

Abstract
Chronic injury to the kidney causes kidney fibrosis with irreversible loss of functional renal parenchyma and leads to the clinical syndromes of chronic kidney disease (CKD) and end-stage renal disease (ESRD). Regardless of the type of initial injury, kidney disease progression follows the same pathophysiologic processes characterized by interstitial fibrosis, capillary rarefaction and tubular atrophy. Myofibroblasts play a pivotal role in fibrosis by driving excessive extracellular matrix (ECM) deposition. Targeting these cells in order to prevent the progression of CKD is a promising therapeutic strategy, however, the cellular source of these cells is still controversial. In recent years, a growing amount of evidence points to resident mesenchymal cells such as pericytes and perivascular fibroblasts, which form extensive networks around the renal vasculature, as major contributors to the pool of myofibroblasts in renal fibrogenesis. Identifying the cellular origin of myofibroblasts and the key regulatory pathways that drive myofibroblast proliferation and transdifferentiation as well as capillary rarefaction is the first step to developing novel anti-fibrotic therapeutics to slow or even reverse CKD progression and ultimately reduce the prevalence of ESRD. This review will summarize recent findings concerning the cellular source of myofibroblasts and highlight recent discoveries concerning the key regulatory signaling pathways that drive their expansion and progression in CKD.
Keywords: Pericyte, Myofibroblast, Fibrosis, Interstitium, Capillary rarefaction, Transforming growth factor beta, TGF-β, Wingless/Int (Wnt) signaling, Platelet derived growth factor, PDGF, Hedgehog signaling, Matrix producing cells, Chronic kidney disease, CKD, End-stage renal disease, ESRD, Pathobiology
Introduction
Chronic kidney disease (CKD) is caused by a wide variety of primary renal diseases but regardless of the type of initial injury the stereotyped response of the kidney is characterized by expansion of myofibroblasts and accumulation of extracellular fibrotic matrix. The initial recruitment of myofibroblasts is initially beneficial for the normal repair process after injury, however in the case of chronic injury this response is maladaptive, leading to overabundant synthesis of matrix which destroys normal kidney architecture, causing CKD progression and end-stage renal disease (ESRD). Extracellular matrix is produced both in the interstitial space between tubules (tubulo-interstitium) and in glomeruli where it causes glomerulosclerosis. This fibrotic process occurs in parallel with capillary rarefaction, inflammation and tubular atrophy. In recent years various epidemiologic studies have brought the connection between acute kidney injury and CKD to ESRD progression to widespread attention [1-3]. Because CKD progression is thought to be primarily driven by a gradual worsening of tubulo-interstitial fibrosis, [1] a therapeutic strategy to inhibit the expansion and synthetic capability of myofibroblasts should hold promise in slowing disease progression. Therefore understanding the cellular and molecular mechanisms of kidney fibrosis (recently reviewed in [4, 5]). will help to develop novel targeted therapies. In this review we will discuss the recent literature regarding the cellular source of myofibroblasts and summarize the key cell signaling pathways and pathophysiologic processes that contribute and regulate myofibroblast expansion culminating in the progression of fibrosis and ESRD.
The cellular origin of matrix producing cells
It has been widely accepted that activated fibroblasts called myofibroblasts are the pathologic matrix producing cells in fibrosis across different organs and tissues. In healthy non-injured kidney myofibroblasts are virtually absent, however after injury they expand in a fashion reminiscent of neoplastic disease, destroying kidney architecture and causing renal failure and ESRD. Myofibroblast cells are characterized by a pronounced rough endoplasmic reticulum and a large nucleolus - reflecting their high synthetic and proliferative capability [6]. They express alpha-smooth muscle actin (α-SMA), which is organized in myofilaments and morphologically described as stress-fibers [7].
The source of myofibroblasts in fibrosis is controversial in part because no single marker specifically identifies all myofibroblasts. Work of different groups implicates a variety of cellular origins for myofibroblasts, including epithelial and endothelial cells, circulating fibrocytes of bone-marrow origin, resident fibroblasts, pericytes and perivascular fibroblasts (Figure 1). For many years tubular epithelial cells were thought to be major contributors to the pool of myofibroblasts [8]. This process, called epithelial to mesenchymal transition (EMT), plays an important role in cancer progression and invasion and describes how terminally differentiated epithelial cells de-differentiate into mesenchymal cells with increased migratory potential [9]. Indeed, epithelial injury plays an important role in renal fibrogenesis. Injured epithelial cells start secreting pro-fibrotic cytokines and growth factors such as TGF-β1, CTGF, PDGF, Sonic/ Indian Hedgehog and FGF, that promote inflammation, immune response, and the activation, transformation and proliferation of pathogenic myofibroblasts [10]. Selective ablation of tubular epithelial cells using a genetic diphtheria toxin receptor strategy results in tubulo-interstitial fibrosis, providing direct evidence that tubular injury is a primary cause of interstitial fibrosis [11]. Many publications reported EMT as a source of kidney myofibroblasts using immunostaining for co-localization of epithelial and mesenchymal markers or lineage tracing techniques [12-14], recently endothelial cells (EndoMT) have also been reported as source of renal myofibroblasts [15]. There is no doubt that injured epithelial cells de-differentiate and express some mesenchymal markers of more primitive states, including vimentin and FSP1, however little convincing data exists that tubular epithelial cell cross the basement membrane and trans-differentiate into myofibroblasts [16]. Indeed, most recent publications provide evidence against EMT as a source of myofibroblasts in kidney [17].
Fig. 1. The cellular source of fibrosis driving myofibroblasts.
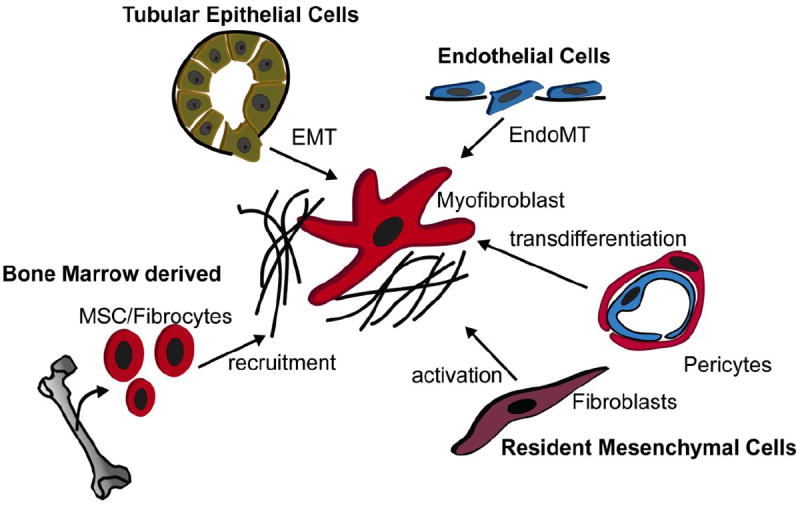
Multiple origins of myofibroblasts have been discussed in renal fibrogenesis including endothelial cells via endothelial to mesenchymal transition (EndoMT), epithelial cells via epithelial to mesenchymal transition (EMT), bone marrow derived cells including circulating fibrocytes and mesenchymal stem cells (MSC) and resident mesenchymal cells as fibroblasts and pericytes.
Circulating bone marrow derived progenitors, as fibrocytes and mesenchymal stem cells, have also been discussed as contributors to kidney myofibroblasts. Whereas bone-marrow transplantation work using Collagen-1α1 or Collagen-1α2 reporter chimeras [18, 19] shows that there is no significant contribution of circulating bone marrow derived cells to the renal myofibroblast pool a recent publication reports bone-marrow cells contribute to as many as 35% to the renal myofibroblast pool [20]. Bone-marrow transplantation experiments using α-SMA-RFP donors and wildtype recipients resulted in 35% overlay of RFP with α-SMA immunostaining after UUO[20]. As bone-marrow mesenchymal stromal cells (MSC) express α-SMA the authors concluded that these cells are the bone-marrow cells that contribute to the renal myofibroblast pool. Despite this intriguing result, this paper failed to show that transplanted MSC engraft and that the α-SMA expressing cells after transplantation are indeed MSC (i.e. fluorescence activated cell sorting of RFP+ cells with co-staining of MSC marker pattern and tri-lineage differentiation capability). Since no single marker for MSC is currently reported that would allow for proper genetic lineage tracing experiments, the gold standard approach for identifying cell hierarchies, more studies are needed to prove that BM-MSC contribute to the myofibroblast pool in models of fibrotic kidney injury. Also, experiments are needed to exclude that the possibility that injected bone marrow cells home to the kidney after transplantation. Lineage tracing experiments with whole bone-marrow transplantation of wildtype mice with a fraction of sorted fluorescent-MSC will provide more definitive evidence to support the hypothesis that bone marrow cells contribute to the renal myofibroblast pool.
The most straighforward and traditional explanation is that local resident mesenchymal cells such as fibroblasts are the major source of myofibroblasts in kidney fibrosis [21]. It is our opinion that this hypothesis is the most plausible one. In recent years another resident mesenchymal cell has become the focus of fibrosis research, the pericyte or perivascular fibroblast that is abundant throughout the kidney and forms an extensive network around capillaries [17, 19, 22, 23]. We performed genetic lineage analysis using an inducible CreERT2 driven by the FoxD1 locus to genetically label and track interstitial pericytes [17]. FoxD1 is expressed in the metanephric mesenchyme from gestational day E11.5 and FoxD1+ cells give rise to pericytes and perivascular fibroblasts, vascular smooth muscle cells and mesangial cells but they do not have epithelial or endothelial potential [24, 25]. We demonstrated that after unilateral ureteral obstruction, these genetically labeled FoxD1+ pericytes and perivascular fibroblasts acquire α-SMA expression and are the major contributor to the myofibroblast pool in fibrosis [17, 26]. Pericytes have subsequently also come into the focus of groups interested in scarring of other tissues. Goritz et al. demonstrated that pericytes become myofibroblasts in spinal cord scarring [22] and Dulauroy et al. reported that the majority of myofibroblasts in muscle and dermal fibrogenesis are derived from PDGFRα+, ADAM12+ pericytes [27].
Recently LeBleu et al. reported that pericytes do not contribute to renal fibrosis [20]. In their study they performed lineage tracing experiments using NG2-YFP and PDGFRβ-RFP mice showing a significant increase of NG2- and PDGFRβ-expressing cells after unilateral ureteral obstruction [20]. However, they also generated mice where viral thymidine kinase is expressed under the control of the NG2 promoter or the PDGFRβ promoter in order to ablate these cells while they are proliferating. They show that ablation of proliferating NG2 or PDGFRβ expressing cells after induction of renal fibrosis did not result in a reduction of kidney fibrosis [20]. As ablation of α-SMA expressing cells did reduce kidney fibrosis by 50% authors concluded that resident fibroblasts are the major source of kidney myofibroblasts [20]. Importantly, this study confirms that myofibroblasts derived through EMT do not play an important role in fibrosis. However, it also contradicts other work showing that PDGFRβ + cells produce extracellular matrix and contribute to the renal myofibroblast pool [17, 28] and also our own experiments showing that the majority if not all α-SMA expressing cells co-label with PDGFRβ in murine kidney fibrosis models (data not shown). Part of this confusion arises from how pericytes are defined: PDGFRβ and NG2 are not in fact specific pericyte markers and are expressed by other renal cell-types [29, 30], PDGFRβ for example has been reported to be expressed in the rat kidney-fibroblast cell-line NRK49F [31] suggesting that resident kidney fibroblasts express PDGFRβ. This raises two important questions: Are all Foxd1-derived stromal cells pericytes, or just a subset and could this explain these discordant results, and also could mosaic expression of the thymidine kinase transgenic allele have influenced the interpretation of the ablation experiments [20]? Clearly, further studies are needed to better define the cellular source of myofibroblasts in kidney fibrosis, with particular focus on rigorous definition of markers that distinguish interstitial resident fibroblasts, pericytes, perivascular fibroblasts and bone-marrow derived fibroblasts.
Capillary rarefaction triggers fibrosis progression
Peritubular capillary rarefaction (Figure 2 A) is, in addition to interstitial fibrosis and tubular atrophy, another hallmark of CKD [32, 33]. It is thought that peritubular capillary rarefaction is a major driver of kidney fibrosis and CKD progression because it may result in reduced nutrient and oxygen supply to tubular cells, which increases organ injury [33-35]. In humans, loss of peritubular capillaries correlates with the severity of fibrosis [36, 37]. One hypothesis for the proposed mechanism underlying the progression of capillary rarefaction during fibrogenesis is that pericytes are important for vascular stabilization and regulation, and are directly attached to the endothelial cells of the renal peritubular capillary bed. However, after injury pericytes detach themselves and become ECM secreting myofibroblasts, thus causing instability of the capillary bed and subsequent rarefaction [33, 38, 39] (Figure 2 B). Recent work demonstrates that after injury kidney pericytes upregulate the expression of a disintegrin and metalloprotease with thrombospondin motifs-1 (ADAMTS1) while downregulating its inhibitor, tissue inhibitor of metalloproteinase 3 (TIMP3)[38]. TIMP3 was able to stabilize three-dimensional tubular networks of primary kidney pericytes and human umbilical cord endothelial cells (HUVECs), whereas ADAMTS1 destabilized these capillary networks [38]. Furthermore, knockout of Timp3 in mice resulted in a spontaneous fibrotic phenotype and ischemic kidney injury associated with decreased capillary density with increased interstitial fibrosis compared to wild type mice [38]. Another example of endothelial pericyte crosstalk may exist between the EphrinB4 receptor and ephrinB2 ligand as endothelial cells of mice lacking the intracellular signaling domain of ephrinB2 showed decreased proliferative and migratory potential, whereas pericytes of these mice showed enhanced proliferation and migration [32]. More studies are needed to evaluate the important signaling pathways between pericytes and endothelial cells. However, targeting the interplay between these two cell-types appears to be a promising future therapeutic strategy to slow CKD progression in humans.
Fig. 2. Capillary rarefaction in kidney fibrosis.
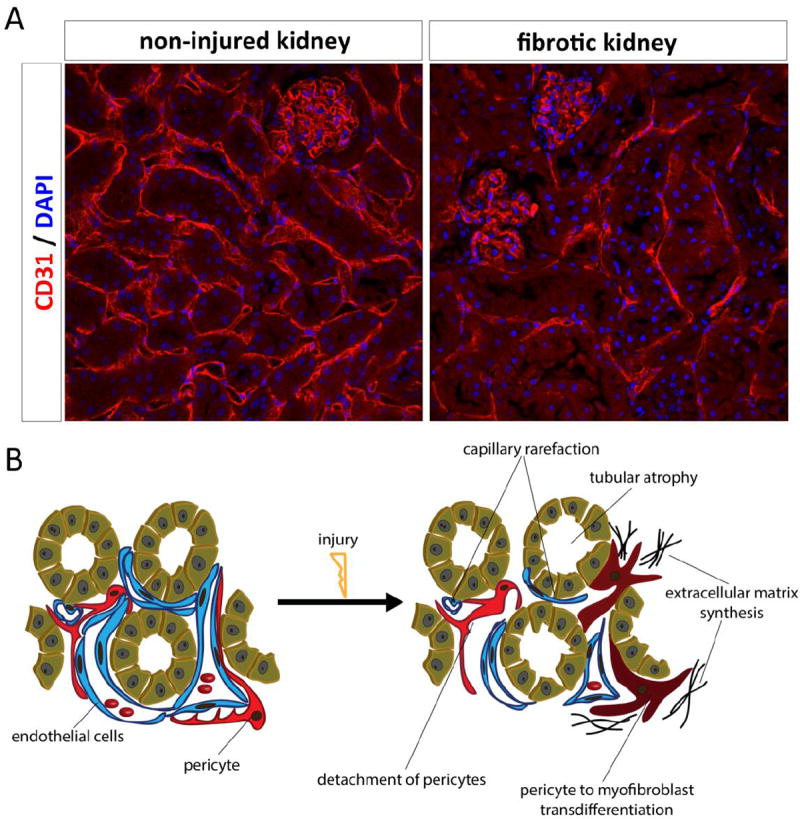
A: Staining for CD31 in a healthy kidney and a fibrotic kidney 8 weeks after ischemia demonstrates a dramatically rarefaction of peritubular capillaries. B: Development of fibrogenesis and capillary rarefaction in the kidney.
Multiple signaling pathways are involved myofibroblast transdifferentiation and expansion
Research over the last few decades provides strong evidence that multiple signaling pathways are involved in myofibroblast transdifferentiation and expansion. We will focus on recent developments within four pathways, Hedgehog-Gli (Hh-Gli), Transforming Growth factor beta (TGF-β), Platelet Derived Growth Factor (PDGF) and Wingless/Int (Wnt). Other important pathways in myofibroblast recruitment and expansion have been described, such as Notch, connective tissue growth factor (CTGF), endothelin and the renin-angiotensin-aldosterone system but these are outside the scope of the current review.
Transforming growth factor beta (TGF-β)
TGF-β ligands are ubiquitously secreted proteins that exist in at least three isoforms (called TGFβ1, TGFβ2 and TGFβ3) and actively contribute to the regulation of organ development, tumorigenesis, immune system, and fibrosis. The action of transforming-growth-factor (TGF)-beta is vital for the induction of fibrosis and its related pathogenetic effects in different organ systems like arthritis [40], diabetic nephropathy [41], idiopathic pulmonary fibrosis [42], and myocardial fibrosis [43]. TGF-β is secreted by, and can signal to, essentially all cells with wide-ranging effects [44]. It can regulate the immune system and constrain the proliferation of various cell types [45]. TGFβ coordinates extracellular matrix (ECM) production where its overexpression causes fibrosis [46, 47]. Cells secrete separate TGFβ homodimer and propeptide-derived homodimers where both homodimers are non-covalently linked [44]. The latter homodimer prevents TGFβ binding to its receptors, thus it is called latency-associated peptide (LAP) [48]. Another layer in the cell membrane control of TGFβ signaling is through latent TGFβ binding protein (LTBP), which is a microfibril-associated protein that tethers the TGFβ-LAP complex to the ECM [49] and is critical for its proper placement in the ECM [50]. TGFβ ligands binding to its receptor triggers assembly of a complex whose components include Serine/Threonine protein kinases [51]. In this receptor complex, type II receptors phosphorylate the type I components, which then propagate the signal by phosphorylating SMAD2 and SMAD3 [52]. These form a complex with SMAD4 which enters the nucleus and recognize the DNA motif CAGAC (the SMAD-binding element (SBE)) [53].
TGFβ can also activate other pathways that are collectively referred to as non-canonical TGFβ signaling, including the MAPK, PI3K, and RHO pathways. TGFβ can induce epithelial to mesenchymal transition (EMT) which has consequences for disease progression in cancer [54]. A Phase I trial with fresolimumab, a human monoclonal antibody that inactivates all forms of transforming growth factor-β (TGF-β), showed promising effectiveness in patients with resistant focal segmental glomerulosclerosis [55]. Eli Lilly is investigating a TGFβ1 ligand-selective blocking antibody, LY2382770, in a Phase II trial for kidney fibrosis in diabetic nephropathy. An alternative approach to suppress TGFβ signaling is gene transfer of antagonizing signaling molecules, such as the inhibitory SMAD7. Using this technique, a group of researchers showed that forced expression of SMAD7 inhibits fibrosis in models of diabetic kidney disease [56]. In Idiopathic Pulmonary Fibrosis, the progressive fibrotic reaction is linked to epithelium-dependent fibroblast activation where TGFβ plays a vital role [57]. Perfinidone, which inhibits TGFβ in vitro, induced improvement in lung function in pooled data from 2 phase III clinical trials [58]. Renal fibrosis is thought to be driven by TGFβ where it induces ECM production and ultimately kidney failure [59, 60]. Target blockade of TGFβ pathway by overexpressing smad7 resulted in decreased fibrosis in a UUO model in rats [61]. Topical application of P144, peptide inhibitor of TGFβ1, reduced skin fibrosis in an established mouse model of scleroderma [62]. Fibrotic diseases induced by TGFβ are complex and show individual genetic predisposition that necessitates investigating surrogate markers of TGFβ activation before initiating treatment that is often systemic [63]. Further investigation is required to understand the therapeutic utility of TGFβ pathway inhibition in fibrotic disease states.
Wingless/Int (Wnt) signaling
The Wnt/β-catenin signaling pathway is an evolutionarily conserved signaling pathway that regulates a variety of cellular outcomes in development and disease. The roles of Wnt/β-catenin signaling in embryogenesis, adult tissue homeostasis, and stem cell renewal are well known [64]; however, the consequences of inappropriate Wnt/β-catenin signaling to fibrogenesis are now under intensive investigation. Wnt proteins bind a heterodimeric receptor complex, consisting of a Frizzled (Fz) and an LRP5/6 protein. When Fz/LRP is not engaged, GSK3 (serine/Threonine kinase) phosphorylates Axin-bound β-catenin [64]. Axin is a scaffold of the destruction complex, which interacts with the tumor suppressor protein, APC, and regulates the stability of cytoplasmic β-catenin. Consequently, β-catenin is ubiquitinated and directed for prompt destruction by the proteasomes hindering the activation of β-catenin target genes in the nucleus [65]. Upon activation of the canonical Wnt pathway, a crucial first step in Wnt signal transduction is binding of Axin to the cytoplasmic tail of LRP6 [66]. This causes dismantling of the destruction complex, allowing β-catenin to accumulate in the cytoplasm and then translocate to the nucleus to bind to its target genes [67]. Cutaneous wound healing studies in mice reveal that β-catenin signaling is activated as a consequence of injury [68]. In another study, non-restricted activation of β-catenin using a stabilized mutant that resists degradation was sufficient to induce excessive collagen synthesis, mimicking aggressive fibromatoses in humans [69]. In a mouse model of Duchenne Muscular Dystrophy, loss of dystrophin leads to upregulation of Wnt-activity in the serum, which then promotes expansion of Sca1+ stromal cells and leads to skeletal muscle fibrosis [70]. In both systemic sclerosis fibrotic lungs and idiopathic pulmonary fibrosis (IPF), fibroblasts express less secreted frizzled-related protein (SFRP1) when compared with controls [71].
SFRPs structurally mimic Wnt frizzled receptors and can prevent β-catenin signaling by acting as Wnt-decoy receptors [72]. Many studies have identified Wnt pathway inhibitors for potential therapeutic use. ICG-001 interacts with cyclic AMP response element binding (CREB)-binding protein (CBP) and specifically blocks the β-catenin activation [73]. In lung fibrosis, ICG-001 reduced the fibrotic phenotype after tracheal bleomycin instillation [74]. Pyrvinium, a potent small molecule Wnt inhibitor, promotes wound repair and remodeling in a coronary artery ligation model of myocardial infarction [75]. In mouse models of kidney fibrosis, many wnt genes are upregulated and the canonical Wnt/β-catenin pathway is activated. [76-78]. Inhibiting the Wnt pathway by gene delivery of DKK1 attenuates renal fibrosis and inhibits myofibroblast activation in a model of obstructive nephropathy[77]. The Liu group has gone on to show that targeted inhibition of the Wnt pathway by IG-001 hindered progression of interstitial fibrosis in UUO, providing a clear translational strategy for treatment of chronic kidney disease [79]. Our lab has shown that exogenous Wnt4 drives myofibroblast differentiation of a pericyte-like cell line [78]. We produced a mouse model with targeted activation of canonical Wnt/β-catenin signaling in interstitial pericytes and fibroblasts. Spontaneous myofibroblast differentiation in the absence of injury was observed in their kidneys [78]. Conversely, inhibition of Wnt with DKK-1 was shown to inhibit pericyte proliferation, activation, and differentiation independent of canonical β-catenin signaling. The authors suggest that activated LRP-6 closely associates with PDGFR, TGFβR, and CTGF receptors at the cell membrane and Wnts may modulate these signaling pathways to modulate pericyte activity [80]. In rat models of chronic renal allograft failure, both the canonical Wnt and Wnt-Ca2+ pathways were differentially modulated which resulted in increased fibronectin expression and activation of fibroblasts through TGFβ1[81]. Evidently, more research is needed to understand the pathophysiology of Wnt signaling in fibrosis and define appropriate therapeutic targets.
Platelet derived growth factor (PDGF) signaling
The PDGF growth factor system is one of the most well studied signaling pathways in the kidney (thoroughly reviewed in [82, 83]) and consists of four ligand isoforms (PDGF-A, -B, -C and – D) and two receptors (PDGFR-α and −β). The entire PDGF system including all four PDGF isoforms and both receptors are upregulated in various models of renal disease including unilateral ureteral obstruction, Thy 1.1 glomerulonephritis, lupus and ischemia-reperfusion injury [82]. There is strong evidence that the PDGF system is involved in regulation of cell-proliferation and migration of myofibroblasts [84]. It has been reported that anti-PDGF-C treatment ameliorates fibrosis in the mouse unilateral ureteral obstruction model [85] and inhibition of PDGF-D prevented renal scarring in a glomerulonephritis model [83]. Chen et al. reported recently that in kidney fibrosis all four PDGF isoforms are induced broadly throughout kidney, with both receptors expressed by pericytes and myofibroblasts. Inhibition of PDGF signaling using an antibody against either receptor PDGFR-α or −β attenuated macrophage infiltration and fibrosis while using a combination of both anti PDGFR-α and −β antibodies did not show an additional effect [28]. Moreover treatment with the PDGFR tyrosine kinase inhibitor imatinib did show the same effect with reduced fibrosis and macrophage infiltration [28]. Altogether, there are multiple lines of evidence that the PDGF system represents a viable therapeutic target in renal fibrogenesis.
Hedgehog-signaling
In 1980 Nusslein-Vollhard et al. identified Hh performing genetic screens in Drosophila [86]. Since then, work of many groups reported the involvement of the Hh signaling pathway in development and disease (recently reviewed in [87]). The Hh family of proteins include three ligands: sonic hedgehog (Shh), Indian hedgehog (Ihh) and desert hedgehog (Dhh). The Hh ligands act by binding to their membrane receptor Patched (Ptc), thereby releasing a tonic inhibition of Ptc on the transmembrane protein Smoothened (Smo), activated Smo translocated into the primary cilium, accumulates and increases the cilia dwell time of Suppressor-of-fused (SUFU) and the Gli proteins Gli2 and Gli3[87]. This process finally leads to the dissociation of the Gli-SUFU complex and the transport of full-length Gli2 and Gli3 into the nucleus where the transcription of hedgehog target genes, including Gli1 and Ptch1 is induced [87]. Gli proteins are the primary effectors of Hh signaling and all Gli proteins (Gli1, Gli2 and Gli3) contain an activator domain at their c-terminus, whereas only Gli2 and Gli3 have an N-terminal repressor domain [87].
Emerging evidence implicates a critical role of the Hh pathway in solid organ fibrosis. There are strong lines of evidence that in cancer and fibrosis Hh ligands are secreted by epithelial cells and signal to the surrounding interstitial mesenchymal cells. In carcinogenesis, for example, Hh ligands from cancer cells act on adjacent stromal cells to promote the tumor micro environment [88]. In liver fibrosis it has been reported that injured hepatocytes and cholangiocytes secrete Hh ligands and Hh responsive hepatic stellate cells undergo transdifferentiation into myofibroblasts and proliferate [89]. A similar mechanism might exist in lung fibrogenesis with injured epithelial cells secreting Shh signaling to interstitial cells that express the receptor Ptch1 [90].[91].
In renal fibrosis we, and others, have recently shown that injured tubular-epithelial cells secrete Hh ligands Ihh and Shh that signal to Hh responsive interstitial pericytes/fibroblasts [92, 93]. As several drugs that antagonize the Hh pathway are already in clinical development, primarily as treatment options in cancer [94], these agents might be promising to treat fibrotic kidney disease. Targeting canonical Hh signaling via inhibition of smoothened with the cyclopamine derivate IPI-926, which has improved half-life and increased potency when compared to cyclopamine had no effect on the severity of kidney fibrosis in our hands[92]. However, others reported that inhibition of smoothened with cyclopamine reduced kidney fibrosis [93]. The reasons for this remain unclear and more studies are needed to dissect the role of canonical Hh signaling in renal fibrosis.
However, the cancer literature suggests that non-canonical activation of Gli through alternate pathways such as PDGF, EGF and TGF signaling might be important [95]. As all of these pathways are activated during renal fibrogenesis an agent that acts downstream in the Hh pathway directly at the Gli protein might be superior to inhibition of Ptch1.
Therapeutic development
As mentioned, tubulo-interstitial fibrosis is a hallmark of all chronic kidney diseases, including glomerulopathies. There are a variety of biological mechanisms underlying the progression of fibrosis and unfortunately, these mechanisms are incompletely understood, and pose a challenge to targeted drug development for this disease. Overall, few treatment regimens have shown a reduction in tubulo-interstital fibrosis in human renal disease and little evidence exists to show that existing treatments can induce regeneration of kidney. Clinical trials in CKD and ESRD patients are difficult and often under-powered [96] and it is unclear whether inhibition of fibrosis will effect hard endpoints such as mortality (randomized controlled clinical trials that effect mortality in ESRD have been reviewed previously [96]). Although not necessarily anti-fibrotic, treatment of chronic kidney diseases with angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs) has shown significant renoprotective effect in randomized clinical trials [97]. In the Benazepril trial the primary benefits observed were reduction in protein excretion, and a decrease in the percentage of patients with a doubling of serum creatinine or progression to dialysis [98]. Likewise, in the Ramipril Efficacy in Nephropathy (REIN) trial the ACE inhibitor ramipril was shown to slow the rate of decline of renal function as measured by GFR, and the patients treated with ramipril had significant decrease in the incidence of end-stage renal disease compared to patients receiving placebo and other anti-hypertensive drugs [99]. As noted with other CKD trials testing ACE inhibition, the patients who benefit most are those with more pronounced proteinuria [100]. While it is not known if fibrotic readouts are altered in CKD patients treated with ACE inhibitors or ARBs, there are reports of reduction in fibrosis and regeneration of glomerular tissue in rodent models of kidney disease [101].
Therapies targeting the renin-angiotensin system are widely accepted for use in CKD, however therapeutics addressing other cell signaling pathways are currently being developed, although some potentially promising candidates have suffered high profile failures. For instance, bardoxolone methyl, developed by Reata/Abbot, is an Nrf2 pathway inducer that was shown to have beneficial effects in animal models of acute kidney injury and was tested to treat diabetic nephropathy clinical trials [102, 103]. Preliminary results in the BEAM trial showed that the drug could significantly increase eGFR, however this was tempered by the observation that bardoxolone also significantly increased albuminuria, adverse events, and produced a trend for increased systolic blood pressure [104]. A larger pivotal clinical trial (the BEACON trial, NCT01351675) testing bardoxolone in patients with diabetic nephropathy was terminated early due to higher rates of mortality and serious adverse events in patients treated with the drug [105].
Attempts at treating diabetic kidney disease were also made by Fibrogen, LLC with a human monoclonal antibody (mAB) against CTGF. This phase II trial has been terminated (NCT0913393) along with a phase I trial in FSGS (NCTNCT00782561). This anti-CTGF mAB is still being tested in patients with liver fibrosis and idiopathic pulmonary fibrosis. Other current trials for kidney disease include a phase II trial that is currently recruiting patients with diabetic nephropathy or glomerulosclerosis in order to test an anti-TGFβ mAB being developed by Lilly called LY2382770 (NCT01113801). Another class of therapies being developed includes the endothelin receptor antagonists. Endothelin-1 is the primary endothelin and acts by binding to two GPCR receptors called endothelin type A (ETA) and endothelin type B (ETB). In the kidney, ETA activation causes preferential vasoconstriction the efferent arteriole, while ETB activation increases tubular urine output and sodium excretion. Therefore, selective ETA inhibition is hypothesized to provide a benefit in terms of decreased proteinuria in CKD patients and indeed this has been supported in animal studies [106]. The ASCEND trial tested a selective ETA antagonist called avosentan in patients with diabetic nephropathy (DN) against placebo and all patients were treated with standard therapy regimens for treatment of DN including ACE inhibitors and ARBs [107]. While the avosentan produced a substantial reduction in urine albumin to creatinine (ACR) ratio compared to the control arm there was also a 40-80% increase in the rate of death, a 50% increase in cardiovascular events, and a three times greater risk of congestive heart failure. Due to the increased risk of heart failure and cardiovascular events attributed to fluid overload the trial was terminated early [108]. It is possible ETA antagonist may show a benefit with altered dosing as the high dose used in this trial may have resulted in ETB antagonism and fluid retention. Adding a diuretic to treatment with an ETA antagonist may also limit serious adverse events as patients with fluid overload were sensitive to loop diuretics [106]. Additional ETA selective/angiotensin II receptor antagonists are being tested in CKD as Retrophin, LLC is testing RE-021 vs. the ARB irbesartan in a phase II trial in patients with FSGS and an ACR of 1.0g/g (NCT01613118, not yet recruiting). Finally, therapeutics against B-cell activating factor, or BAFF, are being tested in other kidney diseases. BAFF is associated with a broad range of B-Cell mediated autoimmune disorders and BAFF inhibitors have been shown to play a role in decreasing B-cells and positively effect various diseases [109, 110]. GlaxoSmithKline is running a phase II trial with the anti-BAFF mAB called belimumab in membranous glomerulonephritis and Anthera Pharmaceuticals recently initiated in a phase II clinical trial titled BRIGHT-SC to test their BAFF inhibitor called blisibimod in IgA nephropathy [111-113].
Conclusion
The origin of myofibroblasts remains hotly debated, however we believe the preponderance of data support resident mesenchymal cells such as pericytes and perivascular fibroblasts, as the major myofibroblast progenitor pool. Whether some of these cells are actually resident fibroblasts is not yet determined. Defining the core signaling pathways driving myofibroblast transdifferentation, proliferation, and maintenance will guide the search for therapeutics that may halt or reverse fibrotic diseases. While clinical research has proved difficult and still no drug is approved in the U.S. to directly treat fibrosis, continued study of myofibroblast biology will ultimately lead to new effective therapies to halt kidney fibrosis.
Acknowledgments
This work was supported by NIH DK088923 and an Established Investigator Award of the American Heart Association to Benjamin D. Humphreys, by a fellowship from the Deutsche Forschungsgemeinschaft to Rafael Kramann (KR 4073/1-1), by a research fellowship from the National Kidney Foundation to Derek P. DiRocco (2011-D000691), and by a fellowship from the American Society of Nephrology (Ben J. Lipps Research Fellowship) to Omar H. Maarouf.
Benjamin D. Humphreys is funded by a grant from Evotec AG, with the goal of discovering new therapeutic targets to treat kidney fibrosis.
Footnotes
Conflict of Interest
Rafael Kramann, Derek P. DiRocco, and Omar H. Maarouf declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any human studies performed by any of the authors. The animal study was performed after approval by the Harvard University Institutional Animal Care and Use Committees.
References
Recently published papers of particular interest have been highlighted as:
-
*
Of importance
-
**
Of major importance
- 1.Venkatachalam MA, Griffin KA, Lan R, et al. Acute kidney injury: a springboard for progression in chronic kidney disease. Am J Physiol Renal Physiol. 2010;298(5):F1078–94. doi: 10.1152/ajprenal.00017.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ishani A, Xue JL, Himmelfarb J, et al. Acute kidney injury increases risk of ESRD among elderly. J Am Soc Nephrol. 2009;20(1):223–8. doi: 10.1681/ASN.2007080837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coca SG, Yusuf B, Shlipak MG, et al. Long-term risk of mortality and other adverse outcomes after acute kidney injury: a systematic review and meta-analysis. Am J Kidney Dis. 2009;53(6):961–73. doi: 10.1053/j.ajkd.2008.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campanholle G, Ligresti G, Gharib SA, Duffield JS. Cellular mechanisms of tissue fibrosis. 3. Novel mechanisms of kidney fibrosis. Am J Physiol Cell Physiol. 2013;304(7):C591–603. doi: 10.1152/ajpcell.00414.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu Y. Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol. 2011;7(12):684–96. doi: 10.1038/nrneph.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eyden B. The myofibroblast: a study of normal, reactive and neoplastic tissues, with an emphasis on ultrastructure. part 2 - tumours and tumour-like lesions. J Submicrosc Cytol Pathol. 2005;37(3-4):231–96. [PubMed] [Google Scholar]
- 7.Follonier Castella L, Gabbiani G, McCulloch CA, Hinz B. Regulation of myofibroblast activities: calcium pulls some strings behind the scene. Exp Cell Res. 2010;316(15):2390–401. doi: 10.1016/j.yexcr.2010.04.033. [DOI] [PubMed] [Google Scholar]
- 8.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–8. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Otranto M, Sarrazy V, Bonte F, et al. The role of the myofibroblast in tumor stroma remodeling. Cell Adh Migr. 2012;6(3):203–19. doi: 10.4161/cam.20377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Friedman SL, Sheppard D, Duffield JS, Violette S. Therapy for fibrotic diseases: nearing the starting line. Sci Transl Med. 2013;5(167):167sr1. doi: 10.1126/scitranslmed.3004700. [DOI] [PubMed] [Google Scholar]
- 11*.Grgic I, Campanholle G, Bijol V, et al. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int. 2012;82(2):172–83. doi: 10.1038/ki.2012.20. This work demonstrates that injury of tubular epithelial cells promotes fibrosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rastaldi MP, Ferrario F, Giardino L, et al. Epithelial-mesenchymal transition of tubular epithelial cells in human renal biopsies. Kidney Int. 2002;62(1):137–46. doi: 10.1046/j.1523-1755.2002.00430.x. [DOI] [PubMed] [Google Scholar]
- 13.Zeisberg M, Hanai J, Sugimoto H, et al. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med. 2003;9(7):964–8. doi: 10.1038/nm888. [DOI] [PubMed] [Google Scholar]
- 14.Iwano M, Plieth D, Danoff TM, et al. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest. 2002;110(3):341–50. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Phua YL, Martel N, Pennisi DJ, et al. Distinct sites of renal fibrosis in Crim1 mutant mice arise from multiple cellular origins. J Pathol. 2013;229(5):685–96. doi: 10.1002/path.4155. [DOI] [PubMed] [Google Scholar]
- 16.Kriz W, Kaissling B, Le Hir M. Epithelial-mesenchymal transition (EMT) in kidney fibrosis: fact or fantasy? J Clin Invest. 2011;121(2):468–74. doi: 10.1172/JCI44595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17*.Humphreys BD, Lin SL, Kobayashi A, et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol. 2010;176(1):85–97. doi: 10.2353/ajpath.2010.090517. These data reveal using genetic lineage tracing that FoxD1+ resident pericytes, perivascular fibroblasts and interstitial fibroblasts give rise to myofibroblasts in renal fibrogenesis after unilateral ureteral obstruction. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roufosse C, Bou-Gharios G, Prodromidi E, et al. Bone marrow-derived cells do not contribute significantly to collagen I synthesis in a murine model of renal fibrosis. J Am Soc Nephrol. 2006;17(3):775–82. doi: 10.1681/ASN.2005080795. [DOI] [PubMed] [Google Scholar]
- 19.Lin SL, Kisseleva T, Brenner DA, Duffield JS. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am J Pathol. 2008;173(6):1617–27. doi: 10.2353/ajpath.2008.080433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20**.Lebleu VS, Taduri G, O’Connell J, et al. Origin and function of myofibroblasts in kidney fibrosis. Nat Med. 2013 doi: 10.1038/nm.3218. This study employed a number of genetic models to dissect the relative contributions of bone marrow, resident kidney fibroblasts, kidney pericytes, endothelial cells and epithelial cells. They conclude that resident fibroblasts account for the majority of myofofibroblasts, with significant contribution from bone marrow and much less contribution from endothelial or epithelial cells. They did not observe a pericyte origin for myofibroblasts. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Picard N, Baum O, Vogetseder A, et al. Origin of renal myofibroblasts in the model of unilateral ureter obstruction in the rat. Histochem Cell Biol. 2008;130(1):141–55. doi: 10.1007/s00418-008-0433-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22**.Goritz C, Dias DO, Tomilin N, et al. A pericyte origin of spinal cord scar tissue. Science. 2011;333(6039):238–42. doi: 10.1126/science.1203165. These data reveal that pericytes are major players in scarring of the spinal cord after injury. The authors performed lineage tracing using Glast-CreER driver mice and demonstrate that Glast expressing pericytes of the sppinal cord parenchyma expand, gain α-SMA and become myofibroblasts after injury. [DOI] [PubMed] [Google Scholar]
- 23.Humphreys BD. Targeting pericyte differentiation as a strategy to modulate kidney fibrosis in diabetic nephropathy. Semin Nephrol. 2012;32(5):463–70. doi: 10.1016/j.semnephrol.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levinson RS, Batourina E, Choi C, et al. Foxd1-dependent signals control cellularity in the renal capsule, a structure required for normal renal development. Development. 2005;132(3):529–39. doi: 10.1242/dev.01604. [DOI] [PubMed] [Google Scholar]
- 25.Hatini V, Huh SO, Herzlinger D, et al. Essential role of stromal mesenchyme in kidney morphogenesis revealed by targeted disruption of Winged Helix transcription factor BF-2. Genes Dev. 1996;10(12):1467–78. doi: 10.1101/gad.10.12.1467. [DOI] [PubMed] [Google Scholar]
- 26.Grgic I, Duffield JS, Humphreys BD. The origin of interstitial myofibroblasts in chronic kidney disease. Pediatr Nephrol. 2012;27(2):183–93. doi: 10.1007/s00467-011-1772-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27**.Dulauroy S, Di Carlo SE, Langa F, et al. Lineage tracing and genetic ablation of ADAM12(+) perivascular cells identify a major source of profibrotic cells during acute tissue injury. Nat Med. 2012 doi: 10.1038/nm.2848. This work demonstrates using genetic fate mapping, that ADAM12+, PDGFR- α+ pericytes are the major source of myofibroblasts in scaring of skelettal muscle and skin. Genetic ablation of ADAM12+ cells or knockdown of ADAM12 was able to ameliorate the severity of fibrosis. [DOI] [PubMed] [Google Scholar]
- 28.Chen YT, Chang FC, Wu CF, et al. Platelet-derived growth factor receptor signaling activates pericyte-myofibroblast transition in obstructive and post-ischemic kidney fibrosis. Kidney Int. 2011;80(11):1170–81. doi: 10.1038/ki.2011.208. [DOI] [PubMed] [Google Scholar]
- 29.Xiong J, Wang Y, Zhu Z, et al. NG2 proteoglycan increases mesangial cell proliferation and extracellular matrix production. Biochem Biophys Res Commun. 2007;361(4):960–7. doi: 10.1016/j.bbrc.2007.07.113. [DOI] [PubMed] [Google Scholar]
- 30.Boor P, Floege J. The renal (myo-)fibroblast: a heterogeneous group of cells. Nephrol Dial Transplant. 2012;27(8):3027–36. doi: 10.1093/ndt/gfs296. [DOI] [PubMed] [Google Scholar]
- 31.Seikrit C, Henkel C, van Roeyen CR, et al. Biological responses to PDGF-AA versus PDGF-CC in renal fibroblasts. Nephrol Dial Transplant. 2013;28(4):889–900. doi: 10.1093/ndt/gfs509. [DOI] [PubMed] [Google Scholar]
- 32.Kida Y, Duffield JS. Pivotal role of pericytes in kidney fibrosis. Clin Exp Pharmacol Physiol. 2011;38(7):467–73. doi: 10.1111/j.1440-1681.2011.05531.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kida Y, Ieronimakis N, Schrimpf C, et al. EphrinB2 reverse signaling protects against capillary rarefaction and fibrosis after kidney injury. J Am Soc Nephrol. 2013;24(4):559–72. doi: 10.1681/ASN.2012080871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Basile DP. Rarefaction of peritubular capillaries following ischemic acute renal failure: a potential factor predisposing to progressive nephropathy. Curr Opin Nephrol Hypertens. 2004;13(1):1–7. doi: 10.1097/00041552-200401000-00001. [DOI] [PubMed] [Google Scholar]
- 35.Eardley KS, Kubal C, Zehnder D, et al. The role of capillary density, macrophage infiltration and interstitial scarring in the pathogenesis of human chronic kidney disease. Kidney Int. 2008;74(4):495–504. doi: 10.1038/ki.2008.183. [DOI] [PubMed] [Google Scholar]
- 36.Seron D, Alexopoulos E, Raftery MJ, et al. Number of interstitial capillary cross-sections assessed by monoclonal antibodies: relation to interstitial damage. Nephrol Dial Transplant. 1990;5(10):889–93. doi: 10.1093/ndt/5.10.889. [DOI] [PubMed] [Google Scholar]
- 37.Choi YJ, Chakraborty S, Nguyen V, et al. Peritubular capillary loss is associated with chronic tubulointerstitial injury in human kidney: altered expression of vascular endothelial growth factor. Hum Pathol. 2000;31(12):1491–7. doi: 10.1053/hupa.2000.20373. [DOI] [PubMed] [Google Scholar]
- 38.Schrimpf C, Xin C, Campanholle G, et al. Pericyte TIMP3 and ADAMTS1 modulate vascular stability after kidney injury. J Am Soc Nephrol. 2012;23(5):868–83. doi: 10.1681/ASN.2011080851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin SL, Chang FC, Schrimpf C, et al. Targeting endothelium-pericyte cross talk by inhibiting VEGF receptor signaling attenuates kidney microvascular rarefaction and fibrosis. Am J Pathol. 2011;178(2):911–23. doi: 10.1016/j.ajpath.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pohlers D, Beyer A, Koczan D, et al. Constitutive upregulation of the transforming growth factor-beta pathway in rheumatoid arthritis synovial fibroblasts. Arthritis Res Ther. 2007;9(3):R59. doi: 10.1186/ar2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang C, Kim Y, Caramori ML, et al. Cellular basis of diabetic nephropathy: II. The transforming growth factor-beta system and diabetic nephropathy lesions in type 1 diabetes. Diabetes. 2002;51(12):3577–81. doi: 10.2337/diabetes.51.12.3577. [DOI] [PubMed] [Google Scholar]
- 42.Wygrecka M, Zakrzewicz D, Taborski B, et al. TGF-beta1 induces tissue factor expression in human lung fibroblasts in a PI3K/JNK/Akt-dependent and AP-1-dependent manner. Am J Respir Cell Mol Biol. 2012;47(5):614–27. doi: 10.1165/rcmb.2012-0097OC. [DOI] [PubMed] [Google Scholar]
- 43.Bujak M, Frangogiannis NG. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res. 2007;74(2):184–95. doi: 10.1016/j.cardiores.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Munger JS, Sheppard D. Cross talk among TGF-beta signaling pathways, integrins, and the extracellular matrix. Cold Spring Harb Perspect Biol. 2011;3(11):a005017. doi: 10.1101/cshperspect.a005017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li MO, Wan YY, Sanjabi S, et al. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2006;24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- 46.Roberts AB, Sporn MB, Assoian RK, et al. Transforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc Natl Acad Sci U S A. 1986;83(12):4167–71. doi: 10.1073/pnas.83.12.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sime PJ, Xing Z, Graham FL, et al. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J Clin Invest. 1997;100(4):768–76. doi: 10.1172/JCI119590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dennis PA, Rifkin DB. Cellular activation of latent transforming growth factor beta requires binding to the cation-independent mannose 6-phosphate/insulin-like growth factor type II receptor. Proc Natl Acad Sci U S A. 1991;88(2):580–4. doi: 10.1073/pnas.88.2.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hyytiainen M, Penttinen C, Keski-Oja J. Latent TGF-beta binding proteins: extracellular matrix association and roles in TGF-beta activation. Crit Rev Clin Lab Sci. 2004;41(3):233–64. doi: 10.1080/10408360490460933. [DOI] [PubMed] [Google Scholar]
- 50.Ramirez F, Sakai LY. Biogenesis and function of fibrillin assemblies. Cell Tissue Res. 2010;339(1):71–82. doi: 10.1007/s00441-009-0822-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol. 2012;13(10):616–30. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wrana JL, Attisano L, Wieser R, et al. Mechanism of activation of the TGF-beta receptor. Nature. 1994;370(6488):341–7. doi: 10.1038/370341a0. [DOI] [PubMed] [Google Scholar]
- 53.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113(6):685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 54.Derynck R, Akhurst RJ. Differentiation plasticity regulated by TGF-beta family proteins in development and disease. Nat Cell Biol. 2007;9(9):1000–4. doi: 10.1038/ncb434. [DOI] [PubMed] [Google Scholar]
- 55.Trachtman H, Fervenza FC, Gipson DS, et al. A phase 1, single-dose study of fresolimumab, an anti-TGF-beta antibody, in treatment-resistant primary focal segmental glomerulosclerosis. Kidney Int. 2011;79(11):1236–43. doi: 10.1038/ki.2011.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen HY, Huang XR, Wang W, et al. The protective role of Smad7 in diabetic kidney disease: mechanism and therapeutic potential. Diabetes. 2011;60(2):590–601. doi: 10.2337/db10-0403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hawinkels LJ, Ten Dijke P. Exploring anti-TGF-beta therapies in cancer and fibrosis. Growth Factors. 2011;29(4):140–52. doi: 10.3109/08977194.2011.595411. [DOI] [PubMed] [Google Scholar]
- 58.Azuma A. Pirfenidone treatment of idiopathic pulmonary fibrosis. Ther Adv Respir Dis. 2012;6(2):107–14. doi: 10.1177/1753465812436663. [DOI] [PubMed] [Google Scholar]
- 59.Schnaper HW, Hayashida T, Hubchak SC, Poncelet AC. TGF-beta signal transduction and mesangial cell fibrogenesis. Am J Physiol Renal Physiol. 2003;284(2):F243–52. doi: 10.1152/ajprenal.00300.2002. [DOI] [PubMed] [Google Scholar]
- 60.Lan HY. Diverse roles of TGF-beta/Smads in renal fibrosis and inflammation. Int J Biol Sci. 2011;7(7):1056–67. doi: 10.7150/ijbs.7.1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lan HY, Mu W, Tomita N, et al. Inhibition of renal fibrosis by gene transfer of inducible Smad7 using ultrasound-microbubble system in rat UUO model. J Am Soc Nephrol. 2003;14(6):1535–48. doi: 10.1097/01.asn.0000067632.04658.b8. [DOI] [PubMed] [Google Scholar]
- 62.Santiago B, Gutierrez-Canas I, Dotor J, et al. Topical application of a peptide inhibitor of transforming growth factor-beta1 ameliorates bleomycin-induced skin fibrosis. J Invest Dermatol. 2005;125(3):450–5. doi: 10.1111/j.0022-202X.2005.23859.x. [DOI] [PubMed] [Google Scholar]
- 63.Akhurst RJ, Hata A. Targeting the TGFbeta signalling pathway in disease. Nat Rev Drug Discov. 2012;11(10):790–811. doi: 10.1038/nrd3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149(6):1192–205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 65.Aberle H, Bauer A, Stappert J, et al. beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997;16(13):3797–804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mao J, Wang J, Liu B, et al. Low-density lipoprotein receptor-related protein-5 binds to Axin and regulates the canonical Wnt signaling pathway. Mol Cell. 2001;7(4):801–9. doi: 10.1016/s1097-2765(01)00224-6. [DOI] [PubMed] [Google Scholar]
- 67.Li VS, Ng SS, Boersema PJ, et al. Wnt signaling through inhibition of beta-catenin degradation in an intact Axin1 complex. Cell. 2012;149(6):1245–56. doi: 10.1016/j.cell.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 68.Fathke C, Wilson L, Shah K, et al. Wnt signaling induces epithelial differentiation during cutaneous wound healing. BMC Cell Biol. 2006;7:4. doi: 10.1186/1471-2121-7-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cheon SS, Cheah AY, Turley S, et al. beta-Catenin stabilization dysregulates mesenchymal cell proliferation, motility, and invasiveness and causes aggressive fibromatosis and hyperplastic cutaneous wounds. Proc Natl Acad Sci U S A. 2002;99(10):6973–8. doi: 10.1073/pnas.102657399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Trensz F, Haroun S, Cloutier A, et al. A muscle resident cell population promotes fibrosis in hindlimb skeletal muscles of mdx mice through the Wnt canonical pathway. Am J Physiol Cell Physiol. 2010;299(5):C939–47. doi: 10.1152/ajpcell.00253.2010. [DOI] [PubMed] [Google Scholar]
- 71.Hsu E, Shi H, Jordan RM, et al. Lung tissues in patients with systemic sclerosis have gene expression patterns unique to pulmonary fibrosis and pulmonary hypertension. Arthritis Rheum. 2011;63(3):783–94. doi: 10.1002/art.30159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jones SE, Jomary C. Secreted Frizzled-related proteins: searching for relationships and patterns. Bioessays. 2002;24(9):811–20. doi: 10.1002/bies.10136. [DOI] [PubMed] [Google Scholar]
- 73.Eguchi M, Nguyen C, Lee SC, Kahn M. ICG-001, a novel small molecule regulator of TCF/beta-catenin transcription. Med Chem. 2005;1(5):467–72. doi: 10.2174/1573406054864098. [DOI] [PubMed] [Google Scholar]
- 74.Henderson WR, Jr, Chi EY, Ye X, et al. Inhibition of Wnt/beta-catenin/CREB binding protein (CBP) signaling reverses pulmonary fibrosis. Proc Natl Acad Sci U S A. 2010;107(32):14309–14. doi: 10.1073/pnas.1001520107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Saraswati S, Alfaro MP, Thorne CA, et al. Pyrvinium, a potent small molecule Wnt inhibitor, promotes wound repair and post-MI cardiac remodeling. PLoS One. 2010;5(11):e15521. doi: 10.1371/journal.pone.0015521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Surendran K, Simon TC, Liapis H, McGuire JK. Matrilysin (MMP-7) expression in renal tubular damage: association with Wnt4. Kidney Int. 2004;65(6):2212–22. doi: 10.1111/j.1523-1755.2004.00641.x. [DOI] [PubMed] [Google Scholar]
- 77.He W, Dai C, Li Y, et al. Wnt/beta-catenin signaling promotes renal interstitial fibrosis. J Am Soc Nephrol. 2009;20(4):765–76. doi: 10.1681/ASN.2008060566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dirocco DP, Kobayashi A, Taketo MM, et al. Wnt4/beta-Catenin Signaling in Medullary Kidney Myofibroblasts. J Am Soc Nephrol. 2013 doi: 10.1681/ASN.2012050512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hao S, He W, Li Y, et al. Targeted inhibition of beta-catenin/CBP signaling ameliorates renal interstitial fibrosis. J Am Soc Nephrol. 2011;22(9):1642–53. doi: 10.1681/ASN.2010101079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ren S, Johnson BG, Kida Y, et al. LRP-6 is a coreceptor for multiple fibrogenic signaling pathways in pericytes and myofibroblasts that are inhibited by DKK-1. Proc Natl Acad Sci U S A. 2013;110(4):1440–5. doi: 10.1073/pnas.1211179110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.von Toerne C, Schmidt C, Adams J, et al. Wnt pathway regulation in chronic renal allograft damage. Am J Transplant. 2009;9(10):2223–39. doi: 10.1111/j.1600-6143.2009.02762.x. [DOI] [PubMed] [Google Scholar]
- 82.van Roeyen CR, Ostendorf T, Floege J. The platelet-derived growth factor system in renal disease: an emerging role of endogenous inhibitors. Eur J Cell Biol. 2012;91(6-7):542–51. doi: 10.1016/j.ejcb.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 83.Ostendorf T, Eitner F, Floege J. The PDGF family in renal fibrosis. Pediatr Nephrol. 2012;27(7):1041–50. doi: 10.1007/s00467-011-1892-z. [DOI] [PubMed] [Google Scholar]
- 84.Chuang PY, Menon MC, He JC. Molecular targets for treatment of kidney fibrosis. J Mol Med (Berl) 2013;91(5):549–59. doi: 10.1007/s00109-012-0983-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Eitner F, Bucher E, van Roeyen C, et al. PDGF-C is a proinflammatory cytokine that mediates renal interstitial fibrosis. J Am Soc Nephrol. 2008;19(2):281–9. doi: 10.1681/ASN.2007030290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nusslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila. Nature. 1980;287(5785):795–801. doi: 10.1038/287795a0. [DOI] [PubMed] [Google Scholar]
- 87.Briscoe J, Therond PP. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol. 2013 doi: 10.1038/nrm3598. [DOI] [PubMed] [Google Scholar]
- 88.Yauch RL, Gould SE, Scales SJ, et al. A paracrine requirement for hedgehog signalling in cancer. Nature. 2008;455(7211):406–10. doi: 10.1038/nature07275. [DOI] [PubMed] [Google Scholar]
- 89.Choi SS, Omenetti A, Syn WK, Diehl AM. The role of Hedgehog signaling in fibrogenic liver repair. Int J Biochem Cell Biol. 2011;43(2):238–44. doi: 10.1016/j.biocel.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Stewart GA, Hoyne GF, Ahmad SA, et al. Expression of the developmental Sonic hedgehog (Shh) signalling pathway is up-regulated in chronic lung fibrosis and the Shh receptor patched 1 is present in circulating T lymphocytes. J Pathol. 2003;199(4):488–95. doi: 10.1002/path.1295. [DOI] [PubMed] [Google Scholar]
- 91.Mailleux AA, Moshai EF, Crestani B. Sonic Hedgehog signaling in pulmonary fibrosis: a spiky issue? Am J Physiol Lung Cell Mol Physiol. 2013;304(6):L391–3. doi: 10.1152/ajplung.00404.2012. [DOI] [PubMed] [Google Scholar]
- 92.Fabian SL, Penchev RR, St-Jacques B, et al. Hedgehog-Gli pathway activation during kidney fibrosis. Am J Pathol. 2012;180(4):1441–53. doi: 10.1016/j.ajpath.2011.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ding H, Zhou D, Hao S, et al. Sonic hedgehog signaling mediates epithelial-mesenchymal communication and promotes renal fibrosis. J Am Soc Nephrol. 2012;23(5):801–13. doi: 10.1681/ASN.2011060614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sahebjam S, Siu LL, Razak AA. The utility of hedgehog signaling pathway inhibition for cancer. Oncologist. 2012;17(8):1090–9. doi: 10.1634/theoncologist.2011-0450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jenkins D. Hedgehog signalling: emerging evidence for non-canonical pathways. Cell Signal. 2009;21(7):1023–34. doi: 10.1016/j.cellsig.2009.01.033. [DOI] [PubMed] [Google Scholar]
- 96*.Kramann R, Floege J, Ketteler M, et al. Medical options to fight mortality in end-stage renal disease: a review of the literature. Nephrol Dial Transplant. 2012;27(12):4298–307. doi: 10.1093/ndt/gfs400. This review summarizes treatment strategies that have undergone randomized controlled clinical trials (RCTs) in ESRD patients with mortality reduction as a pre-specified study endpoint. Survival improvement in ESRD patients has been demonstrated only in telmisartan, candesartan and carvedilol. [DOI] [PubMed] [Google Scholar]
- 97.Sarafidis PA, Khosla N, Bakris GL. Antihypertensive therapy in the presence of proteinuria. Am J Kidney Dis. 2007;49(1):12–26. doi: 10.1053/j.ajkd.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 98.Maschio G, Alberti D, Janin G, et al. Effect of the angiotensin-converting-enzyme inhibitor benazepril on the progression of chronic renal insufficiency. The Angiotensin-Converting-Enzyme Inhibition in Progressive Renal Insufficiency Study Group. N Engl J Med. 1996;334(15):939–45. doi: 10.1056/NEJM199604113341502. [DOI] [PubMed] [Google Scholar]
- 99.Randomised placebo-controlled trial of effect of ramipril on decline in glomerular filtration rate and risk of terminal renal failure in proteinuric, non-diabetic nephropathy. The GISEN Group (Gruppo Italiano di Studi Epidemiologici in Nefrologia) Lancet. 1997;349(9069):1857–63. [PubMed] [Google Scholar]
- 100.Ruggenenti P, Perna A, Benini R, et al. In chronic nephropathies prolonged ACE inhibition can induce remission: dynamics of time-dependent changes in GFR. Investigators of the GISEN Group. Gruppo Italiano Studi Epidemiologici in Nefrologia. J Am Soc Nephrol. 1999;10(5):997–1006. doi: 10.1681/ASN.V105997. [DOI] [PubMed] [Google Scholar]
- 101.Remuzzi A, Gagliardini E, Sangalli F, et al. ACE inhibition reduces glomerulosclerosis and regenerates glomerular tissue in a model of progressive renal disease. Kidney Int. 2006;69(7):1124–30. doi: 10.1038/sj.ki.5000060. [DOI] [PubMed] [Google Scholar]
- 102.Wu QQ, Wang Y, Senitko M, et al. Bardoxolone methyl (BARD) ameliorates ischemic AKI and increases expression of protective genes Nrf2, PPARgamma, and HO-1. Am J Physiol Renal Physiol. 2011;300(5):F1180–92. doi: 10.1152/ajprenal.00353.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pergola PE, Krauth M, Huff JW, et al. Effect of bardoxolone methyl on kidney function in patients with T2D and Stage 3b-4 CKD. Am J Nephrol. 2011;33(5):469–76. doi: 10.1159/000327599. [DOI] [PubMed] [Google Scholar]
- 104**.Pergola PE, Raskin P, Toto RD, et al. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N Engl J Med. 2011;365(4):327–36. doi: 10.1056/NEJMoa1105351. This study highlights the favorable results from a preliminary study of bardoxolone methyl on kidney function. It is important to note that when bardoxolone methyl was tested in larger phase 3 trials the positive effects of this trial vanished. An example of the necessity to understand a biological outcome in the context of a disease. [DOI] [PubMed] [Google Scholar]
- 105.Zoja C, Benigni A, Remuzzi G. The Nrf2 pathway in the progression of renal disease. Nephrol Dial Transplant. 2013 doi: 10.1093/ndt/gft224. [DOI] [PubMed] [Google Scholar]
- 106.Burnier M, Forni V. Endothelin receptor antagonists: a place in the management of essential hypertension? Nephrol Dial Transplant. 2012;27(3):865–8. doi: 10.1093/ndt/gfr704. [DOI] [PubMed] [Google Scholar]
- 107*.Mann JF, Green D, Jamerson K, et al. Avosentan for overt diabetic nephropathy. J Am Soc Nephrol. 2010;21(3):527–35. doi: 10.1681/ASN.2009060593. This study tested the endothelin receptor antagonis avosentan in patients with diabetic nephropathy. Previous studies have shown that blocking the endothelin pathway, particularly ETA receptors, could produce powerful anti-hypertensive effects that were beneficial in preclinical and clinical models of nephropathy. This clinical study used a dose of avosentan that was higher than previous studies indicated as safe and resulted in trial discontinuation due to increased severe adverse effects and death in the experimental arms compared to the control arms. Deeper understanding of the biology of the endothelin pathway in the context of chronic kidney disease is warranted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kohan DE, Pollock DM. Endothelin antagonists for diabetic and non-diabetic chronic kidney disease. Br J Clin Pharmacol. 2012 doi: 10.1111/bcp.12064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Davidson A. Targeting BAFF in autoimmunity. Curr Opin Immunol. 2010;22(6):732–9. doi: 10.1016/j.coi.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Vincent FB, Morand EF, Mackay F. BAFF and innate immunity: new therapeutic targets for systemic lupus erythematosus. Immunol Cell Biol. 2012;90(3):293–303. doi: 10.1038/icb.2011.111. [DOI] [PubMed] [Google Scholar]
- 111.Stohl W. Biologic differences between various inhibitors of the BLyS/BAFF pathway: should we expect differences between belimumab and other inhibitors in development? Curr Rheumatol Rep. 2012;14(4):303–9. doi: 10.1007/s11926-012-0254-6. [DOI] [PubMed] [Google Scholar]
- 112.McCarthy DD, Kujawa J, Wilson C, et al. Mice overexpressing BAFF develop a commensal flora-dependent, IgA-associated nephropathy. J Clin Invest. 2011;121(10):3991–4002. doi: 10.1172/JCI45563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Xin G, Shi W, Xu LX, et al. Serum BAFF is elevated in patients with IgA nephropathy and associated with clinical and histopathological features. J Nephrol. 2012:0. doi: 10.5301/jn.5000218. [DOI] [PubMed] [Google Scholar]