Figure 2C), consistent with previous observations that the average CV is not altered.17 However, CVT was reduced by >20% in TnT-I79N mice compared to TnT-WT (Figure 2C), which resulted in an increased anisotropy ratio (CVL/CVT). CVT slowing and increased anisotropy was prevented by pretreatment with BLEB (Figure 2D).
Fig. 2. Isolated TnT-I79N hearts exhibit QRS prolongation and slower transverse conduction velocity (CVT).
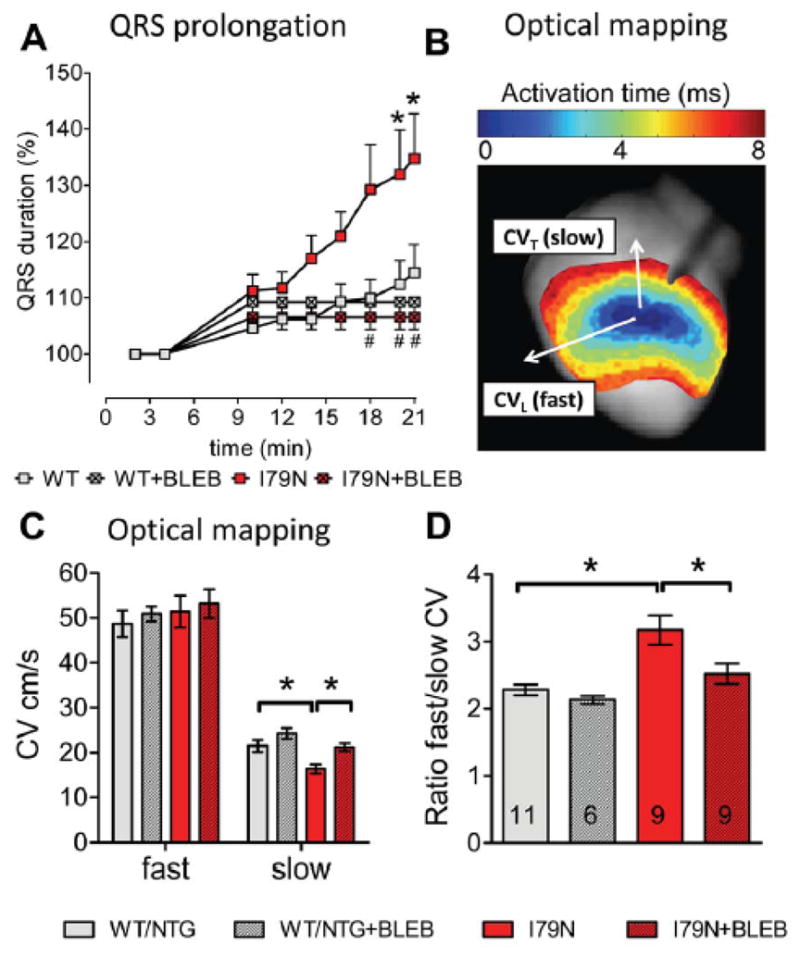
(A) QRS duration determined from volume conducted ECG recordings during brief pacing interruptions (pacing protocol as shown in Supplemental Fig. I); n = 6–19. QRS prolongation was observed in TnT-I79N, but not in TnT-WT and was prevented by blebbistatin (BLEB 3 μM, n=3–18). (C) Longitudinal (fast, CVL) and CVT (slow) conduction velocity and (D) anisotropy ratio in control (WT/NTG) and I79N determined as illustrated in (B). No difference was observed between TnT-WT and NTG mice and data were pooled. N numbers are indicated in columns and are the same for (C) and (D). * p≤0.05; (A) * p≤0.05 vs WT, # p≤0.05 vs I79N
Since the anisotropy ratio and in particular CVT are strongly influenced by the number, activity and spatial arrangement of gap junctions27, 28, we next investigated gap junction properties in detail at the biochemical level.
Rapid changes in Cx43 abundance, solubility and isoform distribution in TnT-I79N hearts
Gap junctions are formed by connexins; Cx43 is the only connexin isoforms expressed in adult ventricular myocardium is Cx43.29 The junctional (=insoluble) Cx43 is found at the cell surface forming gap junction channels at the intercalated disc, while the non-junctional (=soluble) Cx43 is intracellular. Cardiac tissue was fractionated into three fractions (the insoluble fraction was divided into a “low spin” and “high spin” fraction) to measure the relative distribution between junctional (light and dark grey) and non-junctional Cx43 (white) as illustrated in Fig. 3A. The pellet is discarded after each processing step and the supernatant analyzed via Western blot. Alpha-tubulin (α-tub) is completely soluble and illustrates equal loading of all fractions. The distribution of junctional:non-junctional Cx43 before pacing was approximately 50:50 (Fig. 3B) and not significantly different between TnT-WT and TnT-I79N mice. After pacing (see Supplemental Fig. IA for protocol), the control groups (NTG and WT) tended to have increased junctional Cx43, while TnT-I79N tended to have decreased junctional Cx43 (Fig. 3B), although neither result was significantly different from baseline. However, there was a difference between NTG control and TnT-I79N after pacing since they changed into opposite directions. This effect was not observed in the presence of BLEB. Increased solubility of Cx43 has been described in end-stage human ischemic cardiomyopathy 30 and was detected here also after global ischemia (Fig. 3B).
Next, the total expression levels of Cx43 were analyzed in the “low spin” supernatant while loading equal amounts of total protein as shown in Fig. 3C. Cx43 protein levels were the same before pacing (baseline) in both groups, but pacing caused a reduction in Cx43 in the TnT-I79N (94.8±12.2 before pacing and 69.2±5.1 after pacing; Fig. 3D), while Cx43 levels were not affected by pacing in TnT-WT (100±4.8 before pacing and 97.8±15.4 after pacing). BLEB prevented the pacing induced decrease in Cx43 in the TnT-I79N (Fig. 3D).
We also analyzed the Cx43 isoform distribution as in Fig. 1. No difference was observed before pacing, but pacing induced distinct changes in the Cx43-isoform distribution in TnT-I79N: A significant decrease in P2 isoform in TnT-I79N compared to baseline (61.5±5.1 vs 93.7±12.9, respectively), but no change in P1 and a moderate increase in P0 with pacing (136.8±9.3 vs 106.5±10.5, N numbers as in columns). Together, this caused an increase in the P0/P2 ratio in TnT-I79N after pacing that was prevented by BLEB (Fig. 3E) Pacing does not induce changes in TnT-WT and earlier experiments with a slightly different pacing protocol showed no effect of BLEB on Cx43 expression and isoform distribution in TnT-WT (data not shown).
Taken together, at baseline no differences in Cx43 solubility, amount and isoform distribution exist between groups. After rapid pacing, the solubility of Cx43 increases in TnT-I79N and isoform P2 is specifically lost, which is the phospho-isoform considered to be part of the functional gap junctional plaque.24 These results indicate a decrease in Cx43 at the intercalated disc induced by rapid pacing similar to what we observed following Iso treatment in our in vivo model (Fig. 1D–F).
Focal accumulation of Cx43-P0 in TnT-I79N during rapid pacing
To determine the effect of rapid pacing on gap junction localization, frozen heart sections were obtained before and after pacing and immunostained with anti-Cx43 antibodies, specific for either total Cx43 or the Cx43-P0 isoform (Fig. 4A+B). Cx43-P0 isoform staining was barely detectable, but typically homogeneous before pacing (Fig. 4A) or after pacing in TnT-WT or TnT-I79N + BLEB (Fig. 4B top). In TnT-I79N, however, areas of intense staining for P0 isoform were observed after pacing (two examples Fig. 4B bottom). The distribution of Cx43-P0 was non-homogeneous, with small regions of varying shape, size and intensity (“patches”) where Cx43-P0 accumulated (characterized by high P0/Cx43 ratio (Fig. 4D, “red”/“green”)). The “patches” were often found in endocardial regions, while accumulation in epicardial and mid-myocardial tissue appeared less frequent. Areas with Cx43-P0 accumulation had for the most part clear borders with abrupt transition to areas with low level of Cx43-P0 staining.
Fig. 4.
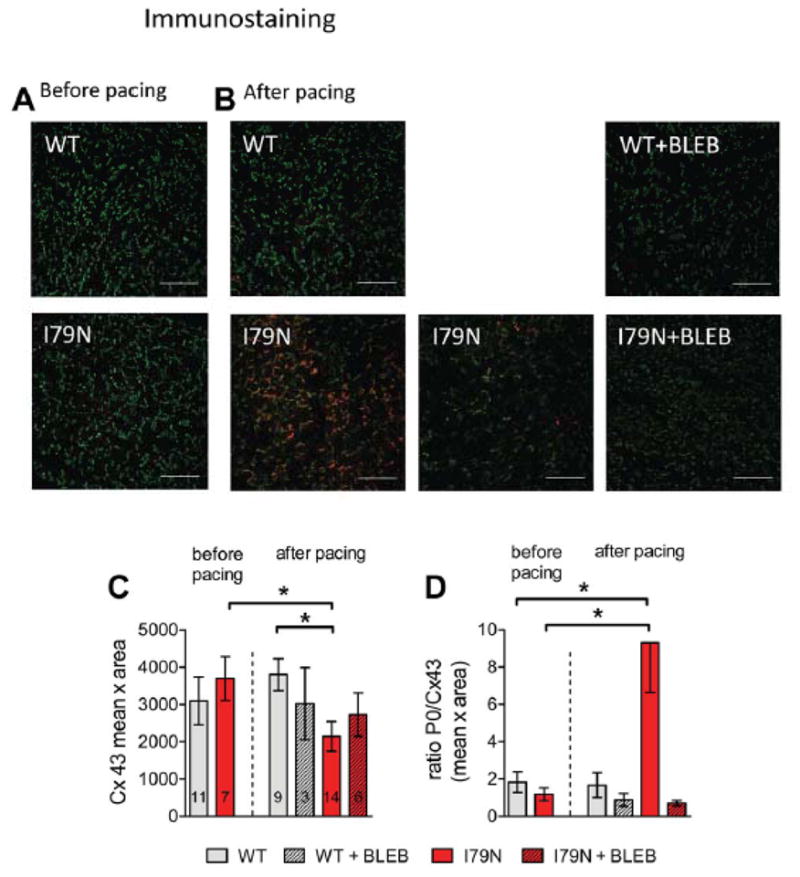
Regional accumulation of dephosphorylated Cx43 isoform P0 in TnT-I79N after pacing. This is not observed in TnT-WT, in TnT-I79N before pacing and is prevented by BLEB. Anti-Cx43 (green) and anti-Cx43-P0 (red) staining of I79N heart sections before (A) and after pacing (B, two examples are shown for I79N after pacing). The scale bar length is 100 μm. (C) Summary data for integrated density (mean intensity x area (pixel)) of Cx43 (N numbers are indicated in columns and are the same for both graphs). See Supplemental Fig. III for examples of anti-Cx43 staining (green) only for all groups. (D) Summary data for the average ratio of Cx43/Cx43-P0 in all groups (C) * p≤0.05
The quantitation of Cx43 expression levels by immunostaining (Fig. 4C, “green”) was consistent with the results predicted from the western blot experiments in Fig. 3. No significant difference between TnT-I79N and control hearts was observed before pacing (integrated Cx43 density 3094±640 vs. 3692±586 in TnT-WT vs TnT-I79N respectively), but after pacing the amount of total Cx43 was lower in TnT-I79N than in TnT-WT hearts (3800±426 in TnT-WT vs. 2143±399 in TnT-I79N; Fig. 4B+C). This difference in the total Cx43 is the result of an opposite trend in TnT-I79N and TnT-WT: the TnT-WT tended to increase Cx43 at the intercalated disc (>20%↑), while the TnT-I79N reduced it during pacing (>40%↓). Importantly, BLEB prevented the pacing-induced changes in Cx43 expression and the formation of Cx43-P0 “patches” (Fig. 4B–D).
Focal accumulation of Cx43-P0 in TnT-I79N in vivo and in control hearts treated with EMD
Next, we similarly investigated gap junctional organization in the mouse cardiac tissue frozen after Iso challenge in vivo (as for Fig. 1). Regions of Cx43-P0 accumulation were detected in TnT-I79N, but not in TnT-WT (Fig. 5A+C). Total Cx43 was decreased by around 50% (Fig. 5B; decreased to 51±9 %), consistent with the Western blot results and the data from isolated paced hearts. Thus, the reduction of junctional Cx43 and the non-homogeneous distribution of Cx43-P0 is also observed in vivo during physiological heart rates and within the same time frame.
Fig. 5. Regional accumulation of Cx43-P0 observed in I79N after Iso injection in vivo and after treatment of isolated control hearts with the Ca2+ sensitizer EMD57033 (EMD, 3 μM).
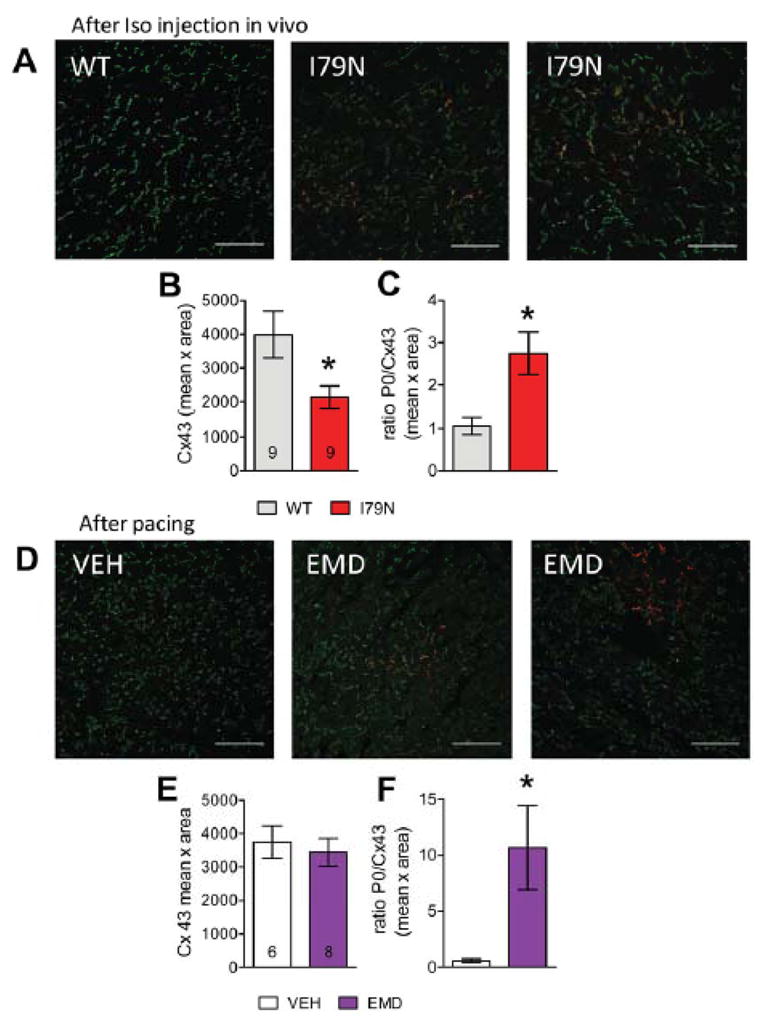
(A) Anti-Cx43 (green) and anti-Cx43-P0 (red) staining of TnT-WT and TnT-79N ventricular tissue after Iso challenge. (B) Summary data for integrated density (mean intensity x area (pixel)) of Cx43 and (C) ratio of Cx43-P0/Cx43. (D) Examples of immunostained tissue from isolated NTG hearts treated with VEH or EMD after rapid pacing. (E+F) Summary data for EMD experiments (for pacing protocol see Supplemental Fig. IA). The scale bar length is100 μm in all images. * p≤0.05 vs WT
To address the role of myofilament Ca2+ sensitization for the arrhythmias and focal changes in Cx43, we studied hearts treated with the Ca2+ sensitizing drug EMD (3 μM). Rapid pacing of EMD treated hearts induced VT and, as in TnT-I79N, VT first occurred after a time delay (see Supplemental Fig. IB). Analogous to the results in TnT-I79N hearts (Figure 4B), EMD caused focal accumulation of Cx43-P0 (“patches”, Fig. 5D, middle and right, see also Fig. 5F). In contrast, the decrease in Cx43 density at the intercalated disc was not observed in EMD treated hearts, indicating that this reduction is not a prerequisite for the development of arrhythmias (Figure 5E). These results indicate that myofilament Ca2+ sensitization, via “patchy” Cx43-P0 accumulation, is likely the key molecular property that is responsible for the increased arrhythmia susceptibility.
We further characterized changes in CV induced by EMD treatment (Supplemental Fig. IV). Optical mapping revealed that the transverse CV decreased within minutes after perfusion with EMD (Supplemental Fig. IVA). This led to increased anisotropy which was prevented by the presence of BLEB (3 μM, Supplemental Fig. IVB). After longer perfusion with EMD we detected conduction abnormalities consistent with the formation of conduction blocks (example shown in Fig. IIIC). Similar to the TnT-I79N hearts, pacing according to the protocol in Supplemental Figure IA led to moderate QRS prolongation over time, while no prolongation was observed with vehicle (VEH) only (Supplemental Fig. IIID).
Myocardial regions with Cx43-P0 accumulation are energetically compromised
What causes the rapid focal Cx43 dephosphorylation detected as Cx43-P0 accumulation? The only other condition that has been described to induce very rapid changes in Cx43 is acute ischemia.31 Ischemia has several downstream effects, but an elegant study by Turner and colleagues 32 demonstrated that the trigger for the Cx43 dephosphorylation during ischemia is the drop in cellular ATP levels. Thus, the Cx43 dephosphorylation observed here is possibly a marker for intracellular ATP depletion. To further confirm that low ATP levels are also the underlying mechanism for Cx43 dephosphorylation in this study, we measured AMP-kinase (AMPK) activation. We assessed cellular ATP levels by testing AMP-kinase (AMPK) activation: When [ATP] drops and subsequently [AMP] rises, AMP binds to AMPK-γ subunit and promotes phosphorylation of the α-subunit in the activation loop at threonine 172 (pAMPK).33–36
Before pacing, Western blotting of whole heart homogenates detected no difference in αAMPK-T172 (pAMPK) phosphorylation and was similarly low in TnT-WT and TnT-I79N (Fig. 6A+B). Rapid pacing increased pAMPK to 225±47% in TnT-I79N and BLEB prevented the increase in pAMPK in TnT-I79N with pacing. TnT-I79N hearts exposed to 21 min of global no-flow ischemia served as a positive control (Fig. 6B+C). Any differences in pAMPK signal were not caused by changes in αAMPK expression due to pacing or ischemia (data not shown).
Fig. 6. Activated AMP-kinase (pAMPK) in areas with dephosphorylated Cx43-P0 accumulation.
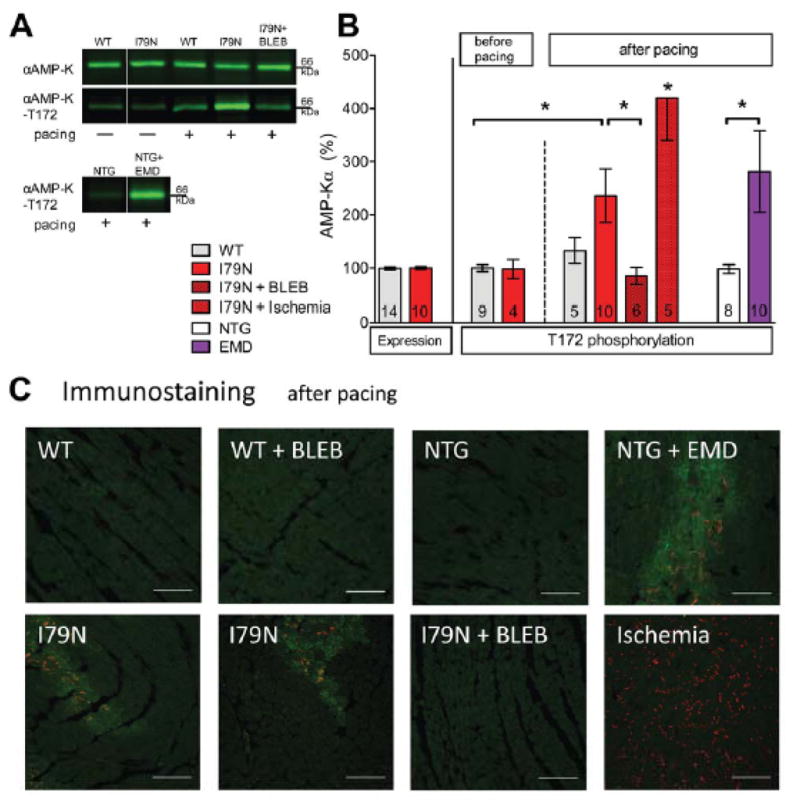
(A+B) Western blot analysis of αAMP-K expression and phosphorylation at threonine 172 (=activation). Representative western blots (A, white line indicates where intermediate lanes were deleted) and summary data (B)). 21 min of global ischemia was included as a positive control. (C) Representative examples showing activated pAMPK –T172 (“green”) in the same regions where Cx43-P0 accumulation (“red”) is observed (TnT-I79N, bottom first and second and NTG+EMD, top forth). Cx43-P0 is more uniformly observed after global ischemia (bottom forth). The scale bar indicates 100 μm. * p≤0.05 as indicated.
After rapid pacing challenge, the tissue distribution of pAMPK was heterogeneous in TnT-I79N hearts, and the highest intensity was found in the regions where Cx43-P0 accumulated (Figure 6C, “green”). No regions with intense pAMPK staining were observed in TnT-WT, NTG or hearts treated with BLEB. No-flow ischemia also activated AMPK, but the distribution was largely homogeneous (Figure 6C, bottom right). These data indicate that the myocardium in the areas of Cx43-P0 accumulation is energetically compromised.
Assessment of gap junctional coupling using Lucifer yellow dye spread
Next we assessed if the accumulation of Cx43-P0 is associated with altered gap junctional function using a modified scrape loading technique.37 Monitoring the passage of fluorescent dyes through gap junctions from one cell to another is a simple and rapid technique to assess intercellular communication. Lucifer yellow (LY) spreads from the site of injury through gap junctions to the neighboring cells, the faster it diffuses the more are cells interconnected. The LY spread distance through intact cardiomyocytes in the longitudinal direction was 364±34 μm and in the transverse direction 135±14 μm in TnT-WT after pacing (Fig. 7A–C+K). For TnT-I79N, sites were classified as Cx43-P0 accumulation negative or positive if Cx43-P0 accumulation was detected along at least two sides (Fig. 7A–K). Sites that could not be clearly classified were not included in the analysis. Consistent with the lower amount of Cx43 found in TnT-I79N compared to TnT-WT after pacing (reduced by about 43%), LY dye spread was reduced in TnT-I79N as well (Cx43-P0 accumulation negative): in the longitudinal direction was 182±18 μm and in the transverse direction 86±10 μm (both p≤0.05 vs TnT-WT). This is a 50% reduction in the longitudinal direction and about 36% reduction in the transverse direction compared to TnT-WT. Intercellular coupling was even further reduced in Cx43-P0 accumulation positive areas: the LY spread was 157±35 in the longitudinal direction (>12%↓; not significant vs Cx43-P0 negative) and, strikingly, only 34±4 μm in the transverse direction (>60%↓, p≤0.05 vs TnT-I79N Cx43-P0 negative). The gap junctional coupling in Cx43-P0 positive areas, when compared to TnT-WT, is reduced >56% in the longitudinal direction and >74% in the transverse direction. Thus, Cx43-P0 identifies areas where significant intercellular uncoupling has occurred.
Fig. 7. Reduced gap junctional coupling assessed by modified scrape loading.

(A,D,G) Representative confocal images of Lucifer yellow (LY, yellow) spread after needle puncture in TnT hearts after pacing. (B,E,H) Corresponding images of non-gap junction permeable Tetramethylrhodamine dextran (TMR, green) that was simultaneously injected. (C, F, J) Composite image of LY, TMR and Cx43-P0 (red). The scale bar length is 100 μm. (K) Summary data of LY spread analysis from 15 sites in TnT-WT (3 hearts) and 17 sites in TnT-I79N hearts (4 hearts). For TnT-I79N only sites were analyzed that were clearly classified as Cx43-P0 negative (I79N P0 neg, n=9) or showed clear Cx43-P0 accumulation along at least two sides (I79N P0 pos, n=8). Dye spread was analyzed along the fiber (longitudinal) or across the fiber (transverse). * p≤0.05 vs. as indicated
DISCUSSION
This study investigates the underlying mechanisms for the increased occurrence of reentrant arrhythmias in myofilament Ca2+ sensitized hearts. Here we show that myofilament Ca2+ sensitization predisposes to the generation of focal energy deprivation during stress. Myocytes in the affected regions display signs of metabolic stress, as evidenced by AMP kinase activation and Cx43-P0 accumulation. To test the dependence of “patchy” Cx43-P0 accumulation on myofilament Ca2+ sensitization, we used different models and protocols: Ca2+ sensitized mutant hearts show this phenomenon in isolated perfused hearts as well as in vivo, it can be induced by Ca2+ sensitizing drugs in controls and is prevented by Ca2+ de-sensitization. Thus, the formation of “patchy” regions with ischemia-like changes during high heart rates appears to be a direct downstream effect of increased myofilament Ca2+ sensitivity. We further demonstrate that gap junctional coupling is reduced in areas with Cx43-P0 accumulation to a sufficient degree to affect CV (see below). The heterogeneous Cx43-P0 accumulation may lead to heterogeneous CV slowing (increased dispersion of CV), which constitutes a highly proarrhythmic state predisposing to conduction block and reentry. These new results are in agreement with our previous report that increased CV dispersion is induced by rapid pacing in Ca2+ sensitized hearts.17 The induction of focal energy deprivation that rapidly decreases gap junctional coupling in Ca2+ sensitized hearts is a novel pro-arrhythmic mechanism that can generate arrhythmia susceptibility in structurally normal hearts within minutes.
Critical role of focal Cx43 dephosphorylation for arrhythmias?
We find a strong association between focal Cx43-P0 accumulation and arrhythmias: The focal Cx43-P0 accumulation is found under all conditions with raised arrhythmia susceptibility, but we did not detect Cx43-P0 accumulation in any conditions without increased arrhythmia susceptibility. In TnT-I79N mice, we find in addition to focal Cx43-P0 accumulation also a decline in Cx43 levels and both may contribute to arrhythmia generation. On the other hand, in EMD treated hearts we only observed focal Cx43-P0 accumulation, not an overall Cx43 decrease (Fig. 5E), but the same type and time of onset for the arrhythmias (Supplemental Fig. IB), indicating the Cx43-P0 accumulation is potentially critical.
How is intercellular coupling affected by Cx43-P0 accumulation? We know that the accumulation of Cx43-P0 is accompanied by gap junction closure e.g. in ischemia.31 Studies by Lampe and colleagues indicate that unphosphorylated S365 is likely the critical feature for Cx43-P0 specific antibody binding.38 Phosphorylation at S365 is lost during hypoxia and this dephosphorylation is at the same time a prerequisite for PKC-dependent phosphorylation at S368, which reduces gap junctional conductivity.39, 40 Here we also directly demonstrate that in areas with Cx43-P0 accumulation the gap junctional coupling is reduced as measured by LY dye spread. It appears that Cx43-P0 is an excellent marker for cellular uncoupling, if the exact molecular change that generates it is responsible for it or not.
Can the patchy gap junctional uncoupling affect conduction velocity? The number of gap junction channels is typically much larger than needed for excitation propagation. This is consistent with our finding that the CVL (this study) or average CV is unaltered.17 Studies using mice with 50% decrease in gap junctions show either no effect on CV 41, 42, or a reduction of approximately 25%.43, 44 A loss of 90% of gap junctions leads to an approximately 50% decrease in CV.45 The reduction in dye spread in this study in the areas with Cx43-P0 accumulation was 56% (longitudinal) and 74% (transverse), which is a range where effects on CV can be expected. Gap junctional uncoupling preferentially affects conduction in the transverse direction.46, 47 It is quite possible that the patchy gap junctional uncoupling may importantly contribute to the decrease in CVT during pacing demonstrated in this study.
A role of focal gap junction changes for reentrant arrhythmias? In general, regional gap junctional uncoupling can produce or unmask preexisting heterogeneities in cellular refractoriness and/or CV that can contribute to slow conduction and reentry. Similar changes in gap junctions occur in hypoxia or ischemia and their role for arrhythmogenesis has been reviewed elsewhere.19, 48, 49 The arrhythmia susceptibility may arise from the increased anisotropy, predisposing to unidirectional conduction block and reentry, even though the exact mechanism is debated.46 Another possible mechanism is that the abrupt excitation spread from poorly coupled into well coupled tissue may facilitate conduction block via a source-sink mismatch and assist in the generation of reentry.50
ATP depletion as cause for Cx43 dephosphorylation
The rapid regional changes in Cx43 phosphorylation in myofilament Ca2+ sensitized hearts and the simultaneous AMPK activation are a striking result of this study. It is well established that ATP depletion is a mechanism that can cause rapid Cx43 dephosphorylation: Metabolic inhibition is frequently utilized experimentally to induce Cx43 dephosphorylation and it reduces gap junctional communication in cortical astrocytes.51, 52 Beardslee et al provided the first evidence for Cx43-P0 accumulation in the intact perfused heart during global ischemia.31 The most direct evidence that ATP depletion is the upstream requirement for Cx43 dephosphorylation was later provided by Turner et al.32 They dissected the individual contributions of hypoxia, extracellular factors (acidosis, build-up of metabolites), glucose and ATP depletion to dephosphorylation of Cx43. They found that Cx43 dephosphorylation closely mirrored fluctuations in cellular ATP levels, requiring >50% of basal ATP levels to maintain Cx43 phosphorylation state. How ATP levels mediate the effect on Cx43 phosphorylation has not been solved. Turner et al. however also provide evidence that pharmacological activation of AMPK does not lead to Cx43 dephosphorylation. This demonstrates that that these two events are not linked and that they are independent indicators of ATP depletion. AMPK phosphorylation primarily relies on AMP binding to cause a conformational change that then promotes increased phosphorylation of T172, but other pathways have been demonstrated. Recent evidence suggests that AMPK activity can be regulated receptor-dependent and dissociated from the energy charge of the cell, e.g. by hormones like adiponectin or oestrogen.53, 54 It is unclear though how such mechanisms would play a role in our study. Thus, the focal accumulation of Cx43-P0 and corresponding elevated levels of pAMPK found in this study both indicate that intracellular [ATP] levels are decreased.
Why is Cx43 dephosphorylation focal?
The focal nature of Cx43 dephosphorylation and AMPK activation indicates that in some areas “demand” exceeded “supply”, but we have not yet elucidated the origins of this mismatch. On one hand, the deficit may lie on the “supply” side and may be due to insufficient perfusion volume, while on the other hand the deficit may result from increased “demand” due to pathologically high energetic requirements of Ca2+ sensitized cardiac muscle. The positive inotropic effect conferred by myofilament Ca2+ sensitization will increase myosin ATPase activity and thereby proportionally increase energy demand.14, 55 Increased heart rates can be expected to further raise energy demand. Moreover, increased “tension cost” (higher energetic cost of force production) has been described for HCM sarcomeric mutations, including TnT mutations.56 It is, however, known that the racemic mixture EMD53998 (50% EMD57033) has no effect on tension cost and actually lowers tension cost in the presence of inorganic phosphate.57 How myofilament Ca2+ sensitivity may impact “supply” is less clear, but the negative lusitropic effect and increased extramural compression may have negative effects on perfusion. In the heart, in contrast to all other organs, coronary flow is slower during phases of contraction than during diastole and thus prolonging the duration of contraction would naturally impinge on coronary flow. Coronary perfusion also greatly relies on pressure waves and the largest is a “suction” wave generated by relaxation of the left ventricle, a critical driver of diastolic coronary blood flow.58 How these effects alone or in combination lead to energy deprivation in some regions and not others requires further studies, but the exact regional balance between raised demand and compromised supply may result in the observed phenotype. It is an intriguing feature of HCM that although the genetic mutation is expressed throughout the ventricle, fibrosis is often patchy and the hypertrophy asymmetrical or even focal.59, 60
Relevance for human disease
There are important implications of this novel mechanism that extends beyond HCM patients and concerns all patients with increased myofilament Ca2+ sensitivity. For example, it is well documented that myofilament Ca2+ sensitivity increases in patients after myocardial infarction.61, 62 More mechanistic insights may also benefit HCM patients by improving risk stratification for arrhythmias, which is currently challenging and unreliable. Arrhythmia risk greatly varies between individual HCM mutation carriers, even between individuals with the same mutation and from the same family. The identification of new risk factors and how different risk factors may act synergistically to produce arrhythmias is clearly needed. Along the same lines, pharmacological strategies to prevent sudden death in HCM patients have not been very successful 63, 64 and comprehensive understanding about the underlying mechanisms may lead to the development of much needed new treatments.
This molecular mechanism will also inform our efforts to develop new positive inotropic drugs for the treatment of heart failure. One new compound that was recently tested in phase 2 clinical trials is omecamtiv mecarbil, a myosin activator that prolongs systolic ejection time and increases stroke volume.65 The mechanism is promising, increasing the number of cross-bridges and enhance contraction without effect on the contraction rate and myocardial oxygen consumption.66 However, at high serum concentrations ischemic episodes were triggered in a few healthy patients, potentially due to the shortening of the diastolic interval. More studies are needed.67
Conclusions
In this study we present evidence that myofilament Ca2+ sensitization can cause focal energy deprivation during stress. This pathway can be activated within minutes and may explain the occurrence of SCD in young HCM patients even before significant structural remodeling occurs. This strongly suggests that lowering myofilament Ca2+ sensitivity to control level may be a promising new strategy to prevent ventricular arrhythmias and SCD. The mouse model and tools used in this study present the unique opportunity to investigate in the future the underlying cause for the regional energy deficit, i.e. the contribution of “supply” and “demand” and test possible treatments.
Interestingly, the two properties that have been both independently proposed to constitute the critical impairment conferred by HCM sarcomeric mutations are A) increased myofilament Ca2+ sensitivity and B) increased energy cost of force production.15, 68–70 As we demonstrate in this study, the pathological effect of increased myofilament Ca2+ sensitivity is linked to focal energy deprivation and this may at least in part unify both theories.
Supplementary Material
Novelty and Significance.
What Is Known?
Increased affinity of myofilaments to Ca2+ is frequently found in inherited and acquired diseases associated with ventricular arrhythmias and sudden cardiac death.
Myofilament Ca2+ sensitization has been shown to cause regional slowing of conduction velocity (CV) and reentrant arrhythmias during stress.
Gap junctions are intercellular channels important to electrically synchronize cells and to facilitate impulse propagation.
Phosphorylation of Connexin 43 (Cx43), the major gap junction protein in cardiac ventricle, regulates intercellular coupling and is sensitive to metabolic changes.
What New Information Does This Article Contribute?
Hearts with increased myofilament Ca2+ sensitivity develop areas of energy deprivation during stress within minutes.
In energy deprived regions, intercellular uncoupling is reduced to a degree that likely affects impulse propagation and may contribute to conduction slowing across the fiber.
Decreasing myofilament Ca2+ sensitivity using the contractile uncoupler blebbistatin prevented the focal energy deprivation, propagation slowing and arrhythmias.
Increased sensitivity of the myofilaments to Ca2+ has been linked to increased arrhythmia susceptibility and may contribute to sudden cardiac death. The present study aimed to determine how myofilament Ca2+ sensitization generates a myocardial substrate that promotes reentry arrhythmias.. We Ca2+ sensitized myofilaments in mice either by expressing a mutant cardiac troponinT or treatment with a Ca2+ sensitizing drug, EMD57033, and in reverse de-sensitized myofilaments using blebbistatin. When hearts were stressed, reentry arrhythmias were observed after a few minutes delay. By measuring Cx43 and AMPK phosphorylation sensitive to cellular ATP levels, we identified myocardial regions of energy deprivation that developed within minutes only in Ca2+ sensitized hearts. In the same regions intercellular coupling via gap junctions is reduced to a degree that is likely to slow excitation spread. This can be predicted to lead to CV heterogeneity, consistent with previously observed regional CV slowing, a very proarrhythmic state. These results provide new mechanistic insights into the genesis of arrhythmias that could facilitate the development of targeted new treatments to prevent arrhythmias.
Acknowledgments
Experiments were performed in part through the use of the VUMC Cell Imaging Shared Resource (supported by NIH grants CA68485, DK20593, DK58404, HD15052, DK59637 and EY08126). We are grateful to Melissa Ryan and Sung In Kim for expert technical assistance.
SOURCES OF FUNDING
This work was supported in part by the American Heart Association Scientist Development Grant 10SDG2640109 (to SH) and an Established Investigator Award 0840071N (to BCK) as well as by the US NIH grants HL88635 (to BCK) and HL71670 (to BCK and FJB).
Nonstandard Abbreviations
- AMPK
AMP-dependent kinase
- α-tub
Alpha-tubulin
- BLEB
(−)-Blebbistatin
- CV
Conduction velocity
- CVL
longitudinal CV
- CVT
transverse CV
- Cx43
Connexin 43
- Cx43-P0
lower phosphorylated and fast migrating isoform of Cx43
- EMD
- HCM
hypertrophic cardiomyopathy
- ICD
Implantable cardioverter-defibrillator
- Iso
Isoproterenol
- LV
left ventricle
- LY
Lucifer yellow
- NTG
Non-transgenic
- pAMPK
phosphorylated (=activated) AMPK at threonine 172
- PVC
Premature ventricular contraction
- SCD
Sudden cardiac death
- TMR
Tetramethylrhodamine dextran
- TnT
Troponin T
- VEH
vehicle (drug solvent)
- VT
Ventricular tachycardia
Footnotes
DISCLOSURES
None.
References
- 1.Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation. 1995;92:785–789. doi: 10.1161/01.cir.92.4.785. [DOI] [PubMed] [Google Scholar]
- 2.Keren A, Syrris P, McKenna WJ. Hypertrophic cardiomyopathy: the genetic determinants of clinical disease expression. Nat Clin Pract Cardiovasc Med. 2008;5:158–168. doi: 10.1038/ncpcardio1110. [DOI] [PubMed] [Google Scholar]
- 3.Maron BJ, Olivotto I, Spirito P, Casey SA, Bellone P, Gohman TE, Graham KJ, Burton DA, Cecchi F. Epidemiology of hypertrophic cardiomyopathy-related death: revisited in a large non-referral-based patient population. Circulation. 2000;102:858–864. doi: 10.1161/01.cir.102.8.858. [DOI] [PubMed] [Google Scholar]
- 4.Shirani J, Pick R, Roberts WC, Maron BJ. Morphology and significance of the left ventricular collagen network in young patients with hypertrophic cardiomyopathy and sudden cardiac death. J Am Coll Cardiol. 2000;35:36–44. doi: 10.1016/s0735-1097(99)00492-1. [DOI] [PubMed] [Google Scholar]
- 5.Elliott P, Spirito P. Prevention of hypertrophic cardiomyopathy-related deaths: theory and practice. Heart. 2008;94:1269–1275. doi: 10.1136/hrt.2008.154385. [DOI] [PubMed] [Google Scholar]
- 6.Elliott PM, Sharma S, Varnava A, Poloniecki J, Rowland E, McKenna WJ. Survival after cardiac arrest or sustained ventricular tachycardia in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 1999;33:1596–1601. doi: 10.1016/s0735-1097(99)00056-x. [DOI] [PubMed] [Google Scholar]
- 7.Theis JL, Bos JM, Theis JD, Miller DV, Dearani JA, Schaff HV, Gersh BJ, Ommen SR, Moss RL, Ackerman MJ. Expression patterns of cardiac myofilament proteins: genomic and protein analysis of surgical myectomy tissue from patients with obstructive hypertrophic cardiomyopathy. Circ Heart Fail. 2009;2:325–333. doi: 10.1161/CIRCHEARTFAILURE.108.789735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maron BJ, Spirito P, Wesley Y, Arce J. Development and progression of left ventricular hypertrophy in children with hypertrophic cardiomyopathy. N Engl J Med. 1986;315:610–614. doi: 10.1056/NEJM198609043151003. [DOI] [PubMed] [Google Scholar]
- 9.Semsarian C, French J, Trent RJ, Richmond DR, Jeremy RW. The natural history of left ventricular wall thickening in hypertrophic cardiomyopathy. Aust N Z J Med. 1997;27:51–58. doi: 10.1111/j.1445-5994.1997.tb00914.x. [DOI] [PubMed] [Google Scholar]
- 10.Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA. 2002;287:1308–1320. doi: 10.1001/jama.287.10.1308. [DOI] [PubMed] [Google Scholar]
- 11.Maron BJ. Sudden death in young athletes. N Engl J Med. 2003;349:1064–1075. doi: 10.1056/NEJMra022783. [DOI] [PubMed] [Google Scholar]
- 12.Maron BJ, McKenna WJ, Danielson GK, Kappenberger LJ, Kuhn HJ, Seidman CE, Shah PM, Spencer WH, 3rd, Spirito P, Ten Cate FJ, Wigle ED Task Force on Clinical Expert Consensus Documents, American College of C, Committee for Practice Guidelines. European Society of C. American College of Cardiology/European Society of Cardiology clinical expert consensus document on hypertrophic cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. J Am Coll Cardiol. 2003;42:1687–1713. doi: 10.1016/s0735-1097(03)00941-0. [DOI] [PubMed] [Google Scholar]
- 13.Green JJ, Berger JS, Kramer CM, Salerno M. Prognostic value of late gadolinium enhancement in clinical outcomes for hypertrophic cardiomyopathy. JACC Cardiovasc Imaging. 2012;5:370–377. doi: 10.1016/j.jcmg.2011.11.021. [DOI] [PubMed] [Google Scholar]
- 14.Huke S, Knollmann BC. Increased myofilament Ca2+-sensitivity and arrhythmia susceptibility. J Mol Cell Cardiol. 2010;48:824–833. doi: 10.1016/j.yjmcc.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knollmann BC, Potter JD. Altered regulation of cardiac muscle contraction by troponin T mutations that cause familial hypertrophic cardiomyopathy. Trends Cardiovasc Med. 2001;11:206–212. doi: 10.1016/s1050-1738(01)00115-3. [DOI] [PubMed] [Google Scholar]
- 16.Schober T, Huke S, Venkataraman R, Gryshchenko O, Kryshtal D, Hwang HS, Baudenbacher FJ, Knollmann BC. Myofilament ca sensitization increases cytosolic ca binding affinity, alters intracellular ca homeostasis, and causes pause-dependent ca-triggered arrhythmia. Circ Res. 2012;111:170–179. doi: 10.1161/CIRCRESAHA.112.270041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baudenbacher F, Schober T, Pinto JR, Sidorov VY, Hilliard F, Solaro RJ, Potter JD, Knollmann BC. Myofilament Ca2+ sensitization causes susceptibility to cardiac arrhythmia in mice. J Clin Invest. 2008;118:3893–3903. doi: 10.1172/JCI36642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sims JJ, Miller AW, Ujhelyi MR. Electrical heterogeneity and arrhythmogenesis: importance of conduction velocity dispersion. J Cardiovasc Pharmacol. 2003;41:795–803. doi: 10.1097/00005344-200305000-00018. [DOI] [PubMed] [Google Scholar]
- 19.Janse MJ, Wit AL. Electrophysiological mechanisms of ventricular arrhythmias resulting from myocardial ischemia and infarction. Physiol Rev. 1989;69:1049–1169. doi: 10.1152/physrev.1989.69.4.1049. [DOI] [PubMed] [Google Scholar]
- 20.Knollmann BC, Blatt SA, Horton K, de Freitas F, Miller T, Bell M, Housmans PR, Weissman NJ, Morad M, Potter JD. Inotropic stimulation induces cardiac dysfunction in transgenic mice expressing a troponin T (I79N) mutation linked to familial hypertrophic cardiomyopathy. J Biol Chem. 2001;276:10039–10048. doi: 10.1074/jbc.M006745200. [DOI] [PubMed] [Google Scholar]
- 21.Westermann D, Knollmann BC, Steendijk P, Rutschow S, Riad A, Pauschinger M, Potter JD, Schultheiss HP, Tschope C. Diltiazem treatment prevents diastolic heart failure in mice with familial hypertrophic cardiomyopathy. Eur J Heart Fail. 2006;8:115–121. doi: 10.1016/j.ejheart.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 22.Knollmann BC, Kirchhof P, Sirenko SG, Degen H, Greene AE, Schober T, Mackow JC, Fabritz L, Potter JD, Morad M. Familial hypertrophic cardiomyopathy-linked mutant troponin T causes stress-induced ventricular tachycardia and Ca2+-dependent action potential remodeling. Circ Res. 2003;92:428–436. doi: 10.1161/01.RES.0000059562.91384.1A. [DOI] [PubMed] [Google Scholar]
- 23.Kleber AG. Gap junctions and conduction of cardiac excitation. Heart Rhythm. 2011;8:1981–1984. doi: 10.1016/j.hrthm.2011.08.007. [DOI] [PubMed] [Google Scholar]
- 24.Musil LS, Cunningham BA, Edelman GM, Goodenough DA. Differential phosphorylation of the gap junction protein connexin43 in junctional communication-competent and -deficient cell lines. J Cell Biol. 1990;111:2077–2088. doi: 10.1083/jcb.111.5.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Solan JL, Lampe PD. Key connexin 43 phosphorylation events regulate the gap junction life cycle. J Membr Biol. 2007;217:35–41. doi: 10.1007/s00232-007-9035-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Solan JL, Lampe PD. Connexin43 phosphorylation: structural changes and biological effects. Biochem J. 2009;419:261–272. doi: 10.1042/BJ20082319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saffitz JE, Kanter HL, Green KG, Tolley TK, Beyer EC. Tissue-specific determinants of anisotropic conduction velocity in canine atrial and ventricular myocardium. Circ Res. 1994;74:1065–1070. doi: 10.1161/01.res.74.6.1065. [DOI] [PubMed] [Google Scholar]
- 28.Spach MS, Heidlage JF, Dolber PC, Barr RC. Electrophysiological effects of remodeling cardiac gap junctions and cell size: experimental and model studies of normal cardiac growth. Circ Res. 2000;86:302–311. doi: 10.1161/01.res.86.3.302. [DOI] [PubMed] [Google Scholar]
- 29.Kaba RA, Coppen SR, Dupont E, Skepper JN, Elneil S, Haw MP, Pepper JR, Yacoub MH, Rothery S, Severs NJ. Comparison of connexin 43, 40 and 45 expression patterns in the developing human and mouse hearts. Cell Commun Adhes. 2001;8:339–343. doi: 10.3109/15419060109080750. [DOI] [PubMed] [Google Scholar]
- 30.Smyth JW, Hong TT, Gao D, Vogan JM, Jensen BC, Fong TS, Simpson PC, Stainier DY, Chi NC, Shaw RM. Limited forward trafficking of connexin 43 reduces cell-cell coupling in stressed human and mouse myocardium. J Clin Invest. 120:266–279. doi: 10.1172/JCI39740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beardslee MA, Lerner DL, Tadros PN, Laing JG, Beyer EC, Yamada KA, Kleber AG, Schuessler RB, Saffitz JE. Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ Res. 2000;87:656–662. doi: 10.1161/01.res.87.8.656. [DOI] [PubMed] [Google Scholar]
- 32.Turner MS, Haywood GA, Andreka P, You L, Martin PE, Evans WH, Webster KA, Bishopric NH. Reversible connexin 43 dephosphorylation during hypoxia and reoxygenation is linked to cellular ATP levels. Circ Res. 2004;95:726–733. doi: 10.1161/01.RES.0000144805.11519.1e. [DOI] [PubMed] [Google Scholar]
- 33.Hardie DG. Minireview: the AMP-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology. 2003;144:5179–5183. doi: 10.1210/en.2003-0982. [DOI] [PubMed] [Google Scholar]
- 34.Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, Hardie DG. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem. 1996;271:27879–27887. doi: 10.1074/jbc.271.44.27879. [DOI] [PubMed] [Google Scholar]
- 35.Frederich M, Balschi JA. The relationship between AMP-activated protein kinase activity and AMP concentration in the isolated perfused rat heart. J Biol Chem. 2002;277:1928–1932. doi: 10.1074/jbc.M107128200. [DOI] [PubMed] [Google Scholar]
- 36.Baron SJ, Li J, Russell RR, 3rd, Neumann D, Miller EJ, Tuerk R, Wallimann T, Hurley RL, Witters LA, Young LH. Dual mechanisms regulating AMPK kinase action in the ischemic heart. Circ Res. 2005;96:337–345. doi: 10.1161/01.RES.0000155723.53868.d2. [DOI] [PubMed] [Google Scholar]
- 37.Sovari AA, Iravanian S, Dolmatova E, Jiao Z, Liu H, Zandieh S, Kumar V, Wang K, Bernstein KE, Bonini MG, Duffy HS, Dudley SC. Inhibition of c-Src tyrosine kinase prevents angiotensin II-mediated connexin-43 remodeling and sudden cardiac death. J Am Coll Cardiol. 2011;58:2332–2339. doi: 10.1016/j.jacc.2011.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sosinsky GE, Solan JL, Gaietta GM, Ngan L, Lee GJ, Mackey MR, Lampe PD. The C-terminus of connexin43 adopts different conformations in the Golgi and gap junction as detected with structure-specific antibodies. Biochem J. 2007;408:375–385. doi: 10.1042/BJ20070550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ek-Vitorin JF, King TJ, Heyman NS, Lampe PD, Burt JM. Selectivity of connexin 43 channels is regulated through protein kinase C-dependent phosphorylation. Circ Res. 2006;98:1498–1505. doi: 10.1161/01.RES.0000227572.45891.2c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lampe PD, TenBroek EM, Burt JM, Kurata WE, Johnson RG, Lau AF. Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J Cell Biol. 2000;149:1503–1512. doi: 10.1083/jcb.149.7.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morley GE, Vaidya D, Samie FH, Lo C, Delmar M, Jalife J. Characterization of conduction in the ventricles of normal and heterozygous Cx43 knockout mice using optical mapping. J Cardiovasc Electrophysiol. 1999;10:1361–1375. doi: 10.1111/j.1540-8167.1999.tb00192.x. [DOI] [PubMed] [Google Scholar]
- 42.Thomas SP, Kucera JP, Bircher-Lehmann L, Rudy Y, Saffitz JE, Kleber AG. Impulse propagation in synthetic strands of neonatal cardiac myocytes with genetically reduced levels of connexin43. Circ Res. 2003;92:1209–1216. doi: 10.1161/01.RES.0000074916.41221.EA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eloff BC, Lerner DL, Yamada KA, Schuessler RB, Saffitz JE, Rosenbaum DS. High resolution optical mapping reveals conduction slowing in connexin43 deficient mice. Cardiovasc Res. 2001;51:681–690. doi: 10.1016/s0008-6363(01)00341-8. [DOI] [PubMed] [Google Scholar]
- 44.Lerner DL, Yamada KA, Schuessler RB, Saffitz JE. Accelerated onset and increased incidence of ventricular arrhythmias induced by ischemia in Cx43-deficient mice. Circulation. 2000;101:547–552. doi: 10.1161/01.cir.101.5.547. [DOI] [PubMed] [Google Scholar]
- 45.Gutstein DE, Morley GE, Tamaddon H, Vaidya D, Schneider MD, Chen J, Chien KR, Stuhlmann H, Fishman GI. Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ Res. 2001;88:333–339. doi: 10.1161/01.res.88.3.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Delmar M, Michaels DC, Johnson T, Jalife J. Effects of increasing intercellular resistance on transverse and longitudinal propagation in sheep epicardial muscle. Circ Res. 1987;60:780–785. doi: 10.1161/01.res.60.5.780. [DOI] [PubMed] [Google Scholar]
- 47.Dhein S, Krusemann K, Schaefer T. Effects of the gap junction uncoupler palmitoleic acid on the activation and repolarization wavefronts in isolated rabbit hearts. Br J Pharmacol. 1999;128:1375–1384. doi: 10.1038/sj.bjp.0702902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De Groot JR, Coronel R. Acute ischemia-induced gap junctional uncoupling and arrhythmogenesis. Cardiovasc Res. 2004;62:323–334. doi: 10.1016/j.cardiores.2004.01.033. [DOI] [PubMed] [Google Scholar]
- 49.Peters NS, Green CR, Poole-Wilson PA, Severs NJ. Cardiac arrhythmogenesis and the gap junction. J Mol Cell Cardiol. 1995;27:37–44. doi: 10.1016/s0022-2828(08)80005-3. [DOI] [PubMed] [Google Scholar]
- 50.Hubbard ML, Henriquez CS. Microscopic variations in interstitial and intracellular structure modulate the distribution of conduction delays and block in cardiac tissue with source-load mismatch. Europace. 2012;14 (Suppl 5):v3–v9. doi: 10.1093/europace/eus267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Contreras JE, Sanchez HA, Eugenin EA, Speidel D, Theis M, Willecke K, Bukauskas FF, Bennett MV, Saez JC. Metabolic inhibition induces opening of unapposed connexin 43 gap junction hemichannels and reduces gap junctional communication in cortical astrocytes in culture. Proc Natl Acad Sci U S A. 2002;99:495–500. doi: 10.1073/pnas.012589799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vergara L, Bao X, Cooper M, Bello-Reuss E, Reuss L. Gap-junctional hemichannels are activated by ATP depletion in human renal proximal tubule cells. J Membr Biol. 2003;196:173–184. doi: 10.1007/s00232-003-0636-9. [DOI] [PubMed] [Google Scholar]
- 53.Oosthuyse T, Bosch AN. Oestrogen’s regulation of fat metabolism during exercise and gender specific effects. Curr Opin Pharmacol. 2012;12:363–371. doi: 10.1016/j.coph.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 54.Akingbemi BT. Adiponectin receptors in energy homeostasis and obesity pathogenesis. Prog Mol Biol Transl Sci. 2013;114:317–342. doi: 10.1016/B978-0-12-386933-3.00009-1. [DOI] [PubMed] [Google Scholar]
- 55.Schober T, Huke S, Venkataraman R, Gryshchenko O, Kryshtal D, Hwang HS, Baudenbacher FJ, Knollmann BC. Myofilament Ca2+ sensitization increases cytosolic Ca2+ binding affinity, alteres intracellular Ca2+ homeostasis and causes pause-dependent Ca2+ triggered arrhythmia. Circulation Research. 2012 doi: 10.1161/CIRCRESAHA.112.270041. in revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Frey N, Brixius K, Schwinger RH, Benis T, Karpowski A, Lorenzen HP, Luedde M, Katus HA, Franz WM. Alterations of tension-dependent ATP utilization in a transgenic rat model of hypertrophic cardiomyopathy. J Biol Chem. 2006;281:29575–29582. doi: 10.1074/jbc.M507740200. [DOI] [PubMed] [Google Scholar]
- 57.Ruegg JC, Strauss JD, Zeugner C, Trayer I. Effect of myosin heavy chain peptides on contractile activation of skinned cardiac muscle fibres. Adv Exp Med Biol. 1993;332:173–180. doi: 10.1007/978-1-4615-2872-2_16. discussion 180–171. [DOI] [PubMed] [Google Scholar]
- 58.Davies JE, Whinnett ZI, Francis DP, Manisty CH, Aguado-Sierra J, Willson K, Foale RA, Malik IS, Hughes AD, Parker KH, Mayet J. Evidence of a dominant backward-propagating “suction” wave responsible for diastolic coronary filling in humans, attenuated in left ventricular hypertrophy. Circulation. 2006;113:1768–1778. doi: 10.1161/CIRCULATIONAHA.105.603050. [DOI] [PubMed] [Google Scholar]
- 59.O’Hanlon R, Grasso A, Roughton M, Moon JC, Clark S, Wage R, Webb J, Kulkarni M, Dawson D, Sulaibeekh L, Chandrasekaran B, Bucciarelli-Ducci C, Pasquale F, Cowie MR, McKenna WJ, Sheppard MN, Elliott PM, Pennell DJ, Prasad SK. Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2010;56:867–874. doi: 10.1016/j.jacc.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 60.Konno T, Chen D, Wang L, Wakimoto H, Teekakirikul P, Nayor M, Kawana M, Eminaga S, Gorham JM, Pandya K, Smithies O, Naya FJ, Olson EN, Seidman JG, Seidman CE. Heterogeneous myocyte enhancer factor-2 (Mef2) activation in myocytes predicts focal scarring in hypertrophic cardiomyopathy. Proc Natl Acad Sci U S A. 2010;107:18097–18102. doi: 10.1073/pnas.1012826107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.de Waard MC, van der Velden J, Bito V, Ozdemir S, Biesmans L, Boontje NM, Dekkers DH, Schoonderwoerd K, Schuurbiers HC, de Crom R, Stienen GJ, Sipido KR, Lamers JM, Duncker DJ. Early exercise training normalizes myofilament function and attenuates left ventricular pump dysfunction in mice with a large myocardial infarction. Circ Res. 2007;100:1079–1088. doi: 10.1161/01.RES.0000262655.16373.37. [DOI] [PubMed] [Google Scholar]
- 62.Boontje NM, Merkus D, Zaremba R, Versteilen A, de Waard MC, Mearini G, de Beer VJ, Carrier L, Walker LA, Niessen HW, Dobrev D, Stienen GJ, Duncker DJ, van der Velden J. Enhanced myofilament responsiveness upon beta-adrenergic stimulation in post-infarct remodeled myocardium. J Mol Cell Cardiol. 2011;50:487–499. doi: 10.1016/j.yjmcc.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 63.McKenna WJ, Behr ER. Hypertrophic cardiomyopathy: management, risk stratification, and prevention of sudden death. Heart. 2002;87:169–176. doi: 10.1136/heart.87.2.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Melacini P, Maron BJ, Bobbo F, Basso C, Tokajuk B, Zucchetto M, Thiene G, Iliceto S. Evidence that pharmacological strategies lack efficacy for the prevention of sudden death in hypertrophic cardiomyopathy. Heart. 2007;93:708–710. doi: 10.1136/hrt.2006.099416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Teerlink JR, Clarke CP, Saikali KG, Lee JH, Chen MM, Escandon RD, Elliott L, Bee R, Habibzadeh MR, Goldman JH, Schiller NB, Malik FI, Wolff AA. Dose-dependent augmentation of cardiac systolic function with the selective cardiac myosin activator, omecamtiv mecarbil: a first-in-man study. Lancet. 2011;378:667–675. doi: 10.1016/S0140-6736(11)61219-1. [DOI] [PubMed] [Google Scholar]
- 66.Shen YT, Malik FI, Zhao X, Depre C, Dhar SK, Abarzua P, Morgans DJ, Vatner SF. Improvement of cardiac function by a cardiac Myosin activator in conscious dogs with systolic heart failure. Circ Heart Fail. 2010;3:522–527. doi: 10.1161/CIRCHEARTFAILURE.109.930321. [DOI] [PubMed] [Google Scholar]
- 67.Dickstein K. Cardiac myosin activation: will theory and practice coincide? Lancet. 2011;378:639–641. doi: 10.1016/S0140-6736(11)61298-1. [DOI] [PubMed] [Google Scholar]
- 68.Abozguia K, Elliott P, McKenna W, Phan TT, Nallur-Shivu G, Ahmed I, Maher AR, Kaur K, Taylor J, Henning A, Ashrafian H, Watkins H, Frenneaux M. Metabolic modulator perhexiline corrects energy deficiency and improves exercise capacity in symptomatic hypertrophic cardiomyopathy. Circulation. 2010;122:1562–1569. doi: 10.1161/CIRCULATIONAHA.109.934059. [DOI] [PubMed] [Google Scholar]
- 69.Crilley JG, Boehm EA, Blair E, Rajagopalan B, Blamire AM, Styles P, McKenna WJ, Ostman-Smith I, Clarke K, Watkins H. Hypertrophic cardiomyopathy due to sarcomeric gene mutations is characterized by impaired energy metabolism irrespective of the degree of hypertrophy. J Am Coll Cardiol. 2003;41:1776–1782. doi: 10.1016/s0735-1097(02)03009-7. [DOI] [PubMed] [Google Scholar]
- 70.Ferrantini C, Belus A, Piroddi N, Scellini B, Tesi C, Poggesi C. Mechanical and energetic consequences of HCM-causing mutations. J Cardiovasc Transl Res. 2009;2:441–451. doi: 10.1007/s12265-009-9131-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.