

Abstract
Objective
To identify the causative mutations in two Chinese families with retinitis pigmentosa (RP), and to describe the associated phenotype.
Methods
Individuals from two unrelated families underwent full ophthalmic examinations. After informed consent was obtained, genomic DNA was extracted from the venous blood of all participants. Linkage analysis was performed on the known genetic loci for autosomal dominant retinitis pigmentosa with a panel of polymorphic markers in the two families, and then all coding exons of the PRP31 premessenger ribonucleic acid processing factor 31 homolog (PRPF31) gene were screened for mutations with direct sequencing of PCR-amplified DNA fragments. Allele-specific PCR was used to validate a substitution in all available family members and 100 normal controls. A large deletion was detected with real-time quantitative PCR (RQ-PCR) using a panel of primers from regions around the PRPF31 gene. Long-range PCR, followed by DNA sequencing, was used to define the breakpoints.
Results
Clinical examination and pedigree analysis revealed two four-generation families (RP24 and RP106) with autosomal dominant retinitis pigmentosa. A significant two-point linkage odd disequilibrium score was generated at marker D19S926 (Zmax=3.55, θ=0) for family RP24 and D19S571 (Zmax=3.21, θ=0) for family RP106, and further linkage and haplotype studies confined the disease locus to chromosome 19q13.42 where the PRPF31 gene is located. Mutation screening of the PRPF31 gene revealed a novel deletion c.1215delG (p.G405fs+7X) in family RP106. The deletion cosegregated with the family’s disease phenotype, but was not found in 100 normal controls. No disease-causing mutation was detected in family RP24 with PCR-based sequencing analysis. RQ-PCR and long-range PCR analysis revealed a complex insertion-deletion (indel) in the patients of family RP24. The deletion is more than 19 kb and encompasses part of the PRPF31 gene (exons 1–3), together with three adjacent genes.
Conclusions
Our results further confirmed that haploinsufficiency is the main mechanism for RP11 and that genomic arrangements may be prevalent in PRPF31 mutations.
Introduction
Retinitis pigmentosa (RP) is the most common form of inherited retinopathy, with an incidence of 1 in 4,000. RP is a clinically and genetically heterogeneous group of retinal dystrophies, characterized by progressive degeneration of the photoreceptors. Clinical features include progressive night blindness, constriction and gradual loss of the peripheral visual field, and eventual loss of visual acuity [1,2]. RP can be inherited as an autosomal recessive (arRP), an autosomal dominant (adRP), or an X-linked recessive (xlRP) pattern [1,2]. To date, at least 23 causative genes have been identified for adRP, 35 for arRP, and two for xlRP (RetNet).
The PRP31 premessenger ribonucleic acid (pre-mRNA) processing factor 31 homolog (PRPF31) gene located in chromosome 19q13.42 was identified as the causative gene of adRP at the RP11 locus [3]. This gene encompasses 14 exons and codes 499 amino acids that ubiquitously express a pre-mRNA splicing factor. To date, 62 mutations of the PRPF31 gene have been recorded in the Human Genome Mutation Database (HGMD), and almost 90% of them are splicing mutations (26%), deletions (39%), nonsense mutations (10%), insertions (8%), and indels (6%). Due to exon skipping and premature stop codons, the majority of mutations result in truncated proteins; therefore, haploinsufficiency was suggested as a mechanism of RP11 evolvement [3-13].
A unique feature of the PRPF31 mutations is bimodal expressivity. Individuals carrying mutations of the PRPF31 gene can either present severe phenotypes at an early age or be completely asymptomatic individuals. This “all or none” form of incomplete penetrance represents the existence of two different wild-type PRPF31 alleles [14-16]. Consequently, an individual carrying a mutant allele and a high expressivity wild-type PRPF31 allele would not display RP symptoms [14-16].
The CCR4-NOT transcription complex, subunit 3 (CNOT3) gene has recently been identified as the main modifier gene responsible for penetrance of PRPF31 mutations. In asymptomatic individuals, the CNOT3 gene was expressed at a low level, allowing high expression of the wild-type PRPF31 [17]. The C allele of single nucleotide polymorphism (SNP) rs4806718 in the intronic region of the CNOT3 gene is associated with the affected status while the T allele is associated with asymptomatic status [17].
In this study, we investigated two Chinese families with adRP. After linkage and haplotyping analysis, the disease-causing gene was mapped to the RP11 region. We identified one novel deletion in one family and one novel complex insertion-deletion (indel) of the PRPF31 gene in the other family. One family with the complex indel showed complete disease penetrance, whereas the other family with the deletion contained an asymptomatic disease gene carrier.
Methods
Participants and deoxyribonucleic acid samples
Thirty individuals including 16 males and 14 females with age range from 5 to 71 years old from two families with RP patients were recruited on a voluntary basis from Beijing Tongren Hospital. Each participant underwent careful ophthalmologic examinations, including best-corrected visual acuity testing using E decimal charts, slit-lamp biomicroscopy, and fundus examination with dilated pupils. Some of the patients had visual field testing and electroretinogram (ERG) examination. ERG was performed (GT-2008V-I ERG system, Guo Te Medical Equipment, Chongqing, China) using corneal ERGjet contact lens electrodes, according to International Society for Clinical Electrophysiology of Vision (ISCEV) standards [18]. The diagnosis of RP was based on the presence of night blindness, typical fundus findings (characteristic retinal pigmentation, vessel attenuation, and waxy pallor of the optic disc), different extent of loss of peripheral visual field, and abnormal ERG. This study adhered to the tenets of the Declaration of Helsinki for research involving human subjects. The Beijing Tongren Hospital Joint Committee on Clinical Investigation approved the study, and informed consent was obtained from all patients. Peripheral blood samples were collected from all the participants by venipuncture in heparinized collecting tubes and stored at 4 °C for less than one week. Genomic DNA was extracted from peripheral blood leukocytes using a genomic DNA extraction and purification kit (Vigorous Whole Blood Genomic DNA extraction Kit, Vigorous Beijing, China), according to the manufacturer’s protocol.
Genotyping and linkage analysis
Linkage analyses were performed with 41 microsatellite markers flanking each of the known adRP loci in the two families (Appendix 1). Short tandem repeat (STR) loci were amplified from family member DNA with fluorescent PCR primers and DNA polymerase in standard multiplex reaction conditions. PCR products were pooled and diluted in water and run on an ABI Prism 373A DNA sequencer (Applied Biosystems Inc., Foster City, CA). Two-point linkage analyses were performed with the LINKAGE program package 5.2, using a dominant model with 100% penetrance for family RP24 and 83.3% (the ratio between symptomatic individuals and all disease-gene carriers) for family RP106.
Mutation screening of the PRP31 premessenger ribonucleic acid processing factor 31 homolog gene
Mutation screening was performed in the two families using direct DNA sequence analysis. The coding regions and the exon–intron boundaries of the PRPF31 gene were amplified with PCR in the patients of the two families. The pairs of primers were used according to a previously published article [4] (Table 1). PCR assays were performed using standard reaction mixtures, and purified PCR products were directly sequenced on an ABI Prism 373A DNA sequencer. Nucleotide sequences were compared with the published cDNA sequence of the PRPF31 gene (GenBank NM_015629). For the PRPF31 gene, cDNA numbering +1 corresponds to A in the ATG translation initiation codon in PRPF31.
Table 1. Primers used in sequencing of the PRPF31 gene and allele-specific PCR analysis.
| Exon/mutation site or SNP | Primer sequence (5'-3') | TM (°C) |
|---|---|---|
| 2,3 |
F: GTCGGGGCAAGTTTTTAGGG |
64 |
| R: GAGATGGGGAGGGGCACAGAGT |
||
| 4 |
F: CCGAGAGGGGGTAGGGATTTAGAT |
64 |
| R: AGGCCAGTGGGGAAGGGAGAGG |
||
| 5 |
F: TTAGGGCCAACCAGCAGAGTC |
64 |
| R: GAGGGGGTCCGAGATGAGC |
||
| 6,7 |
F: GTTCCCGAGCCTCCCCTATCTTCT |
64 |
| R: CGCTCCAGCTCCTCCTCCGACAG |
||
| 8 |
F: CCGGCGGCCTGACCAACC |
64 |
| R: GGGAGGGGCCATGACGCAGTG |
||
| 9 |
F: GCGCGGTTGCTTTGCTGTTA |
64 |
| R: ACTGCCTCCGCCTTGGTAG |
||
| 10,11 |
F: GTGGCGGTGAGGCAGCATTAGGTG |
55 |
| R: CTGGCTGGCTGTGGGGTTGAGGA |
||
| 12,13 |
F: GGGCCTGGTCGCTGA |
64 |
| R: GGGGAGGTACCTGGAGTGG |
||
| 14 |
F: GGTCACAGTTGGGGCCTTCTCCTC |
64 |
| R: TACTGGGCGGTGATCTCGGTCCTG |
||
| 13/p.G405fs+7X |
F: AAGTCGGGCAGTGGC |
60 |
| PRPF31-3A2R |
R: AGGTTCACGCCATTCTC |
|
| INS-R |
R: TAGTCTCATGGACAGGTCTAGCCG |
60 |
| rs4806718 |
F: GCCCTCTCATCCTCCCACTT |
58 |
| R: TGCTGAGTGCTGGGACCT |
Abbreviations: F,forward; R:reverse; Primer sequences with lower case indicate modified nucleotides in order to specifically amplify the mutant sequence; red,mutation-specific nucleotide.
Real-time quantitative polymerase chain reaction and breakpoint analysis
Large deletions were detected using real-time quantitative PCR (RQ-PCR), as described in our previous study, on samples from one affected member of family RP24 with a panel of pairs of primers around the PRPF31 gene (Table 2) [19]. The RQ-PCR reactions were performed on the Rotor-Gene 6000 (Corbett Research, Mortlake, NSW, Australia) in a final volume of 10 µl, containing 300 nM primers and 1 µl (100 ng) genomic DNA, using the EvaGreen PCR MasterMix (Bio-Rad, Hercules, CA). Each assay was performed in triplicate. The relative quantitation (RQ) of the target gene was accomplished using RQ manager software (Bio-Rad) and was calculated with the 2-ddCt method [19]. All experimental samples were normalized using the human glyceraldehyde-3-phosphate dehydrogenase gene as an internal control. Threshold values were set at 0.8–1.3 for normal, 0.45–0.74 for deletions, and 1.6–1.8 for duplication. Experiments were replicated at least twice if a deletion was suspected.
Table 2. Primers informations and results of the real-time quantitative PCR.
| Gene/SNP | Physical position | Primer sequences (Forward 5′-3′) | Primer sequences (Reverse 5′-3′) | RQ results* |
|---|---|---|---|---|
| OSCAR |
54,597,933-54604148 |
|||
|
REN91524 |
54,602,623-54602867 |
GTGCTGGGATTACAGACGTTGG |
ACCAGGAACTTCTGAGGCAGAG |
1.2 |
|
REN91525 |
54,602,851-54603100 |
CCTCAGAAGTTCCTGGTCTTCAGT |
AAAGAAACTAGTCCCTCAACCTCCT |
1.18 |
|
REN91526 |
54,602,890-54603139 |
CCACTAAGGGGAATGAGAAAAGAA |
AGAGTGAGAGTCTGCCTCAGACAA |
0.4 |
|
REN91528 |
54,603,636-54603883 |
CCAGGGAACAATGATCGTAGAGTT |
AGAGAACACAGATTCCCGAAAGAG |
0.51 |
|
REN91537 |
54,605,887-54606047 |
AGGCTGAGACTAAGGAGATTCCAC |
CTGGGAGTTGTGGTCCCTATG |
0.38 |
| NDUFA3 |
54,606,160-54610280 |
|||
| Exon2 |
54,606,422-54606583 |
GTGGCACGAGAGGGTTAGAG |
GCGATAGGGCTTTAGGGGTA |
0.44 |
|
REN91541 |
54,606,723-54606962 |
GTTCTCAACCCTGCTTATGCCTTA |
CAGGAACTCCGGTGCTAATTAAAG |
0.48 |
|
REN91551 |
54,609,843-54610094 |
CTTGGTCTCCATCTTCTCAGGTTT |
TGAGACCAGCACGTCTCCAG |
0.54 |
| TFPT |
54,610,320-54619055 |
|||
|
REN91555 |
54,610,701-54610956 |
ACTTGCCAACCAAGCCTAACAT |
GGAGGAAAGCTGGAAGAACATCT |
0.48 |
|
REN91573 |
54,614,408-54614644 |
AGGAGGTGCGACTTTAGTTACGAC |
CCGGAACTCAGGTCTTTCTGAC |
0.5 |
| PFPF31 |
54,618,790-54635150 |
|||
| Exon1 |
54,619,151 |
ACGTGAGTCCCTTTCCTCCT |
TCACCGATGACGTCTCACTC |
0.47 |
| Exon2 |
54,621,659-54621835 |
TGAGCTCTTAGCTGATCTCGAA |
CATAGCTTGGCGATGGTCTT |
0.38 |
| Intron3 |
54,622,743-54622989 |
CAGAGCAAGACTCCGTCTCA |
TTGCAATGAGCCAAGAACAC |
1.22 |
| Intron3–1 |
54,623,071-54623280 |
TCACCATGTTGACCAGGCTA |
AAGCAGTGAGTCTCCCGAAA |
1.05 |
| Exon6 |
54,626,833-54627082 |
GAGCTGGGCAACAGCCTGGA |
CCCGGGTGCCTGGTGTG |
1.25 |
| Exon12 |
54,632,417-54632529 |
TACCAGGAGGACCTGGGATT |
GCAGCGTCTTGGAGATCCT |
0.86 |
| Exon14 | 54,634,733-54633836 | GGAGATTGTGAACCCACAGG | ACTCTTCTCGCCCTTGACCT | 0.99 |
Abbreviations: RQ,relative quantity; *, mean value of three times.
The breakpoint region was defined using long-range PCR (Vigorous Biotechnology, Beijing, China) with primers adjacent to the deleted regions and additional nested primers 91925F and PRPF31-3A2R (Table 1 and Table 2 ). PCR products were purified and sequenced.
Allele-specific polymerase chain reaction analysis
The variation found in the sequencing was confirmed with allele-specific PCR (AS-PCR) analysis of the available family members and 100 normal controls [20]. An allele-specific forward primer in the exon 13 of the PRPF31 gene was designed (Table 1). AS-PCR analysis was performed for the genomic DNA arrangement with the 91,525 forward primer and INS-R reversed primer (Table 1).
Single nucleotide polymorphism genotype
SNP rs3760698 located in the deletion region was sequenced using RQ-PCR primers in family RP24. CNOT3 rs4806718 alleles were sequenced with primers (Table 1) in the two families.
Results
Clinical findings
Thirty individuals at risk for dominantly inherited RP in two Chinese families (RP24 and RP106) were examined (Figure 1). Eight individuals in family RP24 and five members in family RP106 were diagnosed with RP (Table 3). All patients had experienced night blindness since early childhood and exhibited characteristic RP fundus appearance including atrophic retinal pigment epithelial (RPE) changes with bone spicule-like pigmentation (Figure 2A,C,G). The patients who were over 30 years old usually developed secondary cataract (Figure 2B,D). ERG testing of the patients showed undetectable rod and cone responses (Figure 2H). An asymptomatic individual III:6 (a 37-year-old man with best corrected visual acuity of 1.0 in both eyes and a normal visual field) was identified in family RP106. His fundi were normal, and his ERG revealed normal rod and cone cell function (Figure 2E,F).
Figure 1.

Family structure and haplotype analysis of two Chinese families with retinitis pigmentosa. Pedigree and haplotype analysis of family RP24 showed “loss of heterozygous” segregation with single nucleotide polymorphism (SNP) rs3760698 (red x). #, 1 indicates the wild-type allele; 2 represents the mutant allele. All markers and SNPs are located on chromosome 19, listed in descending order from the centromeric end.
Table 3. The clinical features of patients of the two families.
| Familial number | Patient number | Age | Onset age of NB | BCV (OD/OS) | Cataract | Fundus apperence* | Visual Field | ERG | PRPF31 mutation |
|---|---|---|---|---|---|---|---|---|---|
| RP24 |
III-1 |
71 |
C |
NLP/NLP |
YES |
NA |
NA |
NA |
INDEL |
| IV-2 |
41 |
C |
0.2/0.3 |
YES |
YES |
Constriction,central 20 degrees |
Undetectable |
INDEL |
|
| III-3 |
59 |
C |
HM/HM |
YES |
YES |
Constriction,central 10 degrees |
Undetectable |
INDEL |
|
| IV-4 |
34 |
C |
0.6/0.5 |
NO |
YES |
Constriction,central 30 degrees |
Undetectable |
INDEL |
|
| IV-5 |
22 |
C |
0.7/0.8 |
NO |
YES |
Constriction,central 30 degrees |
Undetectable |
INDEL |
|
| III-5 |
54 |
C |
HM/HM |
YES |
YES |
Constriction,central 15 degrees |
NA |
INDEL |
|
| IV-6 |
29 |
C |
0.4/0.1 |
YES |
YES |
Constriction,central 20 degrees |
NA |
INDEL |
|
| III-6 |
49 |
C |
0.1/0.1 |
YES |
YES |
Constriction,central 15 degrees |
NA |
INDEL |
|
| RP106 |
II-1 |
64 |
C |
0.1/0.2 |
YES |
YES |
NA |
NA |
p.G405fs+7X |
| III-1 |
40 |
C |
0.2/0.01 |
YES |
YES |
Constriction,central 20 degrees |
Undetectable |
p.G405fs+7X |
|
| III-3 |
32 |
C |
0.4/0.3 |
YES |
YES |
Constriction,central 30 degrees |
Undetectable |
p.G405fs+7X |
|
| II-3 |
60 |
C |
0.1/0.1 |
YES |
YES |
NA |
NA |
p.G405fs+7X |
|
| III-6 |
33 |
NO |
1.2/1.2 |
NO |
NO |
Normal |
Normal |
p.G405fs+7X |
|
| IV-3 | 9 | C | 0.8/0.8 | NO | YES | NA | Undetectable | p.G405fs+7X |
Abbreviations:C,childhood; NB, night blindness; BCV, best corrected vision;OD,right eye; OS,left eye ;HM,hand move; NLP,no light peception; NA, unavailable,*,intraretinal bone spicule pigments in four quadrants.
Figure 2.

Ophthalmological findings in patients from the two families. A: The fundus appearance of the right eye of patient III:3 in family RP24 shows atrophic retinal pigment epithelial changes, attenuation of the retinal vessels, and irregular pigment clumps in the peripheral retina. B: A slit-lamp photograph of the same eye showed dense white opacities located in the central zone of the lens. C: The fundus appearance of the right eye of patient III:1 in family RP106 shows atrophic retinal pigment epithelial changes, attenuation of the retinal vessels, and irregular pigment clumps in the peripheral retina. D: A photograph of the anterior segment of the same eye shows the opacities in the central zone of the lens. E: Fundus appearance of the right eye of individual III:6 from family RP106 displays a normal appearance. F: Electroretinography (ERG) shows normal rod and cone cell responses. G: The fundus appearance of the right eye of patient IV:3 (daughter of individual III:6) presents atrophic retinal pigment epithelial changes. H: The ERG shows extinguished rod and cone cell responses.
Genotyping results
Two families were genotyped with 41 polymorphic markers around the known adRP loci. The mapping results excluded the other known adRP loci (or the results were not informative) with the exception of the PRPF31 gene. The marker results for D19S927 and D19S926 were fully informative for linkage for family RP24, with two-point linkage odd disequilibrium scores of 3.48 (θ=0) and 3.55 (θ=0), respectively. For family RP106, the two-point limit of detection score for D19S571 was 3.21 (θ=0) with penetrance 0.83. Individual III:6 also carried the disease allele (Figure 1).
Mutation analysis
After we sequenced the PRPF31 gene, we identified one novel heterozygous small deletion c.1215delG (p.G405fs+7X) in family RP106 (Figure 3). AS-PCR analyses showed that the novel deletion cosegregated with the RP phenotype in family RP106, and was not detected in 100 normal controls (Figure 3). We did not observe any obvious disease-causing mutation in family RP24. Linkage analysis for markers for PRPF31 showed evidence of linkage; therefore, we considered detecting a large deletion around the PRPF31 region in family RP24 using several real-time quantitative PCR tests. We identified a deletion of at least 19 kb involving part of the PRPF31 and three upstream genes (Table 2).
Figure 3.
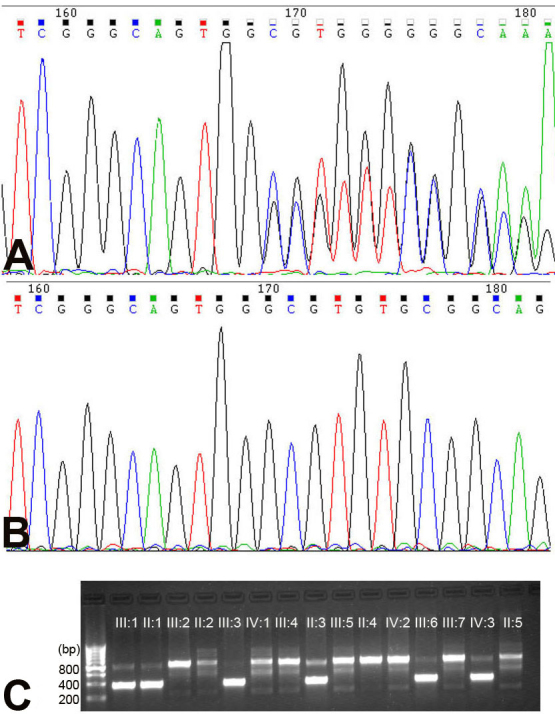
Deoxyribonucleic acid sequence chromatograms and allele-specific polymerase chain reaction analysis on the mutations detected in family RP106. A: The heterozygote sequence (sense strand) shows one-base-deletion c.1215delG (p.G405fs+7X) in patient III:1. B: The sequence shows the corresponding wild- type sequence. C: Allele-specific polymerase chain reaction (AS-PCR) analysis shows the amplified products of the mutation allele (382 bp) cosegregating with patients in this family. The fragments (830 bp), which are the parts of exon 3 of the myocilin (MYOC) gene, were used as the internal control in the AS-PCR analysis.
The RQ-PCR results (Table 2) revealed a normal ratio for REN91525 and intron 3 of the PRPF31 gene; therefore, we applied nested PCR amplifications with two pairs of primers and obtained a PCR fragment of over 700 bp. Sequencing of this product revealed a complex insertion-deletion (indel): a deletion of 19,517 nucleotides and an insertion of about 220 nucleotides. The deletion encompasses exon 1 of osteoclast associated immunoglobulin-like receptor (OSCAR), NADH dehydrogenase [ubiquinone] 1 alpha subcomplex 3 (NDUFA3), TCF3 [E2A] fusion partner (TFPT), and exon 1–3 of the PRPF31 gene (Figure 4). One breakpoint resides within an AluSx repeat in intron 1 of the OSCAR gene, and the other one is located in an AluY repeat in intron 3 of the PRPF31 gene. The origin of the 220 bp insertion appears to be around the OSCAR gene, which is located 25 kb 5′ to the PRPF31 gene. All patients in family RP24 showed “loss of heterozygosis” at rs3760698, which is located in the genomic DNA arrangement region (Figure 1). Sequencing of CNOT3 rs4806718 revealed three patients (III:3, III:5, and III:7) in family RP24 who harbored C/T alleles, whereas the remaining nine patients and the asymptomatic individual carried homozygous T alleles (Figure 1).
Figure 4.
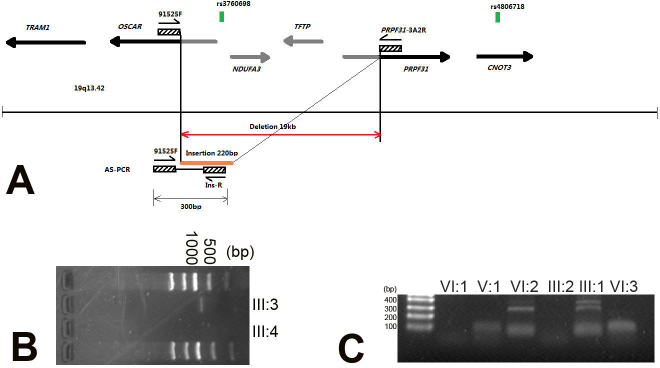
Genomic rearrangement including the PRP31 premessenger ribonucleic acid processing factor 31 homolog gene in family RP24. A: Schematic representation of the genomic region 19q13.42 in proximity to the PRP31 premessenger ribonucleic acid (pre-mRNA) processing factor 31 homolog (PRPF31) gene. B: Nested primers 91525F and PRPF31–3A2R were used to amplify the region across the deletion (the estimated size of a wild-type allele is 19,825 bp); an approximately 700 bp fragment was amplified in patient III:3, and no wild-type alleles were amplified in normal individual III:4. C: Allele-specific polymerase chain reaction (AS-PCR) analysis shows that the amplified products of the mutation allele (315 bp) with primers 91525F and Ins-R cosegregated with patients in this family.
Discussion
In this study, we identified one novel deletion and one novel complex genomic rearrangement in two Chinese families with adRP and characterized the breakpoints of the genomic rearrangement. To date, eight large deletions, ranging from 5 kb to 112 kb, have been identified in PRPF31 [10-13]. Similar to all characterized large deletions of PRPF31, both breakpoints of the large deletion in family RP24 are located in the Alu regions; this suggests that the mechanism underlying this complex structural rearrangement may involve homologous recombination between Alu repeats.
Alu repeats are short, interspersed nuclear elements that account for 10% of the sequenced region of the human genome; however, these repeats account for 26.3% of chromosome 19, which is the most Alu-rich chromosome [21]. This is why genomic arrangements are prevalent round the PRPF31 gene. Of the eight reported large deletions, only one is an insertion-deletion complex genomic DNA rearrangement, which includes an 110 bp deletion and a 640bp insertion [11]. Notably, the 640 bp insertion was also part of the OSCAR gene. Consistent with other reports, no additional phenotype associated with the genetic defects in the NDUFA3, TFPT, or OSCAR were observed in the patients of family RP24 [12,13].
Large deletions or complex chromosome arrangements cannot be revealed with routine PCR-based DNA sequencing. Several techniques, such as RQ-PCR, multiplex ligation-dependent probe (MLPA), and comparative genomic hybridization (CGH) array, are available for detecting genomic aberrations in copy number [22]. A combined strategy involving different techniques applied to genomic DNA analysis could offer the most valuable option for investigating the PRPF31 defect.
The patients of the two families in this study presented clinical features of rod-cone dystrophy in their childhood. The patients in family RP24 who carried the complex genomic DNA arrangement did not show a more severe phenotype, which is consistent with the observations of several other studies [10-13]. In contrast to the incomplete penetrance observed in most adRP families linked to RP11, no asymptomatic mutation/or obligate carriers were identified in family RP24. One large Chinese family carrying a 12 bp deletion was reported with high penetrance [4]. Compared to this high penetrance Chinese family, family RP24, which had 11 affected members, is relatively small, so incomplete penetrance cannot be formally excluded for their novel complex rearrangement.
Similar to most families harboring PRPF31 gene mutations, an incompletely asymptomatic member was observed in family RP106. Recently, Venturini et al. identified the CNOT3 gene as the main modifier gene for determining the penetrance of PRPF31 mutations with a mechanism involving transcriptional repression [17]. The researchers found that the C allele of SNP rs4806718 in the intronic region of the CNOT3 gene was associated with the affected status while the T allele was associated with asymptomatic status [17]. Our genotype results for rs4806718 did not show this kind of association. This may be due to ethnic differences, and further study is needed for confirmation. In conclusion, this study presents two deletions that further support the concept that haploinsufficiency is the main mechanism for RP11 and that genomic arrangements may be prevalent in PRPF31 mutations.
Acknowledgments
We thank the patients and their families for participating in this study. The study was supported by National Natural Science Foundation of China (No. 81170878).
Appendix 1. Markers used in the known ADRP genotyping.
To access the data, click or select the words “Appendix 1.”
References
- 1.Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 2.Ayuso C, Millan JM. Retinitis pigmentosa and allied conditions today: a paradigm of translational research. Genome Med. 2010;2:34. doi: 10.1186/gm155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vithana EN, Abu-Safieh L, Allen MJ, Carey A, Papaioannou M, Chakarova C, Al-Maghtheh M, Ebenezer ND, Willis C, Moore AT, Bird AC, Hunt DM, Bhattacharya SS. A human homolog of yeast pre-mRNA splicing gene, PRPF31, underlies autosomal dominant retinitis pigmentosa on chromosome 19q13.4 (RP11). Mol Cell. 2001;8:375–81. doi: 10.1016/s1097-2765(01)00305-7. [DOI] [PubMed] [Google Scholar]
- 4.Wang L, Ribaudo M, Zhao K, Yu N, Chen Q, Sun Q, Wang L, Wang Q. Novel deletion in the pre-mRNA splicing gene PRPF31 causes autosomal dominant retinitis pigmentosa in a large Chinese family. Am J Med Genet A. 2003;121A:235–9. doi: 10.1002/ajmg.a.20224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martínez-GimenoM Gamundi MJ,Hernan I, Maseras M, Milla E, Ayuso C, Carcia-Sandoval B, Beneyto M, Vilela C, Baiget M, Antinolo G, Carballo M. Mutations in the Pre-mRNA splicing-factor genes PRPF3, PRPF8, and PRPF31 in Spanish families with autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2003;44:2171–7. doi: 10.1167/iovs.02-0871. [DOI] [PubMed] [Google Scholar]
- 6.Sullivan LS, Bowne SJ, Birch DG, Hughbanks-Wheaton D, Heckenlively JR, Lewis RA, Garcia CA, Ruiz RS, Blanton SH, Northrup H, Gire AI, Seaman R, Duzkale H, Spellicy CJ, Zhu J, Shankar SP, Daiger SP. Prevalence of disease-causing mutations in families with autosomal dominant retinitis pigmentosa: a screen of known genes in 200 families. Invest Ophthalmol Vis Sci. 2006;47:3052–64. doi: 10.1167/iovs.05-1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Waseem NH, Vaclavik V, Webster A, Jenkins SA, Bird AD, Bhattacharya SS. Mutations in the gene coding for the pre-mRNA splicing factor, PRPF31, in patients with autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2007;48:1330–4. doi: 10.1167/iovs.06-0963. [DOI] [PubMed] [Google Scholar]
- 8.Ghazawy S, Springell K, Gauba V, McKibbin MA, Inglehearn CF. Dominant retinitis pigmentosa phenotype associated with a new mutation in the splicing factor PRPF31. Br J Ophthalmol. 2007;91:1411–13. doi: 10.1136/bjo.2006.105163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taira K, Nakazawa M, Sato M. Mutation c. 1142 del G in the PRPF31 gene in a family with autosomal dominant retinitis pigmentosa (RP11) and its implications. Jpn J Ophthalmol. 2007;51:45–8. doi: 10.1007/s10384-006-0394-1. [DOI] [PubMed] [Google Scholar]
- 10.Abu-Safieh L, Vithana EN, Mantel I, Holder GE, Pelosini L, Bird AC, Bhattacharya SS. A large deletion in the adRP gene PRPF31: evidence that haploinsufficiency is the cause of disease. Mol Vis. 2006;12:384–8. [PubMed] [Google Scholar]
- 11.Sullivan LS, Bowne SJ, Seaman CR, Blanton SH, Lewis RA, Heckenlively JR, Birch DG, Hughbanks-Wheaton D, Daiger SP. Genomic rearrangements of the PRPF31 gene account for 2.5% of autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2006;47:4579–88. doi: 10.1167/iovs.06-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Köhn L, Bowne SJ, Sullivan LS, Daiger SP, Burstedt MS, Kadzhaev K. Sandgren Ola, Golovleva I. Breakpoint characterization of a novel approximately 59 kb genomic deletion on 19q13.42 in autosomal dominant retinitis pigmentosa with incomplete penetrance. Eur J Hum Genet. 2009;17:651–5. doi: 10.1038/ejhg.2008.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rose AM, Mukhopadhyay R, Webster AR, Bhattacharya SS, Waseem NH. A 112 kb deletion in chromosome 19q13.42 leads to retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2011;52:6597–603. doi: 10.1167/iovs.11-7861. [DOI] [PubMed] [Google Scholar]
- 14.Deery EC, Tithana EN, Newbold RJ, Gallon VA, Bhattacharya SS, Warren MJ, Hunt DM, Wilkie SE. Disease mechanism for retinitis pigmentosa (RP11) caused by mutations in the splicing factor gene PRPF31. Hum Mol Genet. 2002;11:3209–19. doi: 10.1093/hmg/11.25.3209. [DOI] [PubMed] [Google Scholar]
- 15.McGee TL, Devoto M, Ott J, Berson EL, Dryja DP. Evidence that the penetrance of mutations at the RP11 locus causing dominant retinitis pigmentosa is influenced by a gene linked to the homologous RP11 allele. Am J Hum Genet. 1997;61:1059–66. doi: 10.1086/301614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vithana EN, Abu-Safieh L, Pelosini L, Winchester E, Hornan D, Bird AC, Hunt DM, Bustin SA, Bhattacharya SS. Expression of PRPF31 mRNA in patients with autosomal dominant retinitis pigmentosa: a molecular clue for incomplete penetrance? Invest Ophthalmol Vis Sci. 2003;44:4204–9. doi: 10.1167/iovs.03-0253. [DOI] [PubMed] [Google Scholar]
- 17.Venturini G, Rose AM, Shah AZ, Bhattacharya SS, Rivolta C. CNOT3 is a modifier of PRPF31 Mutations in retinitis pigmentosa with incomplete penetrance. PLoS Genet. 2012;8:e1003040. doi: 10.1371/journal.pgen.1003040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marmor MF, Fulton AB, Holder GE, Miyake Y, Brigell M, Bach M. Standard for clinical electroretinography (2008 update). Doc Ophthalmol. 2009;118:69–77. doi: 10.1007/s10633-008-9155-4. [DOI] [PubMed] [Google Scholar]
- 19.Zhang X, Zhang Q, Tong Y, Dai H, Zhao X, Bai F, Xu L, Li Y. Large novel deletions detected in Chinese families with aniridia: correlation between genotype and phenotype. Mol Vis. 2011;17:548–57. [PMC free article] [PubMed] [Google Scholar]
- 20.Ruano G, Kidd KK. Direct haplotying of chromosomal segments from multiple heterozygous via allele-specific PCR amplification. Nucleic Acids Res. 1989;17:8392. doi: 10.1093/nar/17.20.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grover D, Mukerji M, Bhatnagar P, Kannan K, Brahmachari SK. Alu repeat analysis in the complete human genome: trends and variations with respect to genomic composition. Bioinformatics. 2004;20:813–7. doi: 10.1093/bioinformatics/bth005. [DOI] [PubMed] [Google Scholar]
- 22.Armour JAL, Barton DE, Cockburn DJ, Taylor GR. The detection of large deletions or duplications in genomic DNA. Hum Mutat. 2002;20:325–37. doi: 10.1002/humu.10133. [DOI] [PubMed] [Google Scholar]