

Abstract
Purpose
The purpose of this project was to determine the spectrum and frequency of mutations in the small nuclear riboprotein 200 kDa gene (SNRNP200) that cause autosomal dominant retinitis pigmentosa (adRP).
Methods
A well-characterized adRP cohort of 251 families was tested for mutations in the exons and intron/exon junctions of SNRNP200 using fluorescent dideoxy sequencing. An additional 21 adRP families from the eyeGENE® Network were tested for possible mutations. Bioinformatic and segregation analysis was performed on novel variants.
Results
SNRNP200 mutations were identified in seven of the families tested. Two previously reported mutations, p.Arg681Cys and p.Ser1087Leu, were found in two families each. One family had the previously reported p.Arg681His mutation. Two novel SNRNP200 variants, p.Pro682Ser and p.Ala542Val, were also identified in one family each. Bioinformatic and segregation analyses suggested that these novel variants are likely to be pathogenic. Clinical examination of patients with SNRNP200 mutations showed a wide range of clinical symptoms and severity, including one instance of non-penetrance.
Conclusions
Mutations in SNRNP200 caused 1.6% of disease in our adRP cohort. Pathogenic mutations were found primarily in exons 16 and 25, but the novel p.Ala542Val mutation in exon 13 suggests that variation in other genetic regions is also responsible for causing dominant disease. SNRNP200 mutations were associated with a wide range of clinical symptoms similar to those of individuals with other splice-factor gene mutations.
Introduction
Retinitis pigmentosa (RP) is a highly heterogeneous form of inherited retinal degeneration that affects approximately 1.5 million individuals worldwide [1]. Individuals affected with RP initially experience night blindness, followed by progressive loss of visual fields, often culminating in legal or complete blindness. Clinical symptoms of RP include pigmentary bone spicules, optic disc pallor, attenuated blood vessels, and reduced or absent rod and cone electroretinograms (ERGs) [2].
RP is a highly heterogeneous disease that can be inherited in an autosomal dominant, autosomal recessive, X-linked, or digenic fashion and is associated with syndromic diseases such as Bardet-Biedl and Usher syndrome [3]. To date, 23 adRP, 39 arRP, and three XlRP associated genes have been identified, while the genes for several mapped loci are still unknown (RetNet). Proteins from RP-associated genes are involved in a wide variety of cellular processes, including phototransduction, photoreceptor outer segment structure, splicing, transcriptional regulation, and intraflagellar transport, to name a few [4,5].
Mutations in the SNRNP200 gene located on chromosome 2q11.2 were initially identified in 2009 as a cause of the RP33 form of adRP [6]. SNRNP200 encodes Brr2, a 200 kDa helicase involved in the catalytic activation of the RNA spliceosome [6]. Brr2 unwinds the U4/U6 duplex allowing the U4 complex to be released and U6 to bind to U2, and is involved in spliceosome disassembly. Brr2 is thought to be tightly regulated by Pfp8, another adRP-associated protein, and Snu114 GTPase [7]. A recent study suggests that the C-terminal of Prp8 is inserted into the RNA binding tunnel of Brr2 thus inhibiting Brr2 RNA binding and U4/U6 unwinding activity [8].
The hBrr2 protein is composed of two DExD/H box domains, two Sec63 domains, and a helicase superfamily C-terminal domain [9]. Studies have shown that both DExD/H domains bind Prp8 and that disease-associated mutations in Prp8 weaken binding to hBrr2 [10]. The majority of SNRNP200 mutations found to date are located in the first DExD/H domain with the exception of the p.Ser1087Leu mutation. It is reasonable to speculate that the majority of mutations might affect Prp8 interactions, but how the p.Ser1087Leu mutation, located in the first Sec63 domain, causes disease remains to be determined [6,10,11].
The human SNRNP200 gene is composed of 45 exons, which likely has contributed to the small number of reports describing mutation screening in a large number of families [11]. This project examined our well-characterized adRP cohort and additional families from the eyeGENE® Network for mutations in the SNRNP200 gene [12-14]. Seven mutations were identified in our study cohort, two of which are novel, demonstrating that SNRNP200 mutations cause an appreciable fraction of adRP.
Methods
Samples
The patient cohort used in this study has been described in detail previously [12,15-17]. This cohort consists of 251 families with a high likelihood of autosomal dominant RP. Families without previously identified mutations were selected for SNRNP200 testing. In addition, 21 families from the eyeGENE® consortium were tested as part of this study. Similar to the adRP cohort, the eyeGENE® consortium families have a provisional diagnosis of adRP, three affected generations, or two affected generations with male-to-male transmission and no previously identified mutation. DNA from a minimum of one affected individual from each family was tested for mutations.
This study was performed in accordance with the Declaration of Helsinki, and informed written consent was obtained from all participants. This research was approved by the Committee for the Protection of Human Subjects at the University of Texas Health Science Center Houston and by the respective human subjects’ review boards at each participating institution including UC San Francisco and UC Berkeley. Subjects who underwent high-resolution retinal imaging at UC San Francisco and UC Berkeley received a stipend for time spent during the imaging sessions.
Sequencing analysis
PCR primers to amplify each SNRNP200 exon and intron/exon junctions were designed using Primer 3 [18]. Genomic DNA was amplified using selected primers tailed with M13 sequences (Table 1), Amplitaq Gold® 360 Master Mix (Life Technologies, Grand Island, NY), and standard thermocycling conditions with an annealing temperature of 56 °C.
Table 1. SNRNP200 primers.
| Exon | Forward Primer Sequence (5′-3′) | Reverse Primer Sequence (5′-3′) |
|---|---|---|
| 1 |
TGTAAAACGACGGCCAGTCCACATATCCCGCTCAGAAG |
CAGGAAACAGCTATGACCCCTTCGTTTTCGTGGCTCT |
| 2 |
TGTAAAACGACGGCCAGTTTCCATCAGCTGGGGTAACT |
CAGGAAACAGCTATGACCAGCAAAAATGCTGCTCGAAT |
| 3 |
TGTAAAACGACGGCCAGTCCATGAAGCTGAGGAGAGGA |
CAGGAAACAGCTATGACCCTGCCAGCCATCAGTCAAC |
| 4 |
TGTAAAACGACGGCCAGTGGTGTGGCTCTTCACATCCT |
CAGGAAACAGCTATGACCGGGAACTCACTGACATTAAGGTTT |
| 5 |
TGTAAAACGACGGCCAGTGAGAGGTAGTCGGGGAGGAC |
CAGGAAACAGCTATGACCACTAGAGAGCTTGTTTACACTAGAAGG |
| 6 |
TGTAAAACGACGGCCAGTTTGAGGGAGAGGTTCTGTGG |
CAGGAAACAGCTATGACCACCTAGGCATGGGGAAGAAG |
| 7 and 8 |
TGTAAAACGACGGCCAGTAACTTGGTTGCAGGGTGGT |
CAGGAAACAGCTATGACCGGAGGAGGAAATAAGGCAAAA |
| 9 |
TGTAAAACGACGGCCAGTTCTTTCTACCCTGAGGATGGTT |
CAGGAAACAGCTATGACCAGCTTCATGCTGCAATCTTC |
| 10 and 11 |
TGTAAAACGACGGCCAGTGGGGCAGAAAAGGACAAGA |
CAGGAAACAGCTATGACCTTTTGGTCAATGGTTGAAATC |
| 12 |
TGTAAAACGACGGCCAGTATCCTGGCAATCCCACAATA |
CAGGAAACAGCTATGACCGGTGGGGGTGGAAATAAAAA |
| 13 |
TGTAAAACGACGGCCAGTGGGAAATAAGCCCATGGAAG |
CAGGAAACAGCTATGACCTCTTGGAAATGGGGAGAGAA |
| 14 |
TGTAAAACGACGGCCAGTCCAATGCCCTACACTCCAAT |
CAGGAAACAGCTATGACCGCCTACAGAAAAGGCAGCAA |
| 15 |
TGTAAAACGACGGCCAGTGCTTGGGATCCCTTGTCTTT |
CAGGAAACAGCTATGACCCCCAGTAGCACTCCTTGGAA |
| 16 |
TGTAAAACGACGGCCAGTATATTGCTCTTGGGGCAGTG |
CAGGAAACAGCTATGACCGGTGAATCAAACATTCGAAAGAA |
| 17 |
TGTAAAACGACGGCCAGTTGATGGTGCAACTTCTGCTT |
CAGGAAACAGCTATGACCCCTTAACCCATGGGAAACAA |
| 18 |
TGTAAAACGACGGCCAGTAGGGTCTCATGGGAGGAGAT |
CAGGAAACAGCTATGACCTGGCACAAACTTGACATTGC |
| 19 |
TGTAAAACGACGGCCAGTCTGAGGCCAACTGCCTAGAG |
CAGGAAACAGCTATGACCTCTGCTAGCTTTCCAGCATC |
| 20 |
TGTAAAACGACGGCCAGTGGATGCTGGAAAGCTAGCAG |
CAGGAAACAGCTATGACCACAATAGGGACCGACCCACT |
| 21 |
TGTAAAACGACGGCCAGTATTCATGGGAGCCTGTCATT |
CAGGAAACAGCTATGACCTCACATCAAGAGAGCAATCTGAA |
| 22 and 23 |
TGTAAAACGACGGCCAGTGGACAATAATGACATTGGGTCA |
CAGGAAACAGCTATGACCGAGCAGGAAACCACACATGA |
| 24 |
TGTAAAACGACGGCCAGTCTGGCCTGGATGCTCAGA |
CAGGAAACAGCTATGACCAGGATGCACACAGCACACAG |
| 25 |
TGTAAAACGACGGCCAGTCCGTGTGTAGAGTGGCTCAT |
CAGGAAACAGCTATGACCTTTCCCATCAGACCCTTGG |
| 26 |
TGTAAAACGACGGCCAGTAAGCCATGAAGTGGGTGGT |
CAGGAAACAGCTATGACCCACAGGCCTAGGAACAGGAA |
| 27 |
TGTAAAACGACGGCCAGTAGGGAAGACCCTACCCCTTT |
CAGGAAACAGCTATGACCCCAACAGGAGCAAGGGTAAA |
| 28 |
TGTAAAACGACGGCCAGTGAAAAGGTGAGGCTTTGCTG |
CAGGAAACAGCTATGACCCGGGACAAAGTGCAAGACAT |
| 29 |
TGTAAAACGACGGCCAGTCTTGCTGGGGACAGAGTGAT |
CAGGAAACAGCTATGACCAGACCTCCCAAGTGCCTGAG |
| 30 |
TGTAAAACGACGGCCAGTTGATCCATGTCCCAGTCC |
CAGGAAACAGCTATGACCAAAGCCTTCATACGGAATCA |
| 31 |
TGTAAAACGACGGCCAGTTTTGGCATCTCAGGTTTTCC |
CAGGAAACAGCTATGACCCGTCCTCAGACCCAAACATT |
| 32 |
TGTAAAACGACGGCCAGTCTGTTCACAAAGGGGACCTC |
CAGGAAACAGCTATGACCGACCGGGGAGAAGAAAAGAC |
| 33 |
TGTAAAACGACGGCCAGTCGTCTGTGGGGTGTCACATA |
CAGGAAACAGCTATGACCATTCCTGGCAGACACTTTGC |
| 34 |
TGTAAAACGACGGCCAGTTGCACAGCTGGCAAAGTG |
CAGGAAACAGCTATGACCCGCACCCCTCAAGTTTAACA |
| 35 |
TGTAAAACGACGGCCAGTCCATGAGGCAGTGAGCCTAC |
CAGGAAACAGCTATGACCCCCTAACACCTTTTCTTCATCCT |
| 36 |
TGTAAAACGACGGCCAGTAGTTGGGCTTCCTGAGCAT |
CAGGAAACAGCTATGACCTGTGTAAATAAGAGAGGCAGAAGTT |
| 37 |
TGTAAAACGACGGCCAGTAGGTCTCACACAGGGACCAT |
CAGGAAACAGCTATGACCCTATGGAGGCGAGAGGTGAG |
| 38 |
TGTAAAACGACGGCCAGTACGTGATCCTTTGGTGGTTC |
CAGGAAACAGCTATGACCCAGGCAGAGAAGGAGCAGAA |
| 39 |
TGTAAAACGACGGCCAGTATGACACTGCAGGGGACAG |
CAGGAAACAGCTATGACCAGGGATGCCATGTGCTCT |
| 40 |
TGTAAAACGACGGCCAGTCTTTGGCCTGAGCTGGTC |
CAGGAAACAGCTATGACCATAAAAGTGGGCGGAGTTGG |
| 41 |
TGTAAAACGACGGCCAGTGAGGGGTCAGTGGTGCTCTA |
CAGGAAACAGCTATGACCCACCTTCGGAGGAACCATAA |
| 42 |
TGTAAAACGACGGCCAGTTCTTTGCTGCCTGTGTCATT |
CAGGAAACAGCTATGACCTCAGTGATGGGGCAGTGG |
| 43 |
TGTAAAACGACGGCCAGTCCTGTTGGGCACAGCATAAT |
CAGGAAACAGCTATGACCGCCCTTGTACCAAGCACCTA |
| 44 |
TGTAAAACGACGGCCAGTCTGTACCATTTTCATGGTTGC |
CAGGAAACAGCTATGACCGCATGGACACAGGAAGCATT |
| 45 | TGTAAAACGACGGCCAGTAGCATTTGTTCTGGCATGG | CAGGAAACAGCTATGACCGGACAGCACACCTAATGAGC |
PCR products were treated with ExoSap-IT® (Affymetrix, Santa Clara, CA) and sequenced with M13 primers using BigDye® v1.1 (Life Technologies). Sequence reactions were treated with BigDye XTerminator® (Life Technologies) and run on a 3500XL Genetic Analyzer (Life Technologies). Sequence analyses were performed using SeqScape® v2.7 software and novel variants were assessed using polymorphism phenotyping (PolyPhen-2) and Sorting Intolerant From Tolerant (SIFT).
Clinical evaluations
Patients underwent standard ophthalmic evaluations including electroretinograms, and Goldmann visual field evaluation. High-resolution retinal images were obtained using spectral domain optical coherence tomography (SD-OCT) and adaptive optics scanning laser ophthalmoscopy (AOSLO) in each eye of subject UTAD701–01, and images were analyzed using custom-written software to determine cone spacing measures using previously described methods [19-21]. The cone spacing measures were compared with measures from 24 age-similar control subjects and z-scores, or standard deviations from the mean value of control subjects at a given retinal eccentricity, were calculated; z-scores greater than 2 were considered abnormal. For patient UTAD786–01 from the eyeGENE® repository, clinical information was extracted from the eyeGENE® database.
Results
Individuals tested
Seventy-five of the families tested in this study were from our previously described adRP cohort. This cohort is a set of 251 families with a high likelihood of having autosomal dominant RP. At least one individual from most of these families had been tested previously for mutations in the entire coding region of CA4, CRX, FSCN2, IMPDH1, NRL, PRPF31, RDS, RHO, ROM1, RP9, TOPORS, RPGR, and RP2, and the mutation hot spots of RP1, PRPF31, PRPF8, NR2E3, and KLHL7. Mutations were identified in 176 of the families, who were excluded from this study.
Twenty-one families were also selected from the eyeGENE Network for this study. These families met criteria similar to those for the adRP cohort: i) a diagnosis of adRP, ii) a pedigree showing three affected generations or two affected generations with the presence of male-to-male transmission, and iii) negative findings from adRP testing performed earlier in our CLIA-certified DNA diagnostic laboratory.
Sequencing of genomic DNA or whole-genome amplified (WGA) DNA from 96 probands for mutations in SNRNP200 was performed using the “gold standard” for mutation detection, traditional dideoxy sequencing. Potential disease-causing variants were identified in eight of the probands tested. Stock genomic DNAs from these eight probands and any additional available family members were tested independently for the presence of the identified SNRNP200 variant. One variant, p.Gly1162Glu, was dropped from further consideration when its presence was not confirmed in the proband’s genomic DNA. We believe this variant was introduced into the initially tested DNA sample during the whole-genome amplification process as the variant is present in all WGA DNA, but absent from genomic DNA stocks. The remaining seven SNRNP200 variants were verified in genomic DNA and are believed to be pathogenic mutations (Table 2).
Table 2. SNRNP200 mutations identified.
| Family | exon with mutations | DNA change (NM_014014.4) | Protein Change | Previously Reported | SIFT Score | Polyphen Score |
|---|---|---|---|---|---|---|
| RFS048 |
13 |
c.1625 C>T |
p.Ala542Val |
Novel |
0 |
1.00 |
| UTAD565 |
16 |
c. 2041 C>T |
p. Arg681Cys |
Yes [11] |
||
| UTAD728 |
16 |
c. 2041 C>T |
p.Arg681Cys |
Yes [11] |
||
| RFS910 |
16 |
c. 2044 C>T |
p.Pro682Ser |
Novel |
0 |
0.99 |
| UTAD543 |
16 |
c. 2042G>A |
p. Arg681His |
Yes [11] |
||
| UTAD701 |
25 |
c.3260C>T |
p.Ser1087Leu |
Yes [27] |
||
| UTAD786 | 25 | c.3260C>T | p.Ser1087Leu | Yes [20] |
SNRNP200 mutations and phenotypes
A p.Arg681Cys heterozygous mutation was identified in probands from the UTAD565 and UTAD728 families (Figure 1A). This mutation is caused by a C to T substitution at nucleotide 2041 and has been described previously in one family with adRP [11]. Sequencing of three additional affected members and one unaffected member of the UTAD565 family and two additional UTAD728 family members demonstrated that the p.Arg681Cys mutation segregates with disease, further substantiating the mutation’s pathogenicity.
Figure 1.

Families with SNRNP200 mutations. A: Two families with a c.2041C>T, p.Arg681His mutation. B: Two families with a c.3260C>T, p.Ser1087Leu mutation C: Family with a c.2042G>A, p.Arg681His mutation D: Family with c.1625C>T, p.Ala542Val mutation E: Family with a c.2044C>T, p.Pro682Ser mutation. Circles indicate women; squares indicate men. Black filled symbols are affected individuals, grey filled symbols are possibly affected individuals, and open symbols are unaffected individuals. Arrows indicated each family’s proband. Individual ID numbers corresponding to Table 2 are underneath each symbol.
Three members of UTAD565 and three members of UTAD728 were clinically examined (Table 3). The age of onset reported by these family members ranged from 4 to 35 years of age with severely reduced visual fields and non-detectable ERGs seen by the fourth decade of life. One member of the UTAD565 family, UTAD565–00, who, based on pedigree structure, must carry the p.Arg681Cys mutation, was examined at 66 years of age. Extensive clinical assessment of UTAD565–00 showed remarkably normal function of both eyes, including 20/20 visual acuity and full peripheral visual fields to all but a tiny test light (Goldmann Perimeter, I4e white target, slight horizontal constriction to 105° versus 125° normally). Her dark-adapted visual threshold sensitivity was normal centrally for both eyes, ERG responses reached fully normal amplitudes for both eyes (b-wave 440 microvolt dark-adapted and 84 microvolt light-adapted), and even the 30 Hz flicker response amplitude was normal (63 microvolt), although with delayed implicit time (35.2 msec versus normally <32 msec).
Table 3. Clinical Examination of select SNRNP200 patients.
| Patient ID | Age of onset | Age of exam | Visual acuity | Visual field (degrees across horizontal meridian) | Dark-adapted combined / mixed response wave values | Light-adapted response - Flicker response wave values |
|---|---|---|---|---|---|---|
| RFS048–5420 |
5 |
14 |
20/32 OD 20/40 OS |
18° ODS 18° OSS |
8.5 |
2.3 |
| RFS048–4884 |
21 |
40 |
20/40 OD 20/32 OS |
9° ODS* 12° OSS* |
non-detectable (at age 28) |
0.6 (at age 28) |
| RFS048–4879 |
not known |
46 |
20/40 OD 20/32 OS |
not done |
14.6 |
8.6 |
| RFS910–01 |
not known |
50 |
20/40 OD 20/50 OS |
6° ODK <5° OSK |
non-detectable |
non-detectable |
| UTAD543–01 |
13 |
76 |
10/200 OD HM @ 2 feet OS |
5° ODK 7° OSK |
non-detectable |
non-detectable |
| UTAD565–02 |
8 |
59 |
3/200 OD 20/400 OS |
<5° ODK 8° OSK |
non-detectable (at age 40) |
non-detectable (at age 40) |
| UTAD565–01 |
24 |
24 |
20/60 OD 20/50 OS |
35° ODK with temporal island
35° OSK with temporal island |
non-detectable |
2.1 |
| UTAD565–00 |
NA |
66 |
20/20 OD 20/20 OS |
145° ODK 145° OSK |
Within normal limits |
Within normal limits |
| UTAD701–01 |
4 |
34 |
20/63 OD 20/50 OS |
110° ODK+ 105° OSK+ |
severely reducedt |
11.0 |
| UTAD728–01 |
12 |
47 |
20/100 OD 20/160 OS |
20° ODS 20° OSS |
not done |
not done |
| UTAD728–02 |
20 |
54 |
20/640 OD 20/160 OS |
5° ODS 10° OSS |
not done |
not done |
| UTAD728–03 |
35 |
50 |
20/25 OD 20/20 OS |
not done |
not done |
not done |
| UTAD786–01 | 10 | 30 | 20/25 OD 20/25 OS | 100° ODK 75° OSK | not done | not done |
Age of onset was based on patient self-reporting with the exception of UTAD565–02 which was based on medical records. K=kinetic visual field by Goldmann perimeter: III4e unless otherwise noted; S=static visual field by Humphrey perimeter: 30–2, spot size III unless otherwise noted; *Spot size was used V# I4e isopter +V4e isopter tB24Blink artifact from this patient contaminated mixed a and b wave measurements such that an exact value couldn't be determined.
Probands from the UTAD701 and UTAD786 families have a heterozygous p.Ser1087Leu mutation in SNRNP200 (Figure 1B). This mutation has also been described previously and is caused by a C to T substitution at nucleotide 3260. No additional family members were available for testing. Limited clinical data for UTAD786–01 suggest relatively early onset of symptoms, but slow progression of retinal degeneration (Table 3).
UTAD701–01 retained peripheral visual field sensitivity to V4e and III4e size targets, but fields were severely constricted to the central 10° to a I4e stimulus at age 34 (Figure 2B). Visual acuity was reduced to 20/63 in the right eye and 20/50 in the left eye, and the scotopic and photopic ERG responses were severely reduced in each eye (Table 2). High-resolution retinal images using SD-OCT showed cystoid macular edema centrally; the inner segment ellipsoid zone band extended 5 degrees from the fovea nasally and temporally (Figure 2C). AOSLO images of UTAD701–01 showed preserved cone mosaics centrally within cystoid spaces (Figure 2D-E). Where cone mosaics were observed permitting quantitative analysis, cone spacing was increased by 3.5–7.9 standard deviations above the normal mean (Figure 2E).
Figure 2.

Clinical features in UTAD701–01, right eye. A: Color fundus photograph shows retinal vascular attenuation and mild disc pallor; thin white lines outline the retinal region imaged using adaptive optics scanning laser ophthalmoscopy (AOSLO); the thick horizontal white line indicates the spectral domain optical coherence tomography (SD-OCT) location. B: Goldmann visual field testing shows constriction to the I4e target (blue lines). C: Horizontal SD-OCT through the anatomic fovea shows cystoid macular edema near the fovea; the inner segment ellipsoid zone band extends about 5 degrees from the fovea with loss of outer retinal layers at greater eccentricities. D: High-resolution foveal images acquired using adaptive optics scanning laser ophthalmoscopy (AOSLO) reveal walls of cystoid spaces (dark lines). Cones are visible within the cystoid spaces. E: Cone spacing increased by 3.5–7.9 standard deviations above the normal mean (white numbers: standard deviations from normal mean, small white spots and the red circle indicate fixation locus). Scale bars=1°.
The other previously reported mutation detected in our testing was a p.Arg681His missense mutation in the UTAD543 family [11]. The individual examined in this family indicated an onset of symptoms at age 13, and when seen at age 76, had non-detectable ERGs, very restricted visual fields, and slightly more than hand motion acuity (Table 3).
Two novel SNRNP200 mutations were also detected in this study and not observed in any unaffected family members, controls, or databases. A p.Ala542Val SNRNPR200 mutation caused by a C to T substitution at nucleotide 1625 was seen in three affected members of the RFS048 family and was absent in one examined, unaffected at-risk individual (Figure 1D and Table 2). Segregation analysis of this variant along with “probably damaging” PolyPhen and “damaging” SIFT scores strongly suggest that this is the cause of RP in this family.
Clinical examination of the three affected RFS048 family members showed a variable age of onset and progression of symptoms (Figure 3B-C and Table 3). The proband, RFS048–4884, reported onset at 21 years of age progressing to severely reduced photopic flicker responses (<1 µV) and non-detectable scotopic mixed ERG responses by age 28; visual fields were constricted to <15 degrees along the horizontal meridian by age 40. RFS048–5420, the proband’s daughter, reported onset of disease symptoms at age 5 years. Her examination at age 14 showed severely reduced ERG responses (non-detectable dark-adapted rod, 2.3 µV 31 Hz flicker, and 8.5 µV mixed responses), in addition dark-adapted thresholds were elevated 2.4 log units, and visual fields were constricted to <20 horizontal degrees. In contrast, at age 46, the proband’s maternal aunt, RFS048–4879, was more moderately affected, retaining modest albeit significantly reduced ERG function (2.3 µV dark-adapted rod, 8.6 µV 31 Hz flicker, and 14.6 µV mixed responses).
Figure 3.
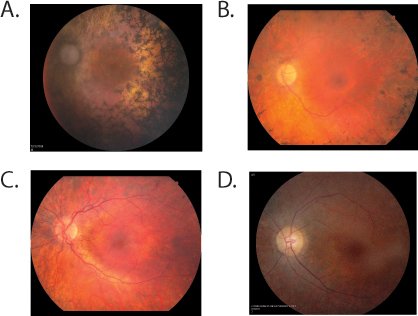
Fundus photographs from individuals with small nuclear riboprotein 200 kDa mutations. A: UTAD565-02 at age 59 with moderatively advanced retinitis pigmentosa . Her right eye fundus shows diffuse atrophy of the optic nerve and the retina outside the macula with heavy pigmentary deposits in the equator and vascular attenuation, while the macula area shows intact retinal pigment epithelium with no foveal reflex. B: RFS048–4884 at age 40 showed severe vascular attenuation and disc pallor. Extensive pigmentary disturbances were seen throughout the peripheral retina. C: RFS048-5420 at age 14 showed moderate vascular attenuation and disc pallor. Moderate pigment clumping was seen in the periphery. D: UTAD701-01 at age 34 shows vascular attenuation, mild disc pallor, and heterogeneous, mottled fundus pigment along the temporal arcades with preserved pigmentation in the central macula.
The second novel SNRNP200 variant observed in this study was a p.Pro682Ser in the RFS910 proband (Figure 1E). This variant has not been seen previously, but mutations in the two adjacent codons, p.Arg681Cys and p.Val683Leu, have been reported previously and, in the case of p.Arg681Cys, also seen in this study [11]. PolyPhen and SIFT scores for the p.Pro682Ser mutation further increase the likelihood that this is a pathogenic mutation (Table 2).
Clinical examination of the RFS910 proband at age 50 showed visual acuity was reduced to 20/40 in the right eye and 20/50 in the left eye, scotopic and photopic ERGs were non-detectible, and dark-adapted visual thresholds were elevated greater than 4 log units (Table 3). A follow-up examination at age 56 indicated a further reduction in visual acuity to 20/80 and 20/250, Goldman visual field perimetry depicted a central field constricted to <5 degrees in the right eye and was not measurable in the left eye.
Conclusions
Our analyses identified five SNRNP200 mutations in seven families with adRP. One of these mutations, p.Arg681His, has been reported previously in only one patient [11]. Identifying an additional adRP family with this mutation strengthens its likelihood of pathogenicity. We also identified two novel SNRNP200 mutations. The p.Pro682Ser mutation is adjacent to previously reported mutations, but the p.Ala542Val mutation that segregates in RFS048 is in a novel exon of the gene not previously associated with pathogenic mutations. These findings establish a SNRNP200 mutation prevalence of 1.6% in our adRP cohort, while the presence of three additional mutations in the small number (n=21) of eyeGENE consortium samples potentially suggests an even higher SNRNP200 mutation frequency.
With the exception of the p.Ser1087Leu mutation, the four remaining mutations identified in this study are located in the first DExD/H box domain. This domain has been shown to interact with Prp8 [9,22]. It is likely that alteration of this binding interaction affects splicesome assembly or function and ultimately causes photoreceptor death.
The individuals and families with SNRNP200 mutations had typical RP symptoms, including reduced or absent ERGs and decreased acuity and visual fields that ranged from mild to severe. Disease onset was reported to occur between 4 and 35 years of age with some individuals having non-detectable ERGs by age 24 and 28 (Table 3). Visual acuity was reduced in a patient with the p.Ser1087Leu mutation who retained peripheral visual field sensitivity to a large target, visual acuity was reduced. The patient had cystoid macular edema and although cones were visible near the fovea, cone spacing was significantly increased, consistent with foveal cone loss. Despite slow progression over 40 years and a preserved peripheral visual field, the fovea showed macular edema and cone loss, although SD-OCT images showed preservation of the inner segment ellipsoid zone band throughout this region.
Three of the seven pedigrees, RFS048, RFS910, and UTAD565, showed evidence of non-penetrance (Figure 1). Although DNA from these non-penetrant individuals was not available for genetic screening, one of the non-penetrant individuals in UTAD565 was examined at age 66. This family member, UTAD565–00 (Figure 1), was diagnosed as having normal vision, although a very small reduction in Goldman visual fields and a slightly delayed implicit time were detected clinically. These findings suggest that mutations in SNRP200, like those of other splicing factor genes such as PRPF31, can be non-penetrant, although often with sub-clinical findings, and raises the possibility that compensatory mechanisms may modify clinical expression of the gene as has been postulated for PRPF31 [23-26].
Acknowledgments
This work was supported through the National Ophthalmic Genotyping and Phenotyping Network or eyeGENE® (Protocol 06-EI-0236), which is funded in part by the National Institutes of Health/National Eye Institute (NIH/NEI), under Contract No. HHS-N-260–2007–00001-C. Dr. Daiger is Director of a CLIA Certified Laboratory in the eyeGENE® Network. We thank the eyeGENE® Network participants and other research families for their valuable contributions to this research. Additional support was from NIH/NEI grant R01EY007142 (S.P.D.), Center and Module grants from the Foundation Fighting Blindness (D.G.B., J.R.H., J.L.D., A.R., and S.P.D.), and grants from Research to Prevent Blindness (unrestricted grant and Physician-Scientist award, J.L.D.). Austin Roorda holds a patent on AOSLO technology through the University of Houston and the University of Rochester (US Patent # 7,118,216). The remaining authors have no commercial interests to disclose. Portions of this research were presented at the annual meeting of the Association for Research in Vision and Ophthalmology (ARVO), Invest. Ophthalmol. Vis. Sci. 2013, 54:E-Abstract 1329-A0023.
References
- 1.Haim M. Epidemiology of retinitis pigmentosa in Denmark. Acta Ophthalmol Scand Suppl. 2002;233:1–34. doi: 10.1046/j.1395-3907.2002.00001.x. [DOI] [PubMed] [Google Scholar]
- 2.Heckenlively JR, Daiger SP. Hereditary Retinal and Choroidal Degenerations. Fifth ed: Churchill Livingston Elsevier; 2007. [Google Scholar]
- 3.Daiger SP, Bowne SJ, Sullivan LS. Perspective on genes and mutations causing retinitis pigmentosa. Arch Ophthalmol. 2007;125:151–8. doi: 10.1001/archopht.125.2.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kennan A, Aherne A, Humphries P. Light in retinitis pigmentosa. Trends Genet. 2005;21:103–10. doi: 10.1016/j.tig.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 5.Ferrari S, Di Iorio E, Barbaro V, Ponzin D, Sorrentino FS, Parmeggiani F. Retinitis pigmentosa: genes and disease mechanisms. Curr Genomics. 2011;12:238–49. doi: 10.2174/138920211795860107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao C, Bellur DL, Lu S, Zhao F, Grassi MA, Bowne SJ, Sullivan LS, Daiger SP, Chen LJ, Pang CP, Zhao K, Staley JP, Larsson C. Autosomal-dominant retinitis pigmentosa caused by a mutation in SNRNP200, a gene required for unwinding of U4/U6 snRNAs. Am J Hum Genet. 2009;85:617–27. doi: 10.1016/j.ajhg.2009.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wahl MC, Will CL, Lührmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136:701–18. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 8.Mozaffari-Jovin S, Wandersleben T, Santos KF, Will CL, Lührmann R, Wahl MC. Inhibition of RNA helicase Brr2 by the C-terminal tail of the spliceosomal protein Prp8. Science. 2013;341:80–4. doi: 10.1126/science.1237515. [DOI] [PubMed] [Google Scholar]
- 9.Liu S, Rauhut R, Vornlocher HP, Lührmann R. The network of protein-protein interactions within the human U4/U6.U5 tri-snRNP. RNA. 2006;12:1418–30. doi: 10.1261/rna.55406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu T, Jin X, Zhang X, Yuan H, Cheng J, Lee J, Zhang B, Zhang M, Wu J, Wang L, Tian G, Wang W. A novel missense SNRNP200 mutation associated with autosomal dominant retinitis pigmentosa in a Chinese family. PLoS ONE. 2012;7:e45464. doi: 10.1371/journal.pone.0045464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benaglio P, McGee TL, Capelli LP, Harper S, Berson EL, Rivolta C. Next generation sequencing of pooled samples reveals new SNRNP200 mutations associated with retinitis pigmentosa. Hum Mutat. 2011;32:E2246–58. doi: 10.1002/humu.21485. [DOI] [PubMed] [Google Scholar]
- 12.Sullivan LS, Bowne SJ, Birch DG, Hughbanks-Wheaton D, Heckenlively JR, Lewis RA, Garcia CA, Ruiz RS, Blanton SH, Northrup H, Gire AI, Seaman R, Duzkale H, Spellicy CJ, Zhu J, Shankar SP, Daiger SP. Prevalence of disease-causing mutations in families with autosomal dominant retinitis pigmentosa (adRP): a screen of known genes in 200 families. Invest Ophthalmol Vis Sci. 2006;47:3052–64. doi: 10.1167/iovs.05-1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bowne SJ, Sullivan LS, Gire AI, Birch DG, Hughbanks-Wheaton D, Heckenlively JR, Daiger SP. Mutations in the TOPORS gene cause 1% of autosomal dominant retinitis pigmentosa (adRP). Mol Vis. 2008;14:922–7. [PMC free article] [PubMed] [Google Scholar]
- 14.Bowne SJ, Sullivan LS, Koboldt DC, Ding L, Fulton R, Abbott RM, Sodergren EJ, Birch DG, Wheaton DH, Heckenlively JR, Liu Q, Pierce EA, Weinstock GM, Daiger SP. Identification of disease-causing mutations in autosomal dominant retinitis pigmentosa (adRP) using next-generation DNA sequencing. Invest Ophthalmol Vis Sci. 2011;52:494–503. doi: 10.1167/iovs.10-6180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sullivan LS, Bowne SJ, Seaman CR, Blanton SH, Lewis RA, Heckenlively JR, Birch DG, Hughbanks-Wheaton D, Daiger SP. Genomic rearrangements of the PRPF31 gene account for 2.5% of autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2006;47:4579–88. doi: 10.1167/iovs.06-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Friedman JS, Ray JW, Waseem N, Johnson K, Brooks MJ, Hugosson T, Breuer D, Branham KE, Krauth DS, Bowne SJ, Sullivan LS, Ponjavic V, Granse L, Khanna R, Trager EH, Gieser LM, Hughbanks-Wheaton D, Cojocaru RI, Ghiasvand NM, Chakarova CF, Abrahamson M, Goring HH, Webster AR, Birch DG, Abecasis GR, Fann Y, Bhattacharya SS, Daiger SP, Heckenlively JR, Andreasson S, Swaroop A. Mutations in a BTB-Kelch protein, KLHL7, cause autosomal-dominant retinitis pigmentosa. Am J Hum Genet. 2009;84:792–800. doi: 10.1016/j.ajhg.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Churchill JD, Bowne SJ, Sullivan LS, Lewis RA, Wheaton DK, Birch DG, Branham KE, Heckenlively JR, Daiger SP. Mutations in the X-linked retinitis pigmentosa genes RPGR and RP2 found in 8.5% of families with a provisional diagnosis of autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2013;54:1411–6. doi: 10.1167/iovs.12-11541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–86. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- 19.Duncan JL, Talcott KE, Ratnam K, Sundquist SM, Lucero AS, Day S, Zhang Y, Roorda A. Cone structure in retinal degeneration associated with mutations in the peripherin/RDS gene. Invest Ophthalmol Vis Sci. 2011;52:1557–66. doi: 10.1167/iovs.10-6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roorda A, Zhang Y, Duncan JL. High-resolution in vivo imaging of the RPE mosaic in eyes with retinal disease. Invest Ophthalmol Vis Sci. 2007;48:2297–303. doi: 10.1167/iovs.06-1450. [DOI] [PubMed] [Google Scholar]
- 21.Duncan JL, Zhang Y, Gandhi J, Nakanishi C, Othman M, Branham KE, Swaroop A, Roorda A. High-resolution imaging with adaptive optics in patients with inherited retinal degeneration. Invest Ophthalmol Vis Sci. 2007;48:3283–91. doi: 10.1167/iovs.06-1422. [DOI] [PubMed] [Google Scholar]
- 22.Pena V, Liu S, Bujnicki JM, Lührmann R, Wahl MC. Structure of a multipartite protein-protein interaction domain in splicing factor prp8 and its link to retinitis pigmentosa. Mol Cell. 2007;25:615–24. doi: 10.1016/j.molcel.2007.01.023. [DOI] [PubMed] [Google Scholar]
- 23.Rio Frio T, Civic N, Ransijn A, Beckmann JS, Rivolta C. Two trans-acting eQTLs modulate the penetrance of PRPF31 mutations. Hum Mol Genet. 2008;17:3154–65. doi: 10.1093/hmg/ddn212. [DOI] [PubMed] [Google Scholar]
- 24.Venturini G, Rose AM, Shah AZ, Bhattacharya SS, Rivolta C. CNOT3 is a modifier of PRPF31 mutations in retinitis pigmentosa with incomplete penetrance. PLoS Genet. 2012;8:e1003040. doi: 10.1371/journal.pgen.1003040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rio Frio T, Wade NM, Ransijn A, Berson EL, Beckmann JS, Rivolta C. Premature termination codons in PRPF31 cause retinitis pigmentosa via haploinsufficiency due to nonsense-mediated mRNA decay. J Clin Invest. 2008;118:1519–31. doi: 10.1172/JCI34211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berson EL, Simonoff EA. Dominant retinitis pigmentosa with reduced penetrance. Further studies of the electroretinogram. Arch Ophthalmol. 1979;97:1286–91. doi: 10.1001/archopht.1979.01020020028006. [DOI] [PubMed] [Google Scholar]
- 27.Zhao C, Bellur DL, Lu S, Zhao F, Grassi MA, Bowne SJ, Sullivan LS, Daiger SP, Chen LJ, Pang CP, Zhao K, Staley JP, Larsson C. Autosomal-dominant retinitis pigmentosa caused by a mutation in SNRNP200, a gene required for unwinding of U4/U6 snRNAs. Am J Hum Genet. 2009;85:617–27. doi: 10.1016/j.ajhg.2009.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]