

Abstract
Objective
To determine the relative contributions of mutations in congenital cataract cases in an Indian population by systematic screening of genes associated with cataract.
Methods
We enrolled 100 congenital cataract cases presenting at the Dr. R. P. Centre for Ophthalmic Sciences, a tertiary research and referral hospital (AIIMS, New Delhi, India). Crystallin, alpha A (CRYAA), CRYAB, CRYGs, CRYBA1, CRYBA4, CRYBB1, CRYBB2, CRYBB3, beaded filament structural protein 1 (BFSP1), gap function protein, alpha 3 (GJA3), GJA8, and heat shock transcription factor 4 gene genes were amplified. Protein structure differences analysis was performed using Discovery Studio (DS) 2.0.
Results
The mean age of the patients was 17.45±16.51 months, and the age of onset was 1.618±0.7181 months. Sequencing analysis of 14 genes identified 18 nucleotide variations. Fourteen variations were found in the crystallin genes, one in Cx-46 (GJA3), and three in BFSP1.
Conclusions
Congenital cataract shows marked clinical and genetic heterogeneity. Five nucleotide variations (CRYBA4:p.Y67N, CRYBB1:p.D85N, CRYBB1:p.E75K, CRYBB1:p.E155K, and GJA3:p.M1V) were predicted to be pathogenic. Variants in other genes might also be involved in maintaining lens development, growth, and transparency. The study confirms that the crystallin beta cluster on chromosome 22, Cx-46, and BFSP1 plays a major role in maintaining lens transparency. This study also expands the mutation spectrum of the genes associated with congenital cataract.
Introduction
Cataracts are the most common cause of visual impairment and account for 10% of all childhood blindness worldwide [1]. The prevalence of cataract, depending on regional socioeconomic development, is 1−6 cases per 10,000 live births in industrialized countries [2,3] and 5−15 per 10,000 in the poorest areas of the world [4,5]. Various etiological factors have been identified, including infection, metabolic disorders, and genetic defects. Hereditary cataracts are clinically highly heterogeneous and show considerable interfamilial and intrafamilial variability [6]. Hereditary congenital cataract may be inherited as autosomal dominant, autosomal recessive, or X-linked traits and thus shows marked genetic heterogeneity.
Congenital cataract is a clinically and genetically heterogeneous disorder [7]. Different mutations in the same gene can cause similar cataract patterns while the highly variable morphologies (total, polar, zonular, and capsular) of cataracts within families suggest that the same mutation in a single gene can lead to different phenotypes [6,8]. To date, more than 40 genetic loci have been linked to congenital cataracts [9]. Among these candidate genes, crystallin and connexin genes represent a major proportion of the mutations identified in congenital cataract and have been associated with cataracts of various morphologies [10], including genes encoding crystallins (crystallin, alpha A [CRYA], CRYB, and CRYG) [11], lens-specific connexins (Cx43, Cx46, and Cx50) [12,13], cytoskeletal structural proteins (beaded filament structural protein 1 [BFSP1]) [14], and heat shock transcription factor 4 gene (HSF4) [15].
The α-, β-, and γ-crystallins represent more than 90% of the lens-soluble proteins in humans, encompassing almost 35% of the mass and accounting for the optical transparency and high refractive index [16,17]. In the human lens, α-crystallin makes up about 40%, β-crystallin 35%, and γ-crystallin 25% of the total crystallin protein. Lamellar and nuclear cataracts are the most common types of hereditary congenital cataract [18,19]. The HSF4 transcription factor is the predominant HSF expressed postnatally in ocular lens [20]. Specific interactions between HSF4 and HSE in the promoters of β-crystallin (causes autosomal dominant congenital cataract when mutated) [21], Hsp70, and Hsp82 have been demonstrated [20]. The aim of our study was to determine the relative contributions of mutations in congenital cataract cases in an Indian population with systematic screening of 14 genes associated with cataract.
Methods
Patient ascertainment and clinical examination
After receiving ethical approval from the institutional review board (IRB#00006862; All India Institute of Medical Sciences, Delhi, India), 100 clinically diagnosed congenital cataract cases from northern India who presented at the Dr. R. P. Centre for Ophthalmic Sciences (AIIMS, New Delhi, India) were enrolled in this study. In this study 69% of the patients were found to male as compared to 31% female. Mean age of the patients is 17.45±16.51 and the mean age of onset of disease was 1.618±0.7181. Affected status was determined by a history of cataract extraction or ophthalmologic examination. A total of 100 normal individuals without any history of ocular disorders were enrolled as controls. Patients with a history of intrauterine infection such as rubella, TORCHES (TOxoplasmosis, Rubella, Cytomegalovirus, HErpes simplex, Syphilis), and traumatic cataract were excluded from the study. Informed consent in accordance with the Declaration of Helsinki was obtained from all participants or their parents.
Polymerase chain reaction and deoxyribonucleic acid sequencing
Brief description for how the blood was drawn and how samples were preserved prior to use is as follow: For DNA isolation 2-3 ml of peripheral blood was collected in EDTA vials from each case. The samples were stored in -80 °C prior to use. Genomic DNA was extracted from the blood samples of the patients with congenital cataract and controls, using an organic method described by Sambrook et al. [22]. The exon-intron regions of all the genes (CRYAA, CRYAB, CRYGs, CRYBA1, CRYBA4, CRYBB1, CRYBB2, CRYBB3, BFSP1, GJA3, GJA8, and HSF4) were amplified. PCR amplifications for all primer sets (Table 1) were performed in a 40 μl volume containing 1.0 μl of 20 mM stock solution for each primer (Eurofins Genomics India, Bangalore, India), 100 ng genomic DNA, 1 unit Taq polymerase (Banglore Genei, Bengaluru, India), 0.1 mM each deoxynucleotide triphosphate (dNTP), and 4 μl 10X PCR buffer (with 15 mM MgCl2). Amplified PCR products were purified using a gel/PCR DNA fragments extraction kit (Geneaid Biotech, Sijhih City, Taiwan). Purified PCR products were sent for sequencing to Molecular Cloning Laboratories (South San Francisco, CA). All sequence variants were compared to the Human Genome Reference Sequence provided by the National Center for Biotechnology Information (NCBI), using ClustalW2 (multiple sequence alignment program for DNA; European Bioinformatics Institute, Wellcome Trust Genome Campus, Hinxton, UK).
Table 1. Oligonucleotides of mutated genes used as primers for PCR amplification.
| Gene Name | Primer Name | Primer Sequence | Anne Temp (°C) | Product Size (bp) |
|---|---|---|---|---|
|
CRYBA4 |
Ex-2-F |
5′-TAGCCCAGTCACTCCTGGAC-3′ |
57 °C |
213 |
| Ex-2-R |
5′-GCCTTGATTGCACCTCTGTG-3′ |
|||
| Ex-3-F |
5′-TTTGCAATCCCTGCTTTACC-3′ |
57 °C |
423 |
|
| Ex-3-R |
5′-ATGGCACCCTCCTACTGTTGG-3′ |
|||
| Ex-4-F |
5′-AAAAATGTCTCCAGCCATCG-3′ |
57 °C |
314 |
|
| Ex-4-R |
5′-AGCTTGAAGTGGCGACATGAG-3′ |
|||
| Ex-5-F |
5′-AAATGGCAAGGTTTCTGGTAC-3′ |
57 °C |
297 |
|
| Ex-5-R |
5′-GCCTCAGTGTTCTCCTCTGG-3′ |
|||
| Ex-6-F |
5′-AGGGAATGGCATGATCAAAG-3′ |
57 °C |
335 |
|
| Ex-6-R |
5′-TGCTGGGTTCACACAGGTTAC-3′ |
|||
|
CRYBB1 |
Ex-2-F |
5′-ACAGGATGTGGGGCTATGAG-3′ |
59 °C |
380 |
| Ex-2-R |
5′-GTGCGGAGGAGTAAGAGGTG-3′ |
|||
| Ex-3-F |
5′-CATTTCACAAACTGTGGCTCA-3′ |
62 °C |
379 |
|
| Ex-3-R |
5′-GGACATAATGTATGTGCCAGGA-3′ |
|||
| Ex-4-F |
5′-GTAGGGAGTGGGGGCTTCTA-3′ |
62 °C |
286 |
|
| Ex-4-R |
5′-CTCCTTCTTGCCCTTGTCAG-3′ |
|||
| Ex-5-F |
5′-GCTCATCTCTCTCGCTCCAC-3′ |
61 °C |
298 |
|
| Ex-5-R |
5′-TCTGATTCTGCCTGTGCTTG-3′ |
|||
| Ex-6-F |
5′-TCAATGAAGGACAGGCTGGT-3′ |
62 °C |
381 |
|
| Ex-6-R |
5′-TCCAGGAGAAATTTTGGCTTT-3′ |
|||
|
CRYBB2 |
Ex-2-F |
5′-CAGAGGGGAGTGGTCTCAAG-3′ |
59 °C |
244 |
| Ex-2-R |
5′-ATGCCAAGCCCATTTTACAG-3′ |
|||
| Ex-3-F |
5′-TCAGCATCCTTTGGGTTCTC-3′ |
59 °C |
299 |
|
| Ex-3-R |
5′-CAAGGGTAGATTCCCCCACT-3′ |
|||
| Ex-4-F |
5′-AACCCTAGGGGTCAACATCA-3′ |
62 °C |
297 |
|
| Ex-4-R |
5′-CTCCAAGGTGGCAGAGAGAG-3′ |
|||
| Ex-5-F |
5′-GAGTGATGTGTGGGACATGC-3′ |
62 °C |
377 |
|
| Ex-5-R |
5′-CAGAGGTCAGCAGAGCACAC-3′ |
|||
| Ex-6-F |
5′-GGCTTCACCCTTCCTAGTGG-3′ |
59 °C |
399 |
|
| Ex-6-R |
5′-CAAAGACCCACAGCAGACAA-3′ |
|||
|
GAJ3 |
Ex-1A-E |
5′-TGCGGACCCGGCACTCAGC-3′ |
62 °C |
383 |
| Ex-1A-R |
5′-TCCATGCGCACGATGTGCAGTCA-3′ |
|||
| Ex-1B-F |
5′-CTGTTCATCTTCCGCATTTTGG-3′ |
62 °C |
603 |
|
| Ex-1B-R |
5′-TCTTCTTCCAGCCCAGGTGGTA-3′ |
|||
| Ex-1C-F |
5′-AAGCTCAAGCAGGGCGTGAC-3′ |
62 °C |
624 |
|
| Ex-1C-R |
5′-CTAGATGGCCAAGTCCTCCGG-3′ |
|||
| BFSP1 | Ex-1-F |
5′-GGGCCTCCGGTGTTTATTTA-3′ |
58 °C |
589 |
| Ex-1-R |
5′-ATCGACAGGGGACCGAGAGAC-3′ |
|||
| Ex-2-F |
5′-AAAGGAGAGGGCATCGTACC-3′ |
58 °C |
238 |
|
| Ex-2-R |
5′-AACCTGCACTTCCACCATTC-3′ |
|||
| Ex-3-F |
5′-CAGGTGGTCTGTGTGCACAT-3′ |
58 °C |
249 |
|
| Ex-3-R |
5′-TCGGCTTACCTGATCAAACC-3′ |
|||
| Ex-4-F |
5′-RGCCATTCCTGTTCTCATCT-3′ |
58 °C |
250 |
|
| Ex-4-R |
5′-GCCCTTCCCTGGGAGTCT-3′ |
|||
| Ex-5-F |
5′-ACCTTCTCTGCCCTTTTCCT-3′ |
58 °C |
227 |
|
| Ex-5-R |
5′-CACCTCCATGAAACATGTGG-3′ |
|||
| Ex-6-F |
5′-CCTTTTCCTGGGTGAGGTCTG-3′ |
58 °C |
366 |
|
| Ex-6-R |
5′-GGCACACAATAGGCACTCAA-3′ |
|||
| Ex-7-F |
5′-CTTGCCCCTGACCTCTGTT-3′ |
58 °C |
199 |
|
| Ex-7-R |
5′-AAGAGAGCCGCTTGGTTTTT-3′ |
|||
| Ex-8-F |
5′-TTCCAACCAGCGTATTTTCTTT-3′ |
58 °C |
699 |
|
| Ex-8-R |
5′-TCAGGGCCTTCCAGCTCT-3′ |
|||
| In5-Ex-7-F |
5′-CATCTTCCAGGGTGTCCAG-3′ |
58 °C | 316 | |
| In5-Ex-7-R | 5′-AAGAGAGCCGCTTGGTTTTT-3′ |
Bioinformatics analysis
MutationTaster, a free, web-based application was used for rapid evaluation of the disease-causing potential of DNA sequence alterations [23]. The Sorting Intolerant From Tolerant (SIFT) analysis tool was also used to predict the functional impact of the missense changes identified in this study [24]. Positions with normalized probabilities <0.05 are predicted to be deleterious, and those ≥0.05 are predicted to be tolerated. Another free, web-based application, PolyPhen-2, structurally analyzes an amino acid polymorphism and predicts whether that amino acid change is likely to be deleterious to protein function [25,26]. A PolyPhen-2 score of >2.0 indicates the change is probably damaging to protein function. Scores of 1.5–2.0 are possibly damaging, and scores of <1.5 are likely benign. Three web-based applications were used to predict the pathogenicity of non-synonymous variations. The variations were considered pathogenic only when the outcome of two out of three applications suggested the variations were disease causing.
Protein modeling
The normal and mutant proteins were analyzed for their structure. Prediction of structure differences between the wild-type and mutant proteins was performed using Discovery Studio (DS) 2.0 (Accelrys, San Diego, CA) [27]. The first step in the homology modeling method is to find a suitable homologous structure (template). Thus, structural differences between the wild-type and mutant were predicted in mutants when a suitable template or homolog structure was present. The model structure of the three mutant proteins was developed and refined with minimization programs in the presence of the CHARMm force field in a manner similar to the structure of the A4 protein.
Statistical analysis
Pearson χ2/Fisher’s exact test was applied to compare the two groups (cases versus controls). P values less than 0.05 were considered significant. All tests were performed with SPSS software for Windows (version 11.5; SPSS Inc., Chicago, IL).
Results and Discussion
A total of 100 patients with sporadic congenital cataract were enrolled in this study. The age range of the patients was 1 month to 3 years. The mean age of onset was 1.618±0.7181 months, as the age at which the disease was first noticed by the child’s parents or by a clinician. Out of 100 patients, 80 had bilateral congenital cataract, and 20 had unilateral congenital cataract. All cases enrolled were sporadic, and the male to female ratio was 2.2:1 (69 men and 31 women). Different forms of cataracts with variable degrees of opacification were observed. Nuclear cataract (72%) was the most prevalent phenotype found. The other phenotypes observed were zonular with nuclear (19%), total cataract (05%), Zonular/lamellar (03%) and anterior polar cataract (01%).
Mutations in more than 40 genetic loci have been linked to congenital cataracts. Of these mutations, approximately half involve crystallins, one-quarter involve connexins, and the remaining involve other genes [9]. Direct sequencing analysis of 14 genes (CRYAA, CRYAB, CRYGs, CRYBA1, CRYBA4, CRYBB1, CRYBB2, CRYBB3, BFSP1, GJA3, GJA8, and HSF4) identified 18 nucleotide variations (Table 2), 14 of which were in crystallin genes (CRYBA4, CRYBB1, and CRYBB2), one in Cx-46 (GJA3), and three in BFSP1. Five nucleotide variations (Figure 1; CRYBA4:p.Y67N, CRYBB1:p.E75K, CRYBB1:p.D85N, CRYBB1:p.E155K, and GJA3:p.M1V) were predicted to be pathogenic in in silico analysis. No variation was detected in the CRYAs, CRYGs, CRYBA1, CRYBB3, GJA8, and HSF4 genes.
Table 2. Nucleotide variations found in congenital cataract patients.
| S.No. | Nucleotide Change | Locus | Codon Change | Amino acid Change | Type of Mutation | POLYPHEN/SIFT /Mutation Taster |
|---|---|---|---|---|---|---|
| 1. |
g.27021536* |
CRYBA4 |
ACG>GCG |
T84A |
NS |
0.823/0.02/PM |
| 2. |
g.27021532* |
CRYBA4 |
GGC>GGA |
G82G |
SYN |
NA |
| 3. |
g.27021497* |
CRYBA4 |
CGA>AGA |
R71R |
SYN |
NA |
| 4. |
g.27021485* # |
CRYBA4 |
TAC>AAC |
Y67N |
NS |
3.084/0.00/DC |
| 5. |
rs5761637T>A^ |
CRYBA4 |
TTT>TTC |
F57F |
SYN |
NA |
| 6. |
rs4276A>G^ |
CRYBA4 |
intronic |
NA |
NA |
NA |
| 7. |
rs73880140C>T^ |
CRYBA4 |
intronic |
NA |
NA |
NA |
| 8. |
rs2071862G>A^ |
CRYBA4 |
intronic |
NA |
NA |
NA |
| 9. |
g.G27008082A* # |
CRYBB1 |
GAC>AAC |
D85N |
NS |
1.689/ 0.02/DC |
| 10. |
g.G27008112A*# |
CRYBB1 |
GAA>AAA |
E75K |
NS |
2.002/0.00/DC |
| 11. |
rs57400078C>A^ |
CRYBB1 |
intronic |
NA |
NA |
NA |
| 12. |
g. A26997943G*# |
CRYBB1 |
GAA>AAA |
E155K |
NS |
2.088/0.00/DC |
| 13. |
g. G25617606A* |
CRYBB2 |
GAT>AAT |
D4N |
NS |
0.552/0.49/DC |
| 14. |
g.G25617414A* |
CRYBB2 |
CAG>CAA |
Q6Q |
SYN |
NA |
| 15. |
c. A178G # |
GJA3 |
ATG>GTG |
M1V |
NS |
2.864/0.00/TP |
| 16. |
g.G17475531A* |
BFSP1 |
GAC>AAC |
D395N |
NS |
1.398/0.01/PM |
| 17. |
g.G17475444A* |
BFSP1 |
GAA>AAA |
E424K |
NS |
0.521/0.92/DC |
| 18. | rs147241220 A>G^ | BFSP1 | CTA>CTG | L44L | SYN | NA |
(Abbrevations: *Novel variations, ^Reported-Ensembl, SYN-synonymous, NS-Non synonymous, A-Not applicable, PM-polymorphism, TP-Truncated protein, DC-disease causing), #-Pathogenic variations
Figure 1.
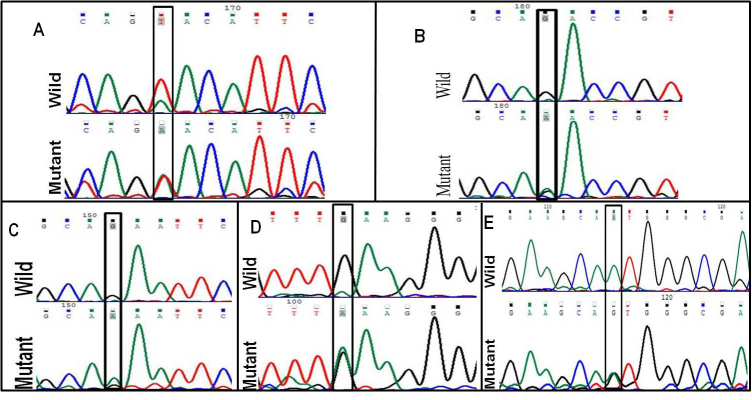
Deoxyribonucleic acid sequence electropherogram of pathogenic variations. (A) crsytallin beta a4, CRYBA4:p.Y67N (T>A), (B) crystalline beta b1, CRYBB1:p.D85N (G>A), (C) CRYBB1:p.E75K (G>A), (D) CRYBB1:p.E155K (A>G), and (E) gap function protein, alpha 3, GJA3:p.M1V (A>G).
Functional changes in crystallin molecular properties could cause the breakdown of the lens microstructure and result in changes in the refractive index and increased light scattering [28]. Out of the 14 variations observed in the crystallin genes (Table 2), eight were observed in the CRYBA4 gene, four in the CRYBB1 gene, and two in the CRYBB2 gene. Billingsley et al. [29] identified CRYBA4 as a cataract gene in a large Indian family with an autosomal dominant cataract phenotype. A total of 102 nucleotide variations (Table 3) have been reported in the CRYBA4 gene, but only three have been associated with cataract [30] (Table 4). Two non-synonymous, novel variations (CRYBA4:p.T84A, CRYBA4:p.Y67N) were found in exon 4 of the CRYBA4 gene. Computational assessment showed CRYBA4:p.Y67N was pathogenic whereas CRYBA4:p.T84A was polymorphic. CRYBA4:p.Y67N was found in two cases affected with bilateral nuclear cataract whereas CRYBA4:p.T84A was found in four patients. Two had zonular cataract, and two had nuclear cataract. None of the variations (CRYBA4:p.T84A, CRYBA4:p.Y67N) were found in the controls. The mutation CRYBA4:p.Y67N located in the neighboring β strand of the N-terminal domain whereas the CRYBA4:p.T84A mutation is situated in the β crystallin A4 protein in the loop region.
Table 3. Genetics variations found in the CRYBA4, CRYBB1, CRYBB2 and GJA8 genes (Ensembl).
| Type of Variants |
Gene Name |
||||
|---|---|---|---|---|---|
| CRYBA4 | CRYBB1 | CRYBB2 | GJA3 | BFSP1 | |
| Stop gained |
1 |
1 |
– |
– |
– |
| Splice site |
2 |
2 |
– |
– |
6 |
| Essential splice site |
3 |
1 |
– |
– |
– |
| Synonymous coding |
9 |
10 |
2 |
29 |
36 |
| Non-synonymous coding |
16 |
15 |
7 |
– |
43 |
| Within non-coding gene |
52 |
– |
– |
– |
53 |
| Frameshift coding |
– |
1 |
– |
2 |
4 |
| Intronic |
54 |
13 |
14 |
3 |
219 |
| 5 prime UTR |
– |
1 |
1 |
– |
9 |
| Upstream |
– |
– |
– |
– |
7 |
| downstream |
– |
– |
– |
– |
5 |
| All | 102 | 42 | 24 | 34 | 339 |
Table 4. Summary of the mutations identified in CRYBA4, CRYBB1, CRYBB2 and GJA3 genes with different congenital cataract phenotypes belonging to different populations (Cat-Map).
| Gene | Exon/ Intron | DNA Change | Coding Change | Pattern of Inheritance | Origin | Cataract Phenotype |
|---|---|---|---|---|---|---|
| CRYBB2 |
Ex2 |
c.5C>T |
p.A2V |
AD |
China |
Congenital posterior subcapsular |
| Ex2 |
c.54G>A |
p.K18KfsX17 |
India |
Congenital zonular |
||
| Ex2 |
c.62T>A |
p.I21N |
China |
Nuclear |
||
| Ex2 |
c.92C>G |
p.S31W |
AD |
China |
Coronary |
|
| Ex4 |
c.177G>C |
p.W59C |
AD |
India |
Total |
|
| Ex5 |
c.383A>T |
p.D128V |
AD |
Germany |
Nuclear, “ring-shaped” cortical |
|
| Ex5 |
c.(433C>T; 440A>G; 449C>T) |
p.(R145W; Q147R; T150M) |
? |
Denmark |
? |
|
| Ex5 |
c.436G>A |
p.V146M |
AD? |
China |
Nuclear (Microcornea) |
|
| Ex6 |
c.452G>C |
p.W151C |
AD |
India |
Central nuclear |
|
| Ex6 |
c.463C>T, c.471C>T |
p.Q155X |
AD |
USA |
Cerulean |
|
| Ex6 |
c.463C>T, c.471C>T |
p.Q155X |
AD |
Switzerland |
Central zonular pulverulent |
|
| Ex6 |
c.463C>T, c.471C>T |
p.Q155X |
AD |
India |
Sutural cerulean |
|
| Ex6 |
c.463C>T, c.471C>T |
p.Q155X |
AD |
China |
Progressive polymorphic |
|
| Ex6 |
c.463C>T, c.471C>T |
p.Q155X |
AD |
Chile |
Variable |
|
| Ex6 |
c.463C>T, c.471C>T |
p.Q155X |
AD |
China |
Progressive polymorphic coronary |
|
| Ex6 |
c.463C>T, c.471C>T |
p.Q155X |
? |
India |
Cortical, pulverulent |
|
| Ex6 |
c.463C>T, c.471C>T |
p.Q155X |
AD |
China |
Cerulean |
|
| Ex6 |
c.477C>A |
p.Y159X |
? |
Denmark |
? |
|
| Ex6 |
c.607G>A |
p.V187M |
AD |
Lesotho |
Nuclear (Strabismus) |
|
| CRYBB1 |
Ex1 |
c.2T>A |
p.M1K |
AR |
Somalia |
Nuclear, pulverulent |
| Ex2 |
c.171delG |
p.G57GfsX107 (p.N58TfsX106) |
AR |
Israel |
Nuclear |
|
| Ex6 |
c.658G>T |
p.G220X |
AD |
USA |
Central sutural pulverulent |
|
| Ex6 |
c.667C>T |
p.Q223X |
AD |
China |
Nuclear progressive |
|
| Ex6 |
c.682T>C |
p.S228P |
AD |
China |
Nuclear (Nystagmus) |
|
| Ex6 |
c.698G>A |
p.R233H |
AD |
China |
Nuclear (Nystagmus) |
|
| Ex6 |
c.757T>C |
p.X253RextX27 |
AD |
UK |
Nuclear cortical riders (Microcornea) |
|
| CRYBA4 |
Ex4 |
c.190G>T |
p.G64W |
China |
Congenital nuclear (Microcornea) |
|
| Ex4 |
c.206T>C |
p.L69P |
AD |
India |
? (Microphthalmia) |
|
| Ex4 |
c.281T>C |
p.F94S |
AD |
India |
Lamellar |
|
|
GJA3 |
Ex2 |
c.-39C>G |
Cx |
China |
Age-related nuclear |
|
| Ex2 |
c.5G>A |
p.G2D |
AD |
China |
Nuclear pulverulent, Posterior polar |
|
| Ex2 |
c.7G>T |
p.D3Y |
AD |
Honduras |
Zonular pulverulent |
|
| Ex2 |
c.32T>C |
p.L11S |
AD |
Denmark |
“Ant-egg” |
|
| Ex2 |
c.56C>T |
p.T19M |
AD |
India |
Posterior polar |
|
| Ex2 |
c.82G>A |
p.V28M |
AD |
India |
Total, anterior capsular, cortical |
|
| Ex2 |
c.96C>A |
p.F32L |
AD |
China |
Nuclear pulverulent |
|
| Ex2 |
c.98G>T |
p.R33L |
AD |
India |
Granular embryonal |
|
| Ex2 |
c.130G>A |
p.V44M |
AD |
China |
Central nuclear (punctate cortical) |
|
| Ex2 |
c.130G>A |
p.V44M |
AD |
USA |
? |
|
| Ex2 |
c.134G>C |
p.W45S |
AD |
China |
Nuclear |
|
| Ex2 |
c.139G>A |
p.D47N |
AD |
China |
Nuclear |
|
| Ex2 |
c.176C>T |
p.P59L |
AD |
USA |
Nuclear punctate |
|
| Ex2 |
c.176C>T |
p.P59L |
AD |
Denmark |
? |
|
| Ex2 |
c.176C>T |
p.P59L |
AD |
China |
? |
|
| Ex2 |
c.188A>G |
p.N63S |
AD |
UK |
Variable pulverulent |
|
| Ex2 |
c.226C>G |
p.R76G |
AD |
India |
Total |
|
| Ex2 |
c.227G>A |
p.R76H |
AD |
Australia |
Nuclear lamellar pulverulent |
|
| Ex2 |
c.227G>A |
p.R76H |
AD |
Denmark |
Lamellar, sutural |
|
| Ex2 |
c.260C>T |
p.T87M |
AD |
India |
“Pearl-box” |
|
| Ex2 |
c.415G>A |
p.V139M |
Cx |
China |
Age-related cortical |
|
| Ex2 |
c.560C>T |
p.P187L |
AD |
UK |
Zonular pulverulent |
|
| Ex2 |
c.559C>T |
p.P187S |
AD |
China |
Nuclear pulverulent |
|
| Ex2 |
c.563A>C |
p.N188T |
AD |
China |
Nuclear pulverulent |
|
| Ex2 |
c.1137insC |
p.S380QfsX87 |
AD |
UK |
Punctate |
|
| Ex2 |
c.1143_1165del23 |
p.381fs*48 |
AD |
China |
Punctate nuclear |
|
| BFSP1 | Ex6 | c.736–957del | p.T246del74fsX6 | AR | India | Cortical progressive, juvenile onset |
For modeling studies, the crystal structure of the human CRYBA4 protein (PDB id: 3LWK) was used as the template. The complete model structure including the missing region of the native human crystallin βA4 as well as its mutant (CRYBA4:p.T84A, CRYBA4:p.Y67N) were developed using the homology modeling approach to study the effect of the mutations on the crystallin’s structure and function. The CRYBA4 protein model structure is dominated by β strands. The two domains interact through intramolecular contacts mediated by loop regions. The missing loop regions were generated from residues Asn83 to Pro87, and residues 180–183 lie in the N-terminal domain and the C-terminal domain, respectively (Figure 2). The model structures of the mutants (CRYBA4:p.T84A, CRYBA4:p.Y67N) were developed by the Build Mutant protocol, and the mutant was refined similarly to the wild-type protein structure.
Figure 2.
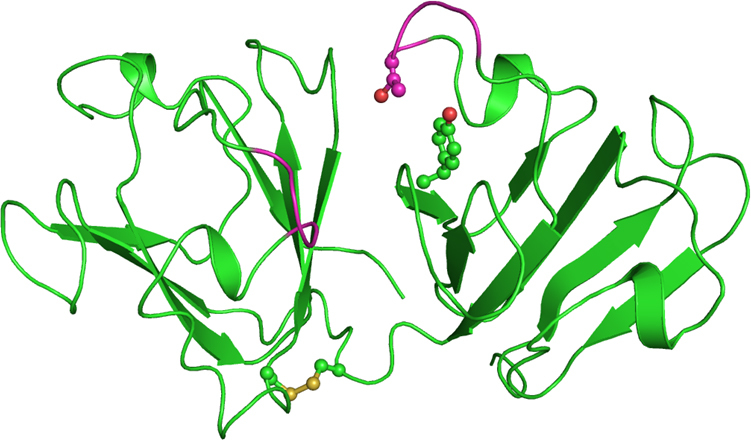
Cartoon representation of the model structure of the wild-type crystallin beta-A4 protein. The disulfide bridge and residues at the mutation site are shown as balls and sticks. The newly generated loops (residue 83–87 and 180–183) are in magenta.
The mutation CRYBA4:p.Y67N does not affect the conformation of the region housing the mutation (Figure 3). Tyrosine (Tyr) is larger and more hydrophobic than Asparagine (Asn). Thus, the bulky Tyr67 side chain restricts the movement of the loop, which imparts structural flexibility. As this protein is functional in its multimeric form, the increased flexibility in the mutant affects the stability of the oligomer as well as interactions with other partner proteins. The mutation (CRYBA4:p.T84A) is present in the modeled loop in the N-terminal domain of human crystallin β-A4. In this loop, two residues, Asn83 and Thr84, are involved in the hydrogen bond–mediated interactions with Gly159 and Gln161 of the C-terminal domain (Figure 4a). These interactions serve as a bridge between the two domains. The substitution of the hydrophobic Alanine (Ala) to Threonine (Thr) disrupts the intramolecular hydrogen bond, which reduces the interdomain contact region as the Ala side chain is incapable of forming hydrogen bonds (Figure 4B). A slight broadening of the loop is also observed as the Asn83-mediated interactions are still conserved (Figure 4A,B). Thus, the mutation can lead to changes in the conformation of protein and its interaction with other partner proteins.
Figure 3.
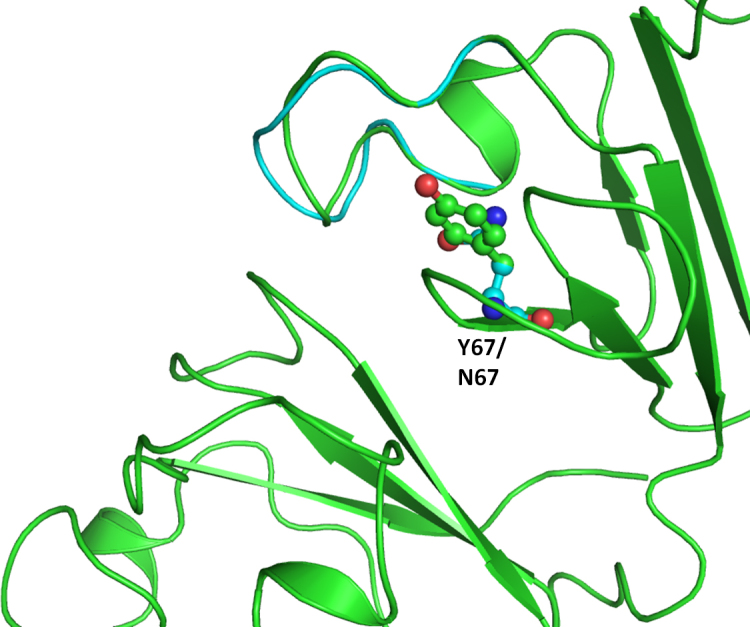
Superimposition of the model structure of the crystallin beta-A4 protein mutant (Tyr67Asn; in cyan) on the wild-type (in green).
Figure 4.
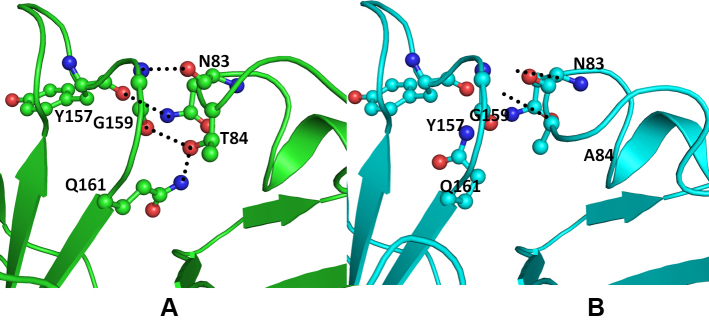
Model structure representation of wild (green) and Thr84Ala mutant (cyan) crystallin beta-A4 (CRYBA4) protein. A: The Thr84 hydroxyl group forms a hydrogen bond with the main chain carbonyl oxygen of Gly159 and the amide nitrogen of the Gln161 side chain. Asn83 is involved in the hydrogen-bonded interaction with the Gly159 main chain nitrogen atom and the Tyr157 main chain oxygen. B: The Ala84 mutant cannot be involved in this interaction. The important interacting residues are rendered as balls and sticks, and the hydrogen bonds are depicted as black dotted lines.
CRYBB1 is a major subunit of the β-crystallins and comprises 9% of the total soluble crystallin in the human lens [31]. A total of 42 nucleotide changes have been reported (Table 3). Of these, seven have been associated with congenital cataract [30] (Table 4). In this study, we detected four nucleotide variations (CRYBB1:p.D85N, CRYBB1:p.E75K, CRYBB1:p.E155K, and CRYBB1:rs57400078) in the CRYBB1 gene. Three novel variations (CRYBB1:p.D85N, CRYBB1:p.E75K, and CRYBB1:p.E155K) were pathogenic according to the in silico analysis (Table 3). Most of the reported CRYBB1 gene mutations occur in exon 6 (Table 4) [30], which encodes the Greek key IV and the COOH-terminal arm [32,33]. The CRYBB1:p.D85N and CRYBB1:p.E75K variations were found in the same patient with nuclear form of cataract whereas the CRYBB1:p.E155K change was detected in seven cases with different cataract morphology. The CRYBB2 gene is one of the most important genes for lens transparency. We identified two novel variations (CRYBB2:p.D4N, CRYBB2:p.Q6Q) in the CRYBB2 gene. The non-synonymous, novel change (CRYBB2:p.D4N) was found in one patient with lamellar cataract.
The crystal structure of the human CRYBB1 protein (PDB ID: 1OKI) [34] was used as the template. The overall folds of CRYBB1 and CRYBA4 are similar (Figure 5) except in the loop region. The N-terminal domain of the CRYBB1 protein harbors mutations (CRYBB1:p.E75K and CRYBB1:p.D85N) whereas CRYBB1:p.E155K occurs on the C-terminal domain. Both mutations are present on the surface of the protein (Figure 5) and are thus exposed to solvents and would be engaged in protein–protein interactions. The Glu75 (an acidic residue), a component of the β strand, is positioned to make two hydrogen bonds with the guanidine group of Arg60 present on the adjacent antiparallel β strand in the wild-type protein (Figure 6A). The mutation CRYBB1:p.E75K alters the environment and charge on the protein surface, disrupting the ionic interaction between Glu75 and Arg60 (Figure 6B). Thus, CRYBB1:p.E75K changes the electrostatic potential of the protein surface, which could affect interactions with other interacting partner proteins.
Figure 5.

Cartoon representation of the crystal structure of the wild-type beta crystallin B1 protein. The residues at the mutation site are shown as balls and sticks.
Figure 6.

Model structure representation of the wild and mutant (Glu75Lys) proteins. A: Beta crystallin B1 (CRYBB1) protein showing the important residues (balls and sticks) and the hydrogen bonds (black dotted lines). B: The contacts are lost in the mutant.
The other change (CRYBB1:p.D85N) occurs in the single turn helix conformation of the CRYBB1 protein. Asp85 is involved in hydrogen bonding with the amide nitrogen of the Asn82 side chain present on the adjacent loop (Figure 7A). Aspartate (Asp) and Asn differ only in the side chain group, with a carboxyl group in the former and an amide in the latter. Thus, in the CRYBB1:p.D85N mutant, the hydrogen bond–mediated interaction is retained (Figure 7B). In the other mutation (CRYBB1:p.E155K) in the CRYBB1 protein, Glu155, located on a surface loop, forms two hydrogen bonds, one with the side chain amide nitrogen and the other with the main chain nitrogen of Asn162 present on the same loop (Figure 8A). Since the change occurs on the protein surface, the elongated side chain does not perturb the protein conformation. However, to accommodate the change and impart stability to the loop, the amide group of Asn162 flips by approximately 180°. This results in the formation of a hydrogen bond with the side chain nitrogen atom of the mutated residue Lys155 (Figure 8B). The change in negatively charged Glu155 with positively charged Lys155 affects the electrostatic potential of the surface, which could be vital for binding with other interacting partners. Thus, the modeling studies indicate that the mutation in the CRYBA4 and CRYBB1 proteins alters the internal conformation of the protein and reduces the stability of the proteins. Thus, the observed mutations could affect the function of the protein, including its ability to bind to its interacting partners.
Figure 7.
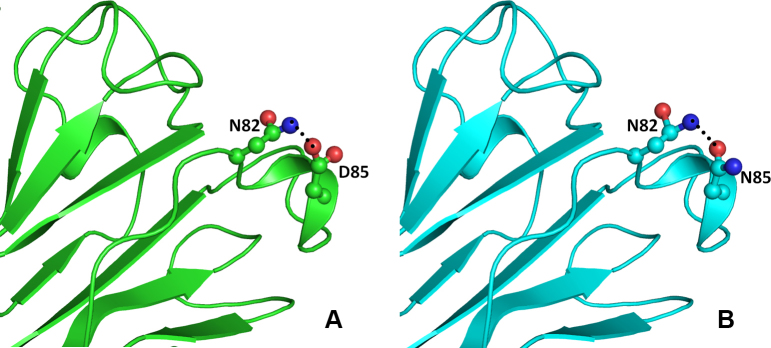
Model structure representation of the wild and mutant (Asp85Asn) proteins. In both structures (A and B), the interaction of residues as balls and sticks and hydrogen bonds as black dotted lines in beta crystallin B1 protein is same.
Figure 8.
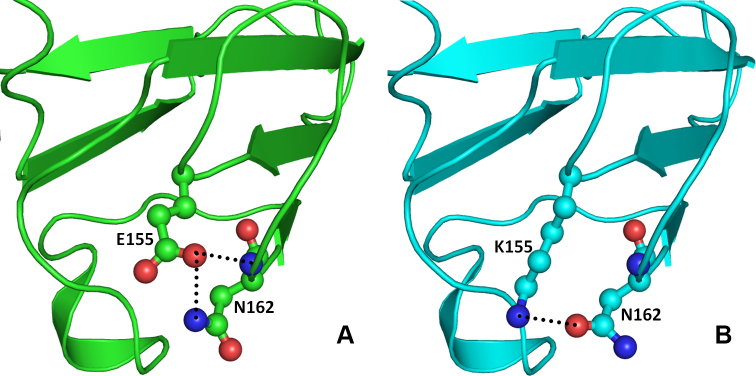
Model structure representation of the wild and mutant (Glu155Lys) proteins. A: Glu155 forms two hydrogen bonds, one with side chain amide nitrogen and another with main chain nitrogen of Asn162 which stabilizes and maintains the loop conformation essential for interactions with other proteins. B: In mutant (Glu155Lys) proteins, Asn162 flips by approximately 180° and forms hydrogen bond with the side chain nitrogen atom of the mutated residue Lys155.
The eye lens is an avascular structure, and intercellular transport of small biomolecules (<1 kDa) is mediated through connexins (Cx) that encode gap junction channel proteins [35,36]. GJA3 encodes a protein containing 435 amino acids and is present in specialized lens fibers, which constitute the majority of the lens [37]. Similar to other connexin proteins, connexin 46 (Cx46) consists of four transmembrane domains (TM1–TM4), two extracellular loops (E1 and E2), a cytoplasmic loop (CL) between TM2 and TM3, and cytoplasmic N-terminal (NT) and C-terminal (CT) domains [38]. We detected a non-synonymous variation GJA3:p.M1V, resulting in the formation of a truncated protein, which leads to opacification in the lens. We found this variation in a patient with anterior polar cataract but not in any of the controls.
BFSP1 and BFSP2 are highly expressed intermediate filaments and when mutated can cause cataract [39]. A total of 339 variations (Table 3) have been reported in BFSP1, but only one has been associated with an autosomal recessive mutation arising from a consanguineous marriage [40] (Table 4) [30]. We detected three nucleotide variations, two of which were novel and non-synonymous (BFSP1:p.D395N, BFSP1:p.E424K); the other one (BFSP1:p.L44L) is reported. None of the mutations were pathogenic. Modeling studies of the non-synonymous changes were not possible due to the lack of a suitable homolog model. The chi-square test and Fisher's exact test were used to compare differences in the frequency of the sequence variants, between the controls and individuals with congenital cataract. Chi-square analysis and Fisher's exact tests did not show any significant difference between the groups for any of the sequence variants.
This study identified variations in 100 patients with congenital cataract in a northern Indian population. Pathogenic changes in the crystallin family accounts for 10% of the population whereas a study in southern Indian patients with congenital cataract [41] reported only 16.6% variations in the crystallin family. Connexins account for 1% of the population compared to the 5.5% reported by the earlier study [41]. Five variations (CRYBA4:p.Y67N, CRYBB1:p.D85N, CRYBB1:E75K, CRYBB1:E155K, GJA3:p.M1V) detected in this study are predicted to cause cataract. This study further confirms that the crystallin beta cluster on chromosome 22, GJA3, and BFSP1 plays a major role in maintaining lens transparency. The disease showed marked clinical and genetics (locus and allelic) heterogeneity. This study also expands the mutation spectrum of the genes associated with congenital cataract. Other genes might be involved in the growth, development, differentiation, and maintenance of lens transparency.
The study of genes related to congenital cataract and knowledge about the molecular mechanisms of their origin, in the near future, could be extended to age-related cataracts, which remain the leading cause of blindness worldwide. The accumulation of information about the physiology of the lens and the factors associated with the formation of senile cataracts acquired through genetic studies of congenital hereditary form could lead to new treatments and techniques to prevent different forms of cataract.
Acknowledgments
Study was financially supported by ICMR (Indian Council of Medical Research, New Delhi, India). Authors thank the patients and their family members for participation. Manoj Kumar is an SRF (senior research fellow awarded by ICMR: 45/19/2009/BMS) gratefully acknowledges the help.
References
- 1.Reddy MA, Francis PJ, Berry V, Bhattacharya SS, Moore AT. Molecular genetic basis of inherited cataract and associated phenotypes. Surv Ophthalmol. 2004;49:300–15. doi: 10.1016/j.survophthal.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Gilbert C, Foster A. Childhood blindness in the context of VISION 2020–the right to sight. Bull World Health Organ. 2001;79:227–32. [PMC free article] [PubMed] [Google Scholar]
- 3.Rahi JS, Sripathi S, Gilbert CE, Foster A. Childhood blindness in India: causes in 1318 blind school students in nine states. Eye (Lond) 1995;9:545–50. doi: 10.1038/eye.1995.137. [DOI] [PubMed] [Google Scholar]
- 4.Apple DJ, Ram J, Foster A, Peng Q. Elimination of cataract blindness: a global perspective entering the new millenium. Surv Ophthalmol. 2000;45:S1–196. [PubMed] [Google Scholar]
- 5.Francis PJ, Berry V, Bhattacharya SS, Moore AT. The genetics of childhood cataract. J Med Genet. 2000;37:481–8. doi: 10.1136/jmg.37.7.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gill D, Klose R, Munier FL, McFadden M, Priston M, Billingsley G, Ducrey N, Schorderet DF, Heon E. Genetic heterogeneity of the Coppock-like cataract: a mutation in CRYBB2 on chromosome 22q11.2. Invest Ophthalmol Vis Sci. 2000;41:159–65. [PubMed] [Google Scholar]
- 7.Scott MH, Hejtmancik JF, Wozencraft LA, Reuter LM, Parks MM, Kaiser-Kupfer MI. Autosomal dominant congenital cataract. Interocular phenotypic variability. Ophthalmology. 1994;101:866–71. doi: 10.1016/s0161-6420(94)31246-2. [DOI] [PubMed] [Google Scholar]
- 8.Héon E, Priston M, Schorderet DF, Billingsley GD, Girard PO, Lubsen N, Munier FL. The gamma-crystallins and human cataracts: a puzzle made clearer. Am J Hum Genet. 1999;65:1261–7. doi: 10.1086/302619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hejtmancik JF. Congenital cataracts and their molecular genetics. Semin Cell Dev Biol. 2008;19:134–49. doi: 10.1016/j.semcdb.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hejtmancik JF, Smaoui N. Molecular genetics of cataract. Dev Ophthalmol. 2003;37:67–82. doi: 10.1159/000072039. [DOI] [PubMed] [Google Scholar]
- 11.Bhat SP. Crystallins, genes and cataract. Prog Drug Res. 2003;60:205–62. doi: 10.1007/978-3-0348-8012-1_7. [DOI] [PubMed] [Google Scholar]
- 12.Goodenough DA. The crystalline lens. A system networked by gap junctional intercellular communication. Semin Cell Biol. 1992;3:49–58. doi: 10.1016/s1043-4682(10)80007-8. [DOI] [PubMed] [Google Scholar]
- 13.Hansen L, Yao W, Eiberg H, Kjaer KW, Baggesen K, Hejtmancik JF, Rosenberg T. Genetic heterogeneity in microcornea-cataract: five novel mutations in CRYAA, CRYGD, and GJA8. Invest Ophthalmol Vis Sci. 2007;48:3937–44. doi: 10.1167/iovs.07-0013. [DOI] [PubMed] [Google Scholar]
- 14.Jakobs PM, Hess JF, FitzGerald PG, Kramer P, Weleber RG, Litt M. Autosomal-dominant congenital cataract associated with a deletion mutation in the human beaded filament protein gene BFSP2. Am J Hum Genet. 2000;66:1432–6. doi: 10.1086/302872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Forshew T, Johnson CA, Khaliq S, Pasha S, Willis C, Abbasi R, Tee L, Smith U, Trembath RC, Mehdi SQ, Moore AT, Maher ER. Locus heterogeneity in autosomal recessive congenital cataracts: linkage to 9q and germline HSF4 mutations. Hum Genet. 2005;117:452–9. doi: 10.1007/s00439-005-1309-9. [DOI] [PubMed] [Google Scholar]
- 16.Graw J. Cataract mutations and lens development. Prog Retin Eye Res. 1999;18:235–67. doi: 10.1016/s1350-9462(98)00018-4. [DOI] [PubMed] [Google Scholar]
- 17.Beby F, Morle L, Michon L. M B, Edery P, Burillon C, Denis P. J Fr Ophtalmol. 2003;26:400–8. The genetics of hereditary cataract. [PubMed] [Google Scholar]
- 18.Amaya L, Taylor D, Russell-Eggitt I, Nischal KK, Lengyel D. The morphology and natural history of childhood cataracts. Surv Ophthalmol. 2003;48:125–44. doi: 10.1016/s0039-6257(02)00462-9. [DOI] [PubMed] [Google Scholar]
- 19.Forster JE, Abadi RV, Muldoon M, Lloyd IC. Grading infantile cataracts. Ophthalmic Physiol Opt. 2006;26:372–9. doi: 10.1111/j.1475-1313.2006.00370.x. [DOI] [PubMed] [Google Scholar]
- 20.Somasundaram T, Bhat SP. Developmentally dictated expression of heat shock factors: exclusive expression of HSF4 in the postnatal lens and its specific interaction with alphaB-crystallin heat shock promoter. J Biol Chem. 2004;279:44497–503. doi: 10.1074/jbc.M405813200. [DOI] [PubMed] [Google Scholar]
- 21.Berry V, Francis P, Reddy MA, Collyer D, Vithana E, MacKay I, Dawson G, Carey AH, Moore A, Bhattacharya SS, Quinlan RA. Alpha-B crystallin gene (CRYAB) mutation causes dominant congenital posterior polar cataract in humans. Am J Hum Genet. 2001;69:1141–5. doi: 10.1086/324158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sambrook J, Russell DW. Purification of nucleic acids by extraction with phenol:chloroform. CSH Protoc. 2006. [DOI] [PubMed] [Google Scholar]
- 23.Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–6. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 24.Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–4. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sunyaev S, Ramensky V, Koch I, Lathe W, 3rd, Kondrashov AS, Bork P. Prediction of deleterious human alleles. Hum Mol Genet. 2001;10:591–7. doi: 10.1093/hmg/10.6.591. [DOI] [PubMed] [Google Scholar]
- 26.Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Inc AS. Discovery Studio Modeling Environment. San Diego. 2007. [Google Scholar]
- 28.Slingsby C, Clout NJ. Structure of the crystallins. Eye (Lond) 1999;13(Pt 3b):395–402. doi: 10.1038/eye.1999.113. [DOI] [PubMed] [Google Scholar]
- 29.Billingsley G, Santhiya ST, Paterson AD, Ogata K, Wodak S, Hosseini SM, Manisastry SM, Vijayalakshmi P, Gopinath PM, Graw J, Heon E. CRYBA4, a novel human cataract gene, is also involved in microphthalmia. Am J Hum Genet. 2006;79:702–9. doi: 10.1086/507712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shiels A, Bennett TM, Hejtmancik JF. Cat-Map: putting cataract on the map. Mol Vis. 2010;16:2007–15. [PMC free article] [PubMed] [Google Scholar]
- 31.Srivastava K, Gupta R, Chaves JM, Srivastava OP. Truncated human betaB1-crystallin shows altered structural properties and interaction with human betaA3-crystallin. Biochemistry. 2009;48:7179–89. doi: 10.1021/bi900313c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mackay DS, Boskovska OB, Knopf HL, Lampi KJ, Shiels A. A nonsense mutation in CRYBB1 associated with autosomal dominant cataract linked to human chromosome 22q. Am J Hum Genet. 2002;71:1216–21. doi: 10.1086/344212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Willoughby CE, Shafiq A, Ferrini W, Chan LL, Billingsley G, Priston M, Mok C, Chandna A, Kaye S, Heon E. CRYBB1 mutation associated with congenital cataract and microcornea. Mol Vis. 2005;11:587–93. [PubMed] [Google Scholar]
- 34.Van Montfort RL, Bateman OA, Lubsen NH, Slingsby C. Crystal structure of truncated human betaB1-crystallin. Protein Sci. 2003;12:2606–12. doi: 10.1110/ps.03265903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kar R, Batra N, Riquelme MA, Jiang JX. Biological role of connexin intercellular channels and hemichannels. Arch Biochem Biophys. 2012;524:2–15. doi: 10.1016/j.abb.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Molina SA, Takemoto DJ. The role of Connexin 46 promoter in lens and other hypoxic tissues. Commun Integr Biol. 2012;5:114–7. doi: 10.4161/cib.18715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pfenniger A, Wohlwend A, Kwak BR. Mutations in connexin genes and disease. Eur J Clin Invest. 2011;41:103–16. doi: 10.1111/j.1365-2362.2010.02378.x. [DOI] [PubMed] [Google Scholar]
- 38.Mathias RT, White TW, Gong X. Lens gap junctions in growth, differentiation, and homeostasis. Physiol Rev. 2010;90:179–206. doi: 10.1152/physrev.00034.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song S, Hanson MJ, Liu BF, Chylack LT, Liang JJ. Protein-protein interactions between lens vimentin and alphaB-crystallin using FRET acceptor photobleaching. Mol Vis. 2008;14:1282–7. [PMC free article] [PubMed] [Google Scholar]
- 40.Ramachandran RD, Perumalsamy V, Hejtmancik JF. Autosomal recessive juvenile onset cataract associated with mutation in BFSP1. Hum Genet. 2007;121:475–82. doi: 10.1007/s00439-006-0319-6. [DOI] [PubMed] [Google Scholar]
- 41.Devi RR, Yao W, Vijayalakshmi P, Sergeev YV, Sundaresan P, Hejtmancik JF. Crystallin gene mutations in Indian families with inherited pediatric cataract. Mol Vis. 2008;14:1157–70. [PMC free article] [PubMed] [Google Scholar]