

Abstract
Background
Genetic epilepsy with febrile seizures plus (GEFS+) is a familial epilepsy syndrome with extremely variable expressivity. The aim of our study was to identify the responsible locus for GEFS+ syndrome in a consanguineous Tunisian family showing three affected members, by carrying out a genome-wide single nucleotide polymorphisms (SNPs) genotyping followed by a whole-exome sequencing. We hypothesized an autosomal recessive (AR) mode of inheritance.
Results
Parametric linkage analysis and haplotype reconstruction identified a new unique identical by descent (IBD) interval of 527 kb, flanking by two microsatellite markers, 18GTchr22 and 15ACchr22b, on human chromosome 22q13.31 with a maximum multipoint LOD score of 2.51. Our analysis was refined by the use of a set of microsatellite markers. We showed that one of them was homozygous for the same allele in all affected individuals and heterozygous in healthy members of this family. This microsatellite marker, we called 17ACchr22, is located in an intronic region of TBC1D22A gene, which encodes a GTPase activator activity. Whole-exome sequencing did not reveal any mutation on chromosome 22q13.31 at the genome wide level.
Conclusions
Our findings suggest that TBC1D22A is a new locus for GEFS+.
Keywords: Febrile seizures, GEFS+, Autosomal recessive, Genome wide SNPs, Linkage analysis, Exome sequencing
Background
Epilepsy is one of the most common serious neurological disorders at worldwide level [1]. Environmental and genetic factors are known to play a role in its pathogenesis. Segregation studies suggest that most epileptic syndromes are complex disorders, but several monogenic forms have also been described. Genetic (formerly named generalized [2]) epilepsy with febrile seizures plus (GEFS+) is such a Mendelian inherited epileptic syndrome. This familial autosomal dominant (AD) epilepsy shows a wide range of phenotypes such as febrile seizures (FS), FS plus (FS+) – defined as FS persisting beyond the age of 6 – as well generalized and partial seizures [2,3]. In some families, most severe epileptic phenotypes have been described such as the severe myoclonic epilepsy of infancy or Dravet syndrome [4-6].
Up to now, eight GEFS+ loci have been registered on OMIM database [7-16]. Three genes are known to be causative for GEFS+ phenotype: SCN1B (MIM#600235;GEFS+1) [7], SCN1A (MIM#182389; GEFS+2) [10] and GABRG2 (MIM#137164; GEFS+3) [11]. Familial forms of pure febrile seizures (FEB) have also been described. Segregation and twin studies of FS suggest a polygenic or multifactorial mode of inheritance [17,18]. In most FEB families, phenotype follows an AD mode of inheritance [19]. To date, according to OMIM (Online Mendelian Inheritance in Man – http://www.ncbi.nlm.nih.gov/omim) database, eleven FEB loci have already been reported [20-30] and only four genes have been linked to FS related phenotypes: SCN1A (MIM#182389; FEB3A) [31], MASS1 or GPR98 (MIM# 602851; FEB4) [32], GABRG2 (MIM#137164; FEB8) [27] and CPA6 (MIM#609562; FEB11) [30]. Finally, two other genes are considered as “susceptibility” genes to FEB syndrome and epilepsy: GABRD (MIM#137163; GEFS+5) [15] and SCN9A (MIM#603415; FEB3B/GEFS+7) [33].
In the present study, we reported the clinical and genetic studies of a GEFS+ consanguineous Tunisian family with an autosomal recessive (AR) mode of inheritance. We used homozygosity mapping, which is a method of choice for localizing genes responsible for AR diseases in consanguineous families [34]. This approach allows identifying identical by descent (IBD) regions inherited from a common ancestor. IBD loci are homozygous in all the affected members but not in other relatives. In the present study we report a new locus on chromosome 22q13.31 in which TBC1D22A gene (TBC1 domain family, member 22A, also known as C22orf4) is located.
Results
Clinical description of consanguineous Tunisian family with GEFS+ patients
The familial pedigree is shown in Figure 1 and main clinical features of affected individuals are described in Table 1. The occurrence of generalized tonico clonic seizures (GTCS), absence seizure and FS+ led us to consider a GEFS+ syndrome in the present family.
Figure 1.

Consanguineous Tunisian GEFS+family pedigree. Legend: A. Haplotype reconstruction for markers on chromosome 22q13.31, which are ordered from centromere to telomere. Markers from the Illumina array are in italic. The IBD haplotype shared by family members is shown in solid lines. Recombinaison events are shown by solid drawbar. Maternal and paternal chromosomes are designated by M and P, respectively. Each phenotype is describing by different colors. B. List of markers used for haplotype reconstruction with physical position. Markers from the Illumina array are in italic.
Table 1.
Clinical characteristics of affected individuals
| Subject (sex, age) * |
FS |
AFS |
EEG | Neurological examination | MRI | Treatment/age | ||
|---|---|---|---|---|---|---|---|---|
| Onset age/remission, age | n | Onset age/remission, age | Type/n | |||||
| IV-2 (F, 13 y) |
4 y/yes, 10 y |
4 |
7 y/8 y |
GTCS/3 |
Normal |
Normal |
Normal |
VPA/7 y-until now |
| IV-3 (M, 18 y) |
2 y/yes, 13 y |
Numerous |
3 y/7 y |
GTCS/1 yearly |
Normal |
Normal |
Normal |
VPA/8 y-12 y |
| 8 y/9 y |
A/numerous |
|||||||
| IV-4 (M, 17 y) | 2 y/no | Numerous | 6 y/11 y | GTCS/5 |
3 Hz generalized SW | Mild mental retardation | Normal | VPA/5 y-until now |
| A/numerous | ||||||||
*All individuals were agree to publish clinical details.
M: Male, F: Female, y: years, FS: Febrile Seizures, AFS: Afebrile Seizures, n: Seizures number, EEG: Electroencephalogram, GTC: Generalized Tonico Clonic Seizures, A: Absence, SW: Spike Wave, MRI: Magnetic Resonance Imaging, VPA: Valproic Acid.
The proband (IV-4) is a 17-year-old boy, born at term after an uneventful pregnancy. At the age of 2, he suffered from a first complex FS. At the age of 6, a febrile GTCS and absence seizures were observed. Due to the continuation of FS, since the age of 5, he has been treated with valproic acid. Neurological examination revealed a mild mental retardation. Standard EEG tracings showed generalized spike waves.
His first cousin, patient IV-3, is an 18-year-old boy, born at term after an uneventful pregnancy, from a consanguineous marriage between first degree cousins. In addition to FS+ which persisted until the age of 13, he experienced, at the age of 3, GTCS. Since the age of 8, absence seizures were also observed. Between the ages of 8 to 12, he was treated with valproic acid. Neurological and EEG exams were normal.
Patient IV-2, first cousin of the two previously described patients, is a 13-year-old girl, born at term after an uneventful pregnancy. She showed FS+ until the age of 10 and she suffered from GTCS since the age of 7. She has been treated by valproic acid since the age of 5. Neurological examination and EEG tracings were normal.
MRI scans were normal in all affected family members.
Genetic linkage and haplotype analysis
We assessed microsatellite markers (Additional file 1: Table S1) of four GEFS+ loci and seven FEB loci, that were known at that time. Haplotype reconstructions and analysis of recombination events allowed excluding all the GEFS+ and FEB loci analyzed in the present family.
A genome wide genotyping was then performed and parametric linkage using the AR mode of inheritance identified a linked region of 10.6 Mb on chromosome 22q13.31 (Additional file 2: Figure FS1) flanked by the two SNPs rs3203726 and rs728592 with a maximum multipoint LOD score of 2.51 (Additional file 1: Table S5). On the same chromosome, positive LOD scores were also observed at six other loci (Additional file 1: Table S6). However, haplotype analysis showed that all these loci exhibited only one or two consecutive homozygous SNPs in every affected individual. Consequently, they are less likely to represent the disease locus. Moreover, due to the number of obligate carriers of the disease (8) compared to affected ones (3), the AD model is not likely.
The 10.6 Mb candidate region was further confirmed by using a set of microsatellite markers (Additional file 1: Table S2). For haplotype reconstructions, we used homozygosity mapping method, which allow localizing homozygous IBD genomic region(s) responsible for AR diseases in consanguineous families. We confirmed the existence of a common homozygous IBD haplotype shared by all the three patients. Therefore, the candidate region was reduced to an interval of 527 kb, flanked by two microsatellite markers, 18GTchr22 and 15ACchr22b. This locus was homozygous in all three affected relatives and heterozygous in other family members (Figure 1A). Within this homozygous IBD locus lie the 9 last exons and the 3’UTR of TBC1D22A which encodes the member 22A of Rab GTPase activator with a TBC1 domain family (Figure 2). Two polymorphic markers, 17ACchr22 and rs2017931, are also displayed within the IBD interval (Figure 1A). The most interesting one is the 17ACchr22 microsatellite marker due to its high variability. Its allele 4, corresponding to 16 AC repeats, was found homozygous in all affected relatives and heterozygous in all other unaffected family members. We estimated allele frequencies of 17ACchr22 in a sample of 70 Tunisian controls. Ten alleles (corresponding to 13 to 22 AC repeats) were observed, and the most common alleles were allele 7 (19 repeats) 25%, allele 8 (20 repeats) 22.1%, allele 5 (17 repeats) 17.9%, and allele 6 (18 repeats) 10.7%. Finally, the IBD allele 4, had a frequency of 7.14% and was never found homozygous in the present Tunisian population (Additional file 1: Table S7).
Figure 2.
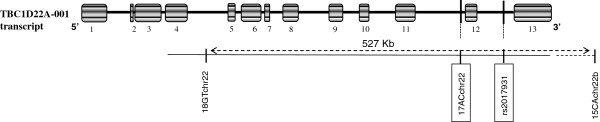
Structure of the TBC1D22A gene according to Ensembl (http://www.ensembl.org/index.html) with localization of markers within and flanking the 527 Kb IBD linked locus.
Array CGH analysis did not reveal any CNV at the genome-wide level in affected individual IV-3.
Mutational analysis
Mutation analysis was performed by whole-exome sequencing in patient IV-3 where 94.5% of the coding part of the RefSeq genes was covered at least 8×, which was sufficient to detect homozygous substitutions. This experiment revealed only three homozygous non-synonymous putative deleterious variations over the entire genome. Two of them were found on chromosome 22q13.31, within the 10.6 Mb linked region: c.5159C > T (S1720L) in the TNRC6B (trinucleotide repeat containing 6B) gene and c.4433C > T (T1478M) (rs8141262) in the CACNA1I (alpha 1I subunit of calcium voltage-dependant channel) gene. Genotyping these two missense variations, in all Tunisian family members, was done by HRM assay and confirmed by Sanger sequencing. These analyses showed that they are not linked to the disease phenotype, since the two other affected cousins (IV-2 and IV-4) were heterozygous for both variations. The third variation, found homozygous in patient IV-3, is located on chromosome 1p36.11: c.604T > C (S202P) (rs6687605) in the LDLRAP1 (low density lipoprotein receptor adaptor protein 1) gene. This polymorphism, genotyped by Sanger sequencing (Additional file 1: Table S3), was also found homozygous in his unaffected cousin IV-1. Therefore, rs6687605 is not linked to GEFS+ phenotype in this family, according to the IBD assumption. Finally, we also performed Sanger sequencing for KCNJ4, since this gene codes for a ion channel protein. This family of genes are often found mutated in several epilepsies. Moreover, this gene is located within one of the 6 chromosome 22 short homozygous regions (Additional file 1: Table S6), which are considered as not linked to the disease (see Genetic linkage and haplotype analysis section). No mutation was observed in this gene.
Discussion
The present study described clinical and molecular investigations on GEFS+ syndrome in a Tunisian consanguineous family. The occurrence of both GTCS and absence in addition to FS+, and in absence of other types of seizures (e.g. myoclonic jerks), led us to consider that GEFS+ phenotype is likely within this family [2]. To our knowledge, it would be the first description of a consanguineous GEFS+ Tunisian family with a putative AR mode of inheritance, a transmission previously described in a Moroccan consanguineous family, showing FS and temporal lobe epilepsy phenotypes [30]. Moreover, all GEFS+ families, described in OMIM database, exclusively show an AD mode of inheritance [7-16]. Using homozygous linkage and whole-exome sequencing method, we mapped the pathogenic locus responsible for the disease within a short interval of chromosome 22q13.31 in the present Tunisian consanguineous family.
We first excluded by linkage analysis all known GEFS+ loci at that time, as well as those associated to FS phenotypes. Epileptic syndromes have also been observed in patients with structural variants [35,36], such as CNVs on chromosome 15q13.3 are known to play an important role in the genetic etiology of idiopathic generalized epilepsy [37]. We also excluded such genomic event in this family.
The 22q13.31 region, where we mapped the disease locus, has been implicated in a complex phenotype with epilepsy in 5-year-old boy with a 7.9 Mb de novo deletion of chromosome 22q13.2-qter [38].
Based on the homozygosity of an IBD locus in patients only, refined mapping allowed reducing the disease locus to the 9 last exons and 3’UTR of TBC1D22A, and even to a shorter interval around the exon 12. However no mutation has been observed by whole-exome sequencing in any of these last exons of TBC1D22A. Homozygous alleles of the two polymorphic markers (17ACchr22 microsatellite and rs2017931), found in all patients, are likely to be non-functional and probably in linkage disequilibrium with the disease mutation in one of the TBC1D22A introns or in 3’UTR. The disease mutation could affect the splicing of TBC1D22A. Unfortunately, we were unable to test this hypothesis in absence of available RNA samples from the affected members of the family.
The first crystal structure of TBC1D22A was done in 2008 showing the diversity of human TBC domain family members [39]. Very recently, Shapshak et al. found that TBC1D22A gene had a differing gene expression profile across patients showing HIV associated dementia [40]. TBC1D22A gene is expressed in brain tissues with a percentage of 2.14% (http://smd.princeton.edu/cgi-bin/source/sourceSearch). Moreover, GWAS suggested an association with human longevity and a SNP (rs5766691) located in TBC1D22A[41], within the 527 kb IBD homozygous interval.
Conclusions
In conclusion, we reported a new locus on chromosome 22q13.31 in a consanguineous Tunisian family with a GEFS+ phenotype with an original AR mode of inheritance. We were not able to identify the disease mutation but restricted the linked interval to a part of TBC1D22A, which is expanded from exon 5 to the 3’UTR of the gene. Further studies will confirm whether this gene is also mutated in other families with GEFS+.
This study was approved by the Research and Ethics Review Board of the Department of Neurology, University Hospital of Geneva and by the Ministry of Higher Education and Scientific and Technological Research of Tunisia.
Methods
Family ascertainment and phenotyping
We recruited a consanguineous Tunisian family extended over four generations, comprising 3 affected and 19 non-affected members, who all originated from Tunis (Tunisia) (Figure 1). The segregation of the trait suggests an AR mode of inheritance: all the affected relatives are on the last generation and one of them (IV-3) is born from first cousins (III-3 and III-4). The two other affected relatives were born from one parent with the same common ancestors as III-3 and III-4. The two other parents (III-1 and III-6) could descend from a putative common ancestor with their spouses because they all originated from a small village located in the North of Tunisia.
All family members were agree to publish clinical details and they were clinically assessed at Charles Nicolle University Hospital (Tunis) by experienced epileptologists (Hela Khiari-Mrabet and Amel Mrabet). Information on FS and afebrile seizures, age at onset, duration, type and number of seizures, intellectual outcome, antiepileptic drug therapy, and seizures outcome were obtained from their parents and case notes when available. Electroencephalographic (EEG) and magnetic resonance imaging (MRI) scans were done for all affected family members. FS+ phenotype was defined as FS persisting beyond the age of 6 years [2]. Generalized tonic clonic seizures (GTCS) and absence seizures were defined according to the criteria established by the “International League against Epilepsy” [42-44].
Tunisian control group
Seventy unrelated healthy Tunisian controls (mean age 48.5 ± 16.5) were recruited at Charles Nicolle University Hospital (Tunis). These individuals did not have any personal and familial history of seizures.
Genomic DNA
Oral and written informed consent was obtained from all participants or their legal representatives. Blood samples were collected and genomic DNA was extracted from peripheral blood leucocytes by phenol/chloroform procedure [45].
Exclusion mapping
Linkage analysis with a panel of 36 microsatellite markers spanning the FEB loci (FEB1, FEB2, FEB3, FEB4, FEB5, FEB6 and FEB11/ETL5) and GEFS+ loci (GEFS+1, GEFS+2, GEFS+3 and GEFS+4) known at that time were firstly explored (Table 2). Primer sequences are listed on Additional file 1: Table S1 and PCR conditions are available as Additional file 1. We excluded all these previously linked loci by linkage analysis.
Table 2.
Tested and excluded known FS and GEFS+ loci
| Locus | Candidate gene | Marker | Genetic position (cM) | Physical position(Mb) | LOD Scores |
|---|---|---|---|---|---|
| FEB1 |
|
D8S553 |
81.50 |
67.10 |
-9.31 |
| |
|
D8S1058 |
90.10 |
73.09 |
-10.43 |
| |
|
D8S279 |
90.20 |
73.15 |
-15.94 |
| FEB2 |
|
D19S424 |
10.97 |
3.18 |
-16.30 |
| |
|
D19S177 |
20.75 |
5.47 |
-12.04 |
| |
|
D19S1034 |
20.75 |
6.06 |
-8.99 |
| |
|
D19S406 |
25.17 |
7.33 |
-13.27 |
| |
|
D19S76 |
25.17 |
7.56 |
-9.23 |
| FEB4 |
MASS1 |
MASS1Int85 |
99.30 |
90.20 |
-12.83 |
| |
|
D5S644 |
104.76 |
95.84 |
-12.95 |
| FEB5 |
|
D6S1620 |
129.10 |
129.99 |
-5.31 |
| |
|
D6S472 |
132.70 |
132.58 |
-9.52 |
| FEB6 |
IMPA2 |
D18S1153 |
34.70 |
10.12 |
-10.08 |
| |
|
IMPA2Int5 |
42.00 |
12.01 |
-16.96 |
| D18S71 |
42.80 |
12.59 |
-9.78 |
||
| GEFS+1 |
SCN1B |
D19S425 |
58.70 |
40.19 |
-11.14 |
| |
|
SCN1BInt1 |
59.00 |
40.21 |
-11.52 |
| |
|
D19S893 |
61.40 |
40.26 |
-11.43 |
| GEFS+2/FEB3 |
SCN1A/SCN2A |
D2S2330 |
175.50 |
166.41 |
-10.71 |
| |
|
D2S2345 |
177.20 |
168.43 |
-6.59 |
| |
|
D2S2314 |
188.90 |
176.57 |
-5.54 |
| GEFS+3 |
GABRG2 |
D5S1465 |
162.00 |
161.35 |
-12.02 |
| |
|
GABRG2Int1 |
162.50 |
161.44 |
-17.29 |
| |
|
GABRG2Int5 |
162.60 |
161.48 |
-17.29 |
| |
|
D5S2576 |
162.63 |
161.51 |
-16.17 |
| |
|
D17S2131 |
162.75 |
161.88 |
-15.99 |
| |
|
D5S422 |
163.90 |
162.09 |
-14.19 |
| GEFS+4 |
|
D2S1360 |
38.33 |
17.36 |
-3.68 |
| |
|
D2S305 |
38.87 |
19.28 |
-3.69 |
| |
|
D2S2342 |
40.47 |
20.19 |
-3.81 |
| FEB11/ETL5 |
CPA6 |
D8S507 |
75.00 |
59.30 |
-5.18 |
| |
|
D8S1812 |
78.30 |
60.85 |
-5.47 |
| |
|
D8S1843 |
78.80 |
62.42 |
-9.12 |
| |
|
D8S544 |
81.00 |
65.75 |
-11.07 |
| |
|
D8S533 |
81.50 |
67.16 |
-11.37 |
| D8S1775 | 85.80 | 68.99 | -11.99 |
Genetic linkage and haplotype analysis
A genome-wide linkage study was performed by using the Illumina Gene Chip Linkage Infinium II HumanLinkage-24 Panel Breadchip® (Illumina, San Diego, CA), which allowed us to genotype 5913 SNPs.
Two-point and multipoint logarithm of odds (LOD) scores were calculated with MERLIN 1.1 program, assuming an AR inheritance with complete penetrance, a disease allele frequency of 0.00001 and a phenocopy rate of 0. The haplotype reconstruction for family members was done manually, regardless of the individual affection status.
To confirm the region with a LOD score of 2.51 found by linkage analysis and in order to refine the mapping, we chose a set of 8 microsatellite markers located at the chromosome 22q13.31 and distributed at average intervals of 0.2 Mb (Figure 1B). According to the linked region defined by positive LOD score, we chose some microsatellite repeats using UCSC database (http://genome.ucsc.edu) by specifying “microsatellite” in “variations and repeats” part. Primer sequences (Additional file 1: Table S2) and PCR conditions are available on Additional file 1.
Genome wide CNVs analysis
We have also performed a copy number variations (CNVs) analysis by using the Agilent Human Genome comparative genomic hybridization (CGH) Microarray Kit 244 K (Agilent, Santa Clara, CA). The slide was scanned on an Agilent DNA microarray scanner. Data were obtained by Agilent Feature extraction software 9 and analyzed with Agilent CGH analytics 3.4 software, using the statistical algorithms zscores and ADM-2 with a sensitivity threshold of 2.5 and 6.0, respectively, and a moving average window of 0.2 Mb. Mapping data were analyzed on the human genome sequence using the NCBI database Build 35, Hg17 (http://www.ncbi.nlm.nih.gov).
Candidate gene mutational analysis
Linkage results showed six other loci with positive LOD scores in the vicinity of the 527 kb IBD region. In one of them lies KCNJ4, which encodes a potassium inwardly-rectifying channel (subfamily J and member 4). According to its physical position and its functional role, this putative candidate gene for epilepsy, was sequenced in the present consanguineous family. We performed PCR for the two exons and splice junctions. We designed flanking exon primers from published sequences with the Primer3 (version.0.4.0) online program (http://bioinfo.ut.ee/primer3-0.4.0/primer3/input.htm). Primer sequences are listed on Additional file 1: Table S3 and PCR conditions are available on Additional file 1.
Exome sequencing
Paired-end sequencing was performed using Illumina GAIIx/HiSeq 2000 instruments (Illumina, San Diego, CA) available at the Department of Genetic Medecine of the School of Medicine from Geneva. Sequence capture was performed using Agilent Sure Select Technology (Agilent, Santa Clara, CA) using either the All Exome kits or custom design corresponding to the linkage intervals and IBD regions. We typically obtain in excess of 30 million read pairs (60 million reads) of 76 nucleotides per GAIIx lane and 100 million reads pairs of 100 bp per HiSeq2000 lane allowing multiplexing of 2–3 exomes per lane.
Alignment of reads and call of single nucleotide variants and small indels were done using the latest version of MAQ/BWA [46] and PinDel [47] softwares. Only genetic variants with high-quality score were further investigated. Variant annotation was performed using the ANNOVAR package [48]. Generation of high-quality data necessary for reliable SNP detection across the targeted regions required 30-fold genomic coverage (equivalent to 15-fold coverage per haplotype).
Each DNA variant was subsequently compared to Single Nucleotide Polymorphism database (dbSNP132, NCBI) and 1000genome project dataset. Only the unique and novel DNA variants were selected for additional filtering based on the nature of the mutation (gene structure location, nucleotide conservation, codon change, de novo occurrence) and familial segregation. Polyphen2 [49] and Sift [50] programs were used to assess the pathogenecity of the filtered variants. If necessary, the final list of potential pathogenic variants were further refined by selecting genes functionally relevant to epilepsy. T1478M of CACNA1I, S1720L of TNRC6B and S202P of LDLRAP1 were further explored by classic Sanger sequencing and high resolution melt (HRM) assay using a Rotor-Gene 6000 instrument (Corbett Life Science, Australia). More details are given in Additional file 1 and in Additional file 1: Table S3 and Additional file 1: Table S4.
Competing interests
The authors declare that they have no competing interests.
Author’s contributions
NB designed the study and wrote the manuscript. FB performed the genome wide CNVs analysis. MG performed the exome sequencing analysis. CBS interpreted the clinical data. HKM and AMr performed the clinical study. ABAE, AM and AS performed supervision of the present study and critical reading of manuscript. All authors have read and approved the final manuscript.
Author’s information
Nejla Belhedi (NB): PhD student; Laboratory of Genetic Immunology and Human Pathologies University Tunis el ManarTunisia and Neurological Department Charles Nicolle Hospital Tunis Tunisia. Frederic Bena (FB): MD, FAMH; Department of Genetic Medecine and Laboratory University Hospitals of Geneva Switzerland. Michel Guipponi (MG): PhD; Department of Genetic Medecine and Laboratory University Hospitals of Geneva Switzerland. Chiraz Souissi-Bouchlaka (CSB): PhD; Laboratory of Genetic Immunology and Human Pathologies University Tunis el ManarTunisia. Hela Khiari-Mrabet (HKM): MD; Neurological Department Charles Nicolle Hospital Tunis Tunisia. Amel Mrabet (AMr): MD; Neurological Department Charles Nicolle Hospital Tunis Tunisia. Amel BenAmmar Elgaaied (ABAE): PhD; Laboratory of Genetic Immunology and Human Pathologies University Tunis el ManarTunisia. Alain Malafosse (AM): MD, PhD; Department of Genetic Medecine and Laboratory University Hospitals of Geneva and Department of Psychiatry University of Geneva Switzerland. Annick Salzmann (AS): PhD; Department of Psychiatry, University of Geneva, Switzerland.
Supplementary Material
Primer Sequences of Microsatellite Markers for Exclusion Mapping. Table S2 Primers Sequences of Microsatellite Markers for Haplotype Analysis on 22q13.31. Table S3 Primers Sequences for Sanger Sequencing. Table S4 Primers Sequences for HRM assay. Table S5 Lod Scores of linked locus on chromosome 22q13.31. Table S6 Loci on Chromosome 22q13.31 with Positive LOD Score. Table S7 17ACchr22 Microsatellite Genotype and Allele Distributions in the Tunisian Control Population.
LOD scores were calculated using Merlin program. A new region was linked on the chromosome 22q with a maximum of LOD score of 2.51.
Contributor Information
Nejla Belhedi, Email: belhedinejla@yahoo.fr.
Frédérique Bena, Email: Frederique.Bena@hcuge.ch.
Amel Mrabet, Email: amemrabet@rns.tn.
Michel Guipponi, Email: Michel.Guipponi@hcuge.ch.
Chiraz Bouchlaka Souissi, Email: chirazsouissi@isajc.rnu.tn.
Hela Khiari Mrabet, Email: helmrabet@yahoo.fr.
Amel Benammar Elgaaied, Email: a_benammarelgaaied@yahoo.fr.
Alain Malafosse, Email: Alain.Malafosse@hcuge.ch.
Annick Salzmann, Email: Annick.Salzmann@hcuge.ch.
Acknowledgments
We are indebted to our patients and their families for their tireless support during our investigations. We thank all members of the Departement of Genetic Medecine and Laboratory, University Hospitals of Geneva, Switzerland, all members of the laboratory of Genetics, Immunology and Human pathologies and all members of Neurological Department, Charles Nicolle Hospital, Tunisia.
References
- Sander JW. The epidemiology of epilepsy revisited. Curr Opin Neurol. 2003;16:165–170. doi: 10.1097/00019052-200304000-00008. [DOI] [PubMed] [Google Scholar]
- Scheffer IE, Berkovic SF. Generalized epilepsy with febrile seizures plus: a genetic disorder with heterogeneous clinical phenotypes. Brain. 1997;120:479–490. doi: 10.1093/brain/120.3.479. [DOI] [PubMed] [Google Scholar]
- Singh R, Scheffer IE, Crossland K, Berkovic SF. Generalized epilepsy with febrile seizures plus: a common childhood-onset genetic epilepsy syndrome. Ann Neurol. 1999;45(1):75–81. doi: 10.1002/1531-8249(199901)45:1<75::AID-ART13>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Scheffer IE, Zhang YH, Jansen FE, Dibbens L. Dravet syndrome or genetic (generalized) epilepsy with febrile seizures plus. Brain Dev. 2009;31:394–400. doi: 10.1016/j.braindev.2009.01.001. [DOI] [PubMed] [Google Scholar]
- Mulley JC, Nelson P, Guerrero S, Dibbens L, Iona X, McMahon JM, Harkin L, Schouten J, Yu S, Berkovic SF, Scheffer IE. A new molecular mechanism for severe myoclonic epilepsy of infancy: exonic deletions in SCN1A. Neurology. 2006;67:1094–1095. doi: 10.1212/01.wnl.0000237322.04338.2b. [DOI] [PubMed] [Google Scholar]
- Ito M, Yamakawa K, Sugawara T, Hirose S, Fukuma G, Kaneko S. Phenotypes and genotypes in epilepsy with febrile seizures plus. Epilepsy Res. 2006;70S:S199–S205. doi: 10.1016/j.eplepsyres.2005.11.028. [DOI] [PubMed] [Google Scholar]
- Wallace RH, Wang DW, Singh R, Scheffer IE, George AL Jr, Phillips HA, Saar K, Reis A, Johnson EW, Sutherland GR, Berkovic SF, Mulley JC. Febrile seizures and generalized epilepsy associated with a mutation in the Na+channel beta1 subunit gene SCN1B. Nat Genet. 1998;19:366–370. doi: 10.1038/1252. [DOI] [PubMed] [Google Scholar]
- Baulac S, Gourfinkel-An I, Picard F, Rosenberg-Bourgin M, Prud’homme JF, Baulac M, Brice A, LeGuern E. A second locus for familial generalized epilepsy with febrile seizures plus maps to chromosome 2q21-q33. Am J Hum Genet. 1999;65:1078–1085. doi: 10.1086/302593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulard B, Guiponni M, Chaigne D, Mouthon D, Buresi C, Malafosse A. Identification of a new locus for generalized epilepsy with febrile seizures plus (GEFS+) on chromosome 2q24-q33. Am J Hum Genet. 1999;65(5):1396–1400. doi: 10.1086/302621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escayg A, MacDonald BT, Meisler MH, Baulac S, Huberfeld G, An-Gourfinkel I, Brice A, LeGuern E, Moulard B, Chaigne D, Buresi C, Malafosse A. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS + 2. Nat Genet. 2000;24:343–435. doi: 10.1038/74159. [DOI] [PubMed] [Google Scholar]
- Baulac S, Huberfeld G, Gourfinkel-An I, Mitropoulou G, Beranger A, Prud’homme JF, Baulac M, Brice A, Bruzzone R, LeGuern E. First genetic evidence of GABA(A) receptor dusfunction in epilepsy: a mutation in the gamma2-subunit gene. Nat Genet. 2001;28(1):46–48. doi: 10.1038/ng0501-46. [DOI] [PubMed] [Google Scholar]
- Poduri A, Wang Y, Gordon D, Barral-Rodriguez S, Barker-Cummings C, Ulgen A, Chitsazzadeh V, Hill RS, Risch N, Hauser WA, Pedley TA, Walsh CA, Ottman R. Novel susceptibility locus at chromosome 6q16.3-22.31 in a family with GEFS+ Neurology. 2009;73:1264–1272. doi: 10.1212/WNL.0b013e3181bd10d3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audenaert D, Claes L, Claeys KG, Deprez L, Van Dyck T, Goossens D, Del-Favero J, Van Paesschen W, Van Broeckhoven C, De Jonghe P. A novel susceptibility locus at 2p24 for generalized epilepsy with febrile seizures plus. J Med Genet. 2005;42:947–952. doi: 10.1136/jmg.2005.031393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baulac S, Gourfinkel-An I, Couarch P, Depienne C, Kaminska A, Dulac O, Baulac M, LeGuern E, Nabbout R. A novel locus for generalized epilepsy with febrile seizures plus in French families. Arch Neurol. 2008;65:943–951. doi: 10.1001/archneur.65.7.943. [DOI] [PubMed] [Google Scholar]
- Dibbens LM, Feng HJ, Richards MC, Harkin LA, Hodgson BL, Scott D, Jenkins M, Petrou S, Sutherland GR, Scheffer IE, Berkovic SF, Macdonald RL, Mulley JC. GABRD encoding a protein for extra- or peri-synaptic GABA-A receptors is a susceptibility locus for generalized epilepsies. Hum Mol Genet. 2004;13:1315–1319. doi: 10.1093/hmg/ddh146. [DOI] [PubMed] [Google Scholar]
- Sugawara T, Tsurubuchi Y, Agarwala KL, Ito M, Fukuma G, Mazaki-Miyazaki E, Nagafuji H, Noda M, Imoto K, Wada K, Mitsudome A, Kaneko S, Montal M, Nagata K, Hirose S, Yamakawa K. A missense mutation of the Na+channel alpha II subunit gene Na(v)1.2 in a patient with febrile and afebrile seizures causes channel dysfunction. Proc Natl Acad Sci U S A. 2001;98:6384–6389. doi: 10.1073/pnas.111065098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg AT, Levy SR, Testa FM, Shinnar S. Classification of childhood epilepsy syndromes in newly diagnosed epilepsy: interrater agreement and reasons for disagreement. Epilepsia. 1999;40:439–444. doi: 10.1111/j.1528-1157.1999.tb00738.x. [DOI] [PubMed] [Google Scholar]
- Tsuboi T. Genetic analysis of febrile convulsions: twin and family studies. Hum Genet. 1987;75(1):7–14. doi: 10.1007/BF00273830. [DOI] [PubMed] [Google Scholar]
- Rich SS, Annegers JF, Hauser WA, Anderson VE. Complex segregation analysis of febrile convulsions. Am J Hum Genet. 1987;41(2):249–257. [PMC free article] [PubMed] [Google Scholar]
- Wallace RH, Berkovic SF, Howell RA, Sutherland GR, Mulley JC. Suggestion of a major gene for familial febrile convulsions mapping to 8q13-21. Am J Med Genet. 1996;33:308–312. doi: 10.1136/jmg.33.4.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EW, Dubovsky J, Rich SS, O’Donovan CA, Orr HT, Anderson VE, Gil-Nagel A, Ahmann P, Dokken CG, Schneider DT, Weber JL. Evidence for a novel gene for familial febrile convulsions, FEB2, linked to chromosome 19p in an extended family from the Midwest. Hum Mol Genet. 1998;7:63–67. doi: 10.1093/hmg/7.1.63. [DOI] [PubMed] [Google Scholar]
- Peiffer A, Thompson J, Charlier C, Otterud B, Varvil T, Pappas C, Barnitz C, Gruenthal K, Kuhn R, Leppert M. A locus for febrile seizures (FEB3) maps to chromosome 2q23-24. Ann Neurol. 1999;46:671–678. doi: 10.1002/1531-8249(199910)46:4<671::AID-ANA20>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Nakayama J, Hamano K, Iwasaki N, Nakahara S, Horigome Y, Saitoh H, Aoki T, Maki T, Kikuchi M, Migita T, Ohto T, Yokouchi Y, Tanaka R, Hasegawa M, Matsui A, Hamaguchi H, Arinami T. Significant evidence for linkage of febrile seizures to chromosome 5q14-q15. Hum Mol Genet. 2000;9:87–91. doi: 10.1093/hmg/9.1.87. [DOI] [PubMed] [Google Scholar]
- Nabbout R, Prud’homme JF, Herman A, Feingold J, Brice A, Dulac O, LeGuern E. A locus for simple pure febrile seizures maps to chromosome 6q22-q24. Brain. 2002;125:2668–2680. doi: 10.1093/brain/awf281. [DOI] [PubMed] [Google Scholar]
- Nakayama J, Yamamoto N, Hamano K, Iwasaki N, Ohta M, Nakahara S, Matsui A, Noguchi E, Arinami T. Linkage and association of febrile seizures to the IMPA2 gene on human chromosome 18. Neurology. 2004;63:1803–1807. doi: 10.1212/01.WNL.0000144499.34164.E0. [DOI] [PubMed] [Google Scholar]
- Hedera P, Ma S, Blair MA, Taylor KA, Hamati A, Bradford Y, Abou-Khalil B, Haines JL. Identification of a novel locus for febrile seizures and epilepsy on chromosome 21q22. Epilepsia. 2006;47:1622–1628. doi: 10.1111/j.1528-1167.2006.00637.x. [DOI] [PubMed] [Google Scholar]
- Audenaert D, Schwartz E, Claeys KG, Claes L, Deprez L, Suls A, Van Dyck T, Lagae L, Van Broeckhoven C, Macdonald RL, De Jonghe P. A novel GABRG2 mutation associated with febrile seizures. Neurology. 2006;67:687–690. doi: 10.1212/01.wnl.0000230145.73496.a2. [DOI] [PubMed] [Google Scholar]
- Nabbout R, Baulac S, Desguerre I, Bahi-Buisson N, Chiron C, Ruberg M, Dulac O, LeGuern E. New locus for febrile seizures with absence epilepsy on 3p and a possible modifier gene on 18p. Neurology. 2007;24:1374–1381. doi: 10.1212/01.wnl.0000260062.02829.e3. [DOI] [PubMed] [Google Scholar]
- Dai XH, Chen WW, Wang X, Zhu QH, Li L, Liu MG, Wang QK, Liu JY. A novel genetic locus for familial febrile seizures and epilepsy on chromosome 3q26.2-q26.33. Hum Genet. 2008;124(4):423–429. doi: 10.1007/s00439-008-0566-9. [DOI] [PubMed] [Google Scholar]
- Salzmann A, Guipponi M, Lyons PJ, Fricker LD, Sapio M, Lambercy C, Buresi C, Ouled Amar Bencheikh B, Lahjouji F, Ouazzani R, Crespel A, Chaigne D, Malafosse A. Carboxypeptidase A6 gene (CPA6) mutations in a recessive familial form of febrile seizures and temporal lobe epilepsy and in sporadic temporal lobe epilepsy. Hum Mutat. 2012;33:124–135. doi: 10.1002/humu.21613. [DOI] [PubMed] [Google Scholar]
- Mantegazza M, Gambardella A, Rusconi R, Schiavon E, Annesi F, Cassulini RR, Labate A, Carrideo S, Chifari R, Canevini MP, Canger R, Franceschetti S, Annesi G, Wanke E, Quattrone A. Identification of an Nav1.1 sodium channel (SCN1A) loss-of-function mutation associated with familial simple febrile seizures. Proc Natl Acad Sci U S A. 2005;102(50):18177–18182. doi: 10.1073/pnas.0506818102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama J, Fu YH, Clark AM, Nakahara S, Hamano K, Iwasaki N, Matsui A, Arinami T, Ptacek LJ. A nonsense mutation of the MASS1 gene in a family with febrile and afebrile seizures. Ann Neurol. 2002;52(5):654–657. doi: 10.1002/ana.10347. [DOI] [PubMed] [Google Scholar]
- Singh NA, Pappas C, Dahle EJ, Claes LR, Pruess TH, De Jonghe P, Thompson J, Dixon M, Gurnett C, Peiffer A, White HS, Filloux F, Leppert MF. A role of SCN9A in human epielpsies, as a cause of febrile seizures and as a potential modifier of Dravet syndrome. PLoS Genet. 2009;5(9):e1000649. doi: 10.1371/journal.pgen.1000649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Botstein D. Homozygosity mapping: a way to map human recessive traits with the DNA of inbred children. Science. 1987;236:1567–1570. doi: 10.1126/science.2884728. [DOI] [PubMed] [Google Scholar]
- Hochstenbach R, Buizer-Voskamp JE, Vorstman JAS, Ophoff RA. Genome arrays for the detection of copy number variations in idiopathic generalized epilepsy and neuropsychiatric disorders: lessons for diagnostic workflow and research. Cytogenet Genome res. 2011;135:174–202. doi: 10.1159/000332928. [DOI] [PubMed] [Google Scholar]
- Mefford HC, Muhle H, Ostertag P, Von Spiczak S, Buysse K, Baker C, Franke A, Malafosse A, Genton P, Thomas P, Gurnett CA, Schreiber S, Bassuk AG, Guipponi M, Stephani U, Helbig I, Eichler EE. Genome-wide copy number variation in epilepsy: novel susceptibility loci in idiopathic generalized and focal epilepsies. PloS Genet. 2010;6(5):e 1000962. doi: 10.1371/journal.pgen.1000962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helbig I, Mefford HC, Sharp AJ, Guipponi M, Fichera M, Franke A, Muhle H, de Kovel C, Baker C, von Spiczak S, Kron KL, Steinich I, Kleefuss-Lie AA, Leu C, Gaus V, Schmitz B, Klein KM, Reif PS, Rosenow F, Weber Y, Lerche H, Zimprich F, Urak L, Fuchs K, Feucht M, Genton P, Thomas P, Visscher F, de Haan GJ, Møller RS, Hjalgrim H, Luciano D, Wittig M, Nothnagel M, Elger CE, Nürnberg P, Romano C, Malafosse A, Koeleman BP, Lindhout D, Stephani U, Schreiber S, Eichler EE, Sander T. 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nat Genet. 2009;41:160–162. doi: 10.1038/ng.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CP, Lin SP, Chern SR, Tsai FJ, Wu PC, Lee CC, Chen YT, Chen WL, Wang W. A de novo 7.9 Mb deletion in 22q13.2/qter in a boy with autistic features, epilepsy, developmental delay, atopic dermatitis and abnormal immunological findings. European J Med Genet. 2010;53:329–332. doi: 10.1016/j.ejmg.2010.06.004. [DOI] [PubMed] [Google Scholar]
- Tempel W, Tong Y, Dimov S, Bochkarev A, Park H. First crystallographic models of human TBC domains in the context of a family-wide structural analysis. Proteins. 2008;71(1):497–502. doi: 10.1002/prot.21885. [DOI] [PubMed] [Google Scholar]
- Shapshak P, Duncan R, Kangueane P, Somboonwit C, Sinnott J, Commins D, Singer E, Levine A. HIV associated dementia and HIV encephalitis II: genes on chromosome 22 expressed in individually microdissected globus pallidus neurons (preliminary analysis) Bioinformation. 2011;6(5):183–186. doi: 10.6026/97320630006183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yashin AI, Wu D, Arbeev KG, Ukraintseva SV. Joint influence of small-effect genetic variants on human longevity. Aging (Albany NY) 2010;2(9):612–620. doi: 10.18632/aging.100191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commission on Classification and Terminology of the International League against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia. 1989;30:389–399. doi: 10.1111/j.1528-1157.1989.tb05316.x. [DOI] [PubMed] [Google Scholar]
- Berg AT, Scheffer IE. New concepts in classification of epilepsies: entering the 21 st century. Epilepsia. 2011;52:1058–1062. doi: 10.1111/j.1528-1167.2011.03101.x. [DOI] [PubMed] [Google Scholar]
- Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised clinical and electroencephalographic classification of epileptic seizures. Epilepsia. 1981;22:489–501. doi: 10.1111/j.1528-1157.1981.tb06159.x. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsh EF, Maniatis T. Molecular cloning: a laboratory manual. NY: Cold spring Harbor; 1989. [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–2871. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. ANNOVAR: Functional annotation of genetic variants from next-generation sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primer Sequences of Microsatellite Markers for Exclusion Mapping. Table S2 Primers Sequences of Microsatellite Markers for Haplotype Analysis on 22q13.31. Table S3 Primers Sequences for Sanger Sequencing. Table S4 Primers Sequences for HRM assay. Table S5 Lod Scores of linked locus on chromosome 22q13.31. Table S6 Loci on Chromosome 22q13.31 with Positive LOD Score. Table S7 17ACchr22 Microsatellite Genotype and Allele Distributions in the Tunisian Control Population.
LOD scores were calculated using Merlin program. A new region was linked on the chromosome 22q with a maximum of LOD score of 2.51.