

To the Editor:
The mapping of SNPs in human genomes has generated a lot of interest from both the biomedical research community and industry. In conjunction with SNP mapping, researchers have shown that haplotypes possess considerably greater potential than the traditional single-SNP approach in disease-gene mapping and in our understanding of complex landscapes of linkage disequilibrium (LD) (Goldstein 2001). In silico methods for haplotype reconstruction have attracted much attention because of their cost-effectiveness and accuracy (Tishkoff et al. 2000) and have played an important role in the definition of human haplotype block structure and in candidate-gene studies of complex traits (Tabor et al. 2002). In a recent publication, Niu et al. (2002) proposed a partition-ligation (PL) strategy and implemented it together with Gibbs sampling, to estimate haplotype phases for a large number of SNPs. Although the resulting program, HAPLOTYPER, has been in high demand from many research groups, a significant portion of researchers are also strongly interested in using an expectation-maximization (EM)–based algorithm. In the present letter, we describe how to combine the PL strategy with the EM algorithm and how to handle the local-mode problem. We also present a fast and robust method of computing the variance of the estimated haplotype frequencies. Some related issues concern the handling of missing data and the multiple imputations of haplotype phases.
The EM algorithm is arguably the most popular statistical algorithm, because of its interpretability and stability. Compared to the Gibbs sampler, the EM approach is a deterministic procedure, requires less computing time, and is easier for convergence check. The output of the EM algorithm, if not trapped in a local mode, is the maximum-likelihood estimate (MLE), which possesses well-established statistical properties. However, the capability of most EM-based approaches is restricted to approximately one dozen loci, because of the memory constraint. A recently developed program, SNPHAP (see David Clayton's Web site [SNPHAP: A Program for Estimating Frequencies of Large Haplotypes of SNPs]), is an exception that, although different from the PL strategy, can handle many more linked loci by using a progressive-extension technique.
The essential steps of the PL strategy (Niu et al. 2002) are as follows: One first breaks down all of the marker loci into stretches of “atomistic” units and then uses either the EM algorithm or the Gibbs sampler to construct haplotypes for each unit and to rebuild the phase hierarchically, through a bottom-up approach. For example, an individual represented in the lipoprotein lipase (LPL) gene SNP data set (Nickerson et al. 1998) has the genotype (01200001000000000100010), where 0 stands for heterozygote and 1 and 2 stand for wild-type and mutant homozygotes, respectively. Since there are 18 heterozygous loci, the standard EM algorithm has to consider 218 possible haplotypes, making it extremely costly for haplotype estimation. Using the PL strategy, we divide the linked loci into four “atomistic” units—(012000), (010000), (000001), and (00010)—and use the EM algorithm to estimate partial haplotypes within each unit. Afterward, two adjacent partial haplotypes are “ligated” by using the EM algorithm again, just like phasing two linked multiallelic markers. The ligation process is repeated until the complete phase is determined.
It is well known that the EM algorithm can be trapped in a local mode. This problem becomes a more serious issue for the PL-EM strategy, because every atomistic haplotype construction or ligation step involves a complete EM algorithm implementation. A naive implementation of the ligation step considers only the partial haplotypes that have nonzero estimated frequencies in the previous EM step. However, it appears that one phase configuration (and the corresponding haplotypes with nonzero estimated frequencies) is more likely when looking only at a partial set of loci, whereas a different configuration is more likely when all loci are taken into consideration. For example, consider the set of individuals with the following genotype data on four loci—(A/A A/A T/T T/T), (A/A A/A T/T T/T), (A/A G/G T/T T/T), (A/A G/G C/C C/C), (A/A G/G C/C C/C), and (A/G A/G T/T T/T). If just the first two loci are concerned, then the EM algorithm estimates the haplotype frequencies as 7/12, 4/12, and 1/12, for (AG), (AA), and (GA), respectively. When all four loci are considered together, however, the EM gives rise to four haplotypes—(AATT), (AGCC), (AGTT), and (GGTT), with frequencies 5/12, 4/12, 2/12, and 1/12, respectively. Thus, had we thrown away the (GG) haplotype prematurely when only the first two SNP markers were analyzed, we would have not been able to reach the MLE.
To overcome this difficulty, we devised a “backup-buffering” strategy during the ligation step. In brief, in addition to keeping in a buffer those partial haplotypes that have EM-algorithm–estimated frequencies greater than a threshold value (e.g., ε=10-5), we also retain in the buffer some partial haplotypes whose estimated frequencies are below ε. The criterion for choosing such a backup partial haplotype is based on the rank of its average estimated frequency over all the EM iterations. The buffer size—that is, the total number of candidate partial haplotypes in a buffer—is kept as a constant in the PL process. Not surprisingly, our simulation study based on the cystic fibrosis data showed that, the larger the buffer size is, the more accurate the phasing results are (for details, see fig. A1 [online only, at J. S. Liu's Web site]).
Niu et al. (2002) observed a modest performance improvement when recombination hotspots were used as the partition sites. Recently, hotspot-detection algorithms, such as a greedy algorithm (Patil et al. 2001) and a dynamic programming approach (Zhang et al. 2002), have been developed. Our PL-EM program can incorporate the information revealed by such algorithms by allowing the user to specify desirable partition points (for details and download of the PL-EM program, see J. S. Liu's Web site). We also conducted an empirical study on the effects that different partition sizes, K, have when hotspot information is absent. Although little difference in phasing performance was observed when three different partition sizes were used—3–4, 5–8, or 9–16 (see fig. A2 [online only, at J. S. Liu's Web site])—we found that the computation time increased sharply when the coarsest partition was used. Overall, K = 5–8 appeared to be a good choice for the atomistic unit size.
Several EM-based algorithms—including HAPLO (Hawley and Kidd 1995), Arlequin (Schneider et al. 2000), and the Mx program (Neale et al. 1999)—provide the variance estimates for the estimated haplotype frequencies. However, since these methods handle no more than ∼20 loci, their variance-estimation method cannot be directly used by the PL-EM program. Instead, we implemented with the PL-EM program a simple and robust approach, to estimate the variances or SEs of the frequencies of those haplotypes that were selected at the final ligation stage.
Let Y be the observed genotype data, Z be the missing phase information, and θ be the vector of haplotype frequencies. As noted by Louis (1982), the Hessian matrix of θ can be computed via an identity analogous to the variance-decomposition rule,
 |
and the variance-covariance matrix of the MLE, 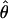 , is the inverse of this matrix evaluated at
, is the inverse of this matrix evaluated at 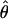 . The first term on the right-hand side of equation (1) can be computed as
. The first term on the right-hand side of equation (1) can be computed as
 |
where m is the number of all candidate haplotypes, ni is the number of occurrences of haplotype i in Z, and the expectation is taken for the ni (which is a function of Z) with θ fixed at the MLE. The second term on the right-hand side needs the variance-covariance matrix of
 |
The calculation of 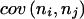 , for example, can be achieved by observing in each individual the probability of the joint occurrence of haplotypes i and j.
, for example, can be achieved by observing in each individual the probability of the joint occurrence of haplotypes i and j.
In the presence of many heterozygous loci, some rare haplotypes with very low frequencies are likely to occur. Then, the inversion of the Hessian matrix becomes computationally burdensome and numerically unstable. Since scientists are mostly concerned with the variance of each 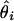 instead of covariances among the
instead of covariances among the 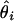 s, we introduce a new, robust method of computing these marginal variances. Take
s, we introduce a new, robust method of computing these marginal variances. Take 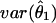 , for example: by applying equation (1) to a reparameterization of the model with θ′=(θ1,1-θ1) and θ′′=(θ2,…,θm)/(1-θ1), we have
, for example: by applying equation (1) to a reparameterization of the model with θ′=(θ1,1-θ1) and θ′′=(θ2,…,θm)/(1-θ1), we have
 |
Thus, 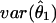 is equal to the reciprocal of the above quantity. Note that the new method and Louis’s method give identical variance estimates if the inversion of the Hessian matrix (eq. [1]) is accurate. Intuitively, the first term on the right-hand side of equation (2) is the standard variance estimate when there is no uncertainty in phasing, and the second term accounts for the loss of information because of unknown phases.
is equal to the reciprocal of the above quantity. Note that the new method and Louis’s method give identical variance estimates if the inversion of the Hessian matrix (eq. [1]) is accurate. Intuitively, the first term on the right-hand side of equation (2) is the standard variance estimate when there is no uncertainty in phasing, and the second term accounts for the loss of information because of unknown phases.
An example of the SE calculation for estimated haplotype frequencies is shown, in table 1, for the LPL data from Nickerson et al. (1998). This example also illustrates that haplotypes can shed new light on population migration and admixture. To better understand the properties of the estimated SEs, we conducted a simulation study using the 12 distinct haplotypes from the β2-adrenergic receptor (β2AR) data set. Assuming that the 12 haplotypes have equal frequencies (1/12), we simulated 100 data sets, each consisting of 90 hypothetical individuals. The PL-EM algorithm was applied to each of the data sets, and a 95% CI for each 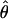 was constructed on the basis of the estimated frequencies and SEs (i.e.,
was constructed on the basis of the estimated frequencies and SEs (i.e., 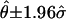 ). The number of times (in 100 trials) that the 95% CI covered the true frequency (θ=1/12) for the 12 haplotypes was 92, 88, 93, 96, 97, 96, 88, 93, 94, 92, 95, and 94, which average to 93.2%. For the purpose of calibration, we note that the average coverage of the true θ was only 93.1% when the haplotype phase information was given.
). The number of times (in 100 trials) that the 95% CI covered the true frequency (θ=1/12) for the 12 haplotypes was 92, 88, 93, 96, 97, 96, 88, 93, 94, 92, 95, and 94, which average to 93.2%. For the purpose of calibration, we note that the average coverage of the true θ was only 93.1% when the haplotype phase information was given.
Table 1.
Application of PL-EM on the LPL Data[Note]
|
Results from |
||||
| Haplotype | ID | Jackson, MS(N=24; k=28) | North Karelia, Finland (N=24; k=20) | Rochester, MN(N=23; k=22) |
| 01000001000000000100000 | H1 | .063 (.035) | .219 (.059) | .348 (.069) |
| 01000001100000000100000 | H2 | … | .073 (.039) | .045 (.031) |
| 00100110100000000000000 | H3 | .146 (.051) | … | .043 (.030) |
| 00100110100000010001100 | H4 | .104 (.044) | … | … |
| 00100110000000010000000 | H5 | .063 (.035) | … | … |
| 10101000011111110011101 | H6 | .063 (.035) | … | … |
| 00100001000000000100000 | H7 | … | .146 (.051) | .023 (.023) |
| 01000110000000000000000 | H8 | .021 (.021) | .125 (.048) | … |
| 00100000111111100011100 | H9 | … | .083 (.040) | … |
| 10111100111111111011111 | H10 | … | … | .087 (.042) |
Note.— The LPL data are based on a study by Nickerson et al. (1998). A total of 88 sites in the 7.9-kb region have been reported among the 71 individuals. Of these 88 biallelic markers, 23 met the following two criteria: (1) minor-allele frequency >20% and (2) marker missing data <2%. Both PL-EM and HAPLOTYPER were applied, to phase the entire 71 subjects by using only these 23 markers. N and k represent the sample size and the number of distinct haplotypes, respectively. Numbers shown in parentheses represent SEs of the frequency estimates. PL-EM appears to output almost the same number of haplotypes as does HAPLOTYPER (k=28, k=20, and k=22 vs. k=28, k=19, and k=22, for the Jackson, North Karelia, and Rochester samples, respectively). The number of distinct haplotypes is greatest in the Jackson sample (African Americans) and is smallest in the North Karelia sample (white Europeans). The Rochester sample shares H1 and H3 with the Jackson sample and shares H1, H2, and H7 with the North Karelia sample, indicating that this American-white population may be the result of admixture between black and European-white populations.
The presence of a significant portion of missing genotypes is a common problem when a great number of linked loci are under investigation. This missing-data problem poses a serious challenge to the existing EM haplotype-inference algorithms, even when the total number of SNP loci is moderate. In the case of missing two allele calls at one locus, for example, all three different genotype configurations—(AA), (Aa), and (aa)—have to be accounted for by the algorithm, which greatly inflates the space of candidate haplotypes. As a consequence, the standard EM algorithm not only needs a lot more memory but also converges much more slowly. The PL-EM algorithm resolves this difficulty seamlessly because of its adoption of the divide-conquer-combine strategy.
It often occurs that, for some individuals with a large number of heterozygous loci, numerous haplotype pairs (each with a nonzero probability) are compatible with their genotype data. In this case, generating all compatible haplotype phases with nontrivial probabilities is more desirable than outputting only the best phase. There is some evidence (X. Lu and J. S. Liu, unpublished data) showing that, by accounting for the phasing uncertainty, one can gain accuracy in LD mapping when using the algorithm BLADE (Liu et al. 2001; this algorithm employs a semi-hidden Markov model and a Markov-chain Monte Carlo method, for inference of the location of the disease mutation among a given set of linked markers with known genetic distances in a case-control setting). To accommodate this need of multiple-haplotype imputation, the PL-EM program can let the user choose to display either the top f most likely phases (if existing) for each individual or all phases with probabilities >0.1.
We evaluated the performances of PL-EM, HAPLOTYPER (Niu et al. 2002), and an enhanced version of PHASE (Stephens et al. 2001), using the angiotensin I–converting enzyme (ACE) data set, the β2AR gene data set, the cystic fibrosis transmembrane conductance regulator (CFTR) gene data set, and data sets produced by coalescence model–based haplotype-simulation software (see the Long Lab's Web site [Tools: Statistical Analysis and Molecular Biology Tools]). All these data sets were constructed in the same way, as described by Niu et al. (2002). The results are summarized in the left panels of figure 1. The PL-EM program’s error rate for individuals’ phasing is comparable to HAPLOTYPER, but is lower than PHASE in the first three cases, which is consistent with the studies described by Niu et al. (2002). For the coalescence simulation, PL-EM and HAPLOTYPER respectively made 35% and 11% more errors than PHASE. Note that Stephens et al. (2001) reported that the EM algorithm made ∼100% more errors than PHASE, indicating that PL-EM performed significantly better than the standard EM algorithm when the coalescence assumption is appropriate.
Figure 1.

Histograms of the average error rates based on either individual phase calls (open bars) or the proportion of incorrectly inferred loci (shaded bars), for ACE (A), β2AR (B), CFTR (C), and coalescence-simulation (D) data. For the ACE data, there are a total of 52 biallelic markers for 11 subjects (Rieder et al. 1999), and 100 independent runs for each algorithm were performed. For the β2AR data, 15 haplotype pairs (each pair corresponding to one subject) were randomly drawn from a total of 10 distinct haplotypes according to their respective frequencies, as shown by Drysdale et al. (2000); this procedure was repeated to generate a total of 100 simulated data sets. For the CFTR data, the 100 data sets were generated by randomly pairing 56 of the 57 complete haplotypes of the 23 linked SNPs in a 1.8-Mb region near the CFTR gene provided by Kerem et al. (1989). The coalescence simulation was done using the Long Lab’s algorithm. A total of 100 replications were performed for a regional size of 10 U of 4Nc, each of which consisted of 20 pairs of unphased chromosomes with 20 linked SNP loci. The error bars are shown as ±1 SE, for the new version of PHASE, HAPLOTYPER, and PL-EM. An asterisk (*) indicates that the old version of PHASE was used for this data set, because its performance is better than that of the new version.
To investigate further how the inference errors were made by the three algorithms, we looked into the following two aspects: (1) how the incorrectly inferred haplotypes differ from the true ones and (2) whether different algorithms made errors on the same individuals. For the first three data sets, PL-EM appeared to produce the least amount of incorrectly inferred loci for those wrongly inferred haplotypes, whereas, for the coalescent-based simulated data, PL-EM and HAPLOTYPER respectively produced 36% and 7% more incorrectly inferred loci than did PHASE (fig. 1, right panels). In the first three cases, most of the errors made by HAPLOTYPER and PL-EM appeared to be a subset of the errors made by PHASE (see fig. A3 [online only, at J. S. Liu's Web site]).
In summary, the PL-EM algorithm can deal with a large number of linked loci that have moderate levels of LD. It is capable of variance estimation, multiple imputation, and the handling of incomplete genotype data. In addition, PL-EM was faster than HAPLOTYPER in these examples, even with the variance estimation. Hence, in practice, if a coalescence model for the population haplotypes is too strong to assume, then PL-EM can be an attractive alternative to HAPLOTYPER, further helping scientists in the haplotype-reconstruction endeavor.
Acknowledgments
We are grateful to Chi-Hse Teng and the two anonymous reviewers for insightful comments. This research was supported in part by the National Science Foundation grants DMS-0094613 and DMS-0104129 and National Institutes of Health grant R01 HG02518-01.
Electronic-Database Information
URLs for data presented herein are as follows:
- J. S. Liu's Web Site, http://www.people.fas.harvard.edu/~junliu/plem/ (for supplemental figs. A1–A3 and detailed documentation and download instructions for the PL-EM algorithm)
- SNPHAP: A Program for Estimating Frequencies of Large Haplotypes of SNPs, http://www-gene.cimr.cam.ac.uk/clayton/software/snphap.txt
- Tools: Statistical Analysis and Molecular Biology Tools, http://hjmuller.bio.uci.edu/~labhome/coalescent.html (for coalescence model–based haplotype-simulation software)
References
- Drysdale CM, McGraw DW, Stack CB, Stephens JC, Judson RS, Nandabalan K, Arnold K, Ruano G, Liggett SB (2000) Complex promoter and coding region β2-adrenergic receptor haplotypes alter receptor expression and predict in vivo responsiveness. Proc Natl Acad Sci USA 97:10483–10488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DB (2001) Islands of linkage disequilibrium. Nat Genet 29:109–211 [DOI] [PubMed] [Google Scholar]
- Hawley ME, Kidd KK (1995) HAPLO: a program using the EM algorithm to estimate the frequencies of multi-site haplotypes. J Hered 86:409–411 [DOI] [PubMed] [Google Scholar]
- Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, et al (1989) Identification of the cystic fibrosis gene: genetic analysis. Science 245:1073–1080 [DOI] [PubMed] [Google Scholar]
- Liu JS, Sabatti C, Teng J, Keats BJ, Risch N (2001) Bayesian analysis of haplotypes for linkage disequilibrium mapping. Genome Res 11:1716–1724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis TA (1982) Finding the observed information matrix when using the EM algorithm. J R Stat Soc B 4:226–233 [Google Scholar]
- Neale MC, Boker S, Xie G, Maes H (1999) Mx: Statistical modeling. Department of Psychiatry, Medical College of Virginia, Richmond [Google Scholar]
- Nickerson DA, Taylor SL, Weiss KM, Clark AG, Hutchinson RG, Stengård J, Salomaa V, Vartiainen E, Boerwinkle E, Sing CF (1998) DNA sequence diversity in a 9.7-kb region of the human lipoprotein lipase gene. Nat Genet 19:233–240 [DOI] [PubMed] [Google Scholar]
- Niu T, Qin ZS, Xu X, Liu JS (2002) Bayesian haplotype inference for multiple linked single-nucleotide polymorphisms. Am J Hum Genet 70:157–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil N, Berno AJ, Hinds DA, Barrett WA, Doshi JM, Hacker CR, Kautzer CR, et al (2001) Blocks of limited haplotype diversity revealed by high-resolution scanning of human chromosome 21. Science 294:1719–1723 [DOI] [PubMed] [Google Scholar]
- Rieder MJ, Taylor SL, Clark AG, Nickerson DA (1999) Sequence variation in the human angiotensin converting enzyme. Nat Genet 22:59–62 [DOI] [PubMed] [Google Scholar]
- Schneider S, Roessli D, Excoffier L (2000) Arlequin: a software for population genetics data analysis. Ver 2.000. Genetics and Biometry Laboratory, Department of Anthropology, University of Geneva, Geneva [Google Scholar]
- Stephens M, Smith NJ, Donnelly P (2001) A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 68:978–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabor HK, Risch NJ, Myers RM (2002) Candidate-gene approaches for studying complex genetic traits: practical considerations. Nat Rev Genet 3:391–397 [DOI] [PubMed] [Google Scholar]
- Tishkoff SA, Pakstis AJ, Ruano G, Kidd KK (2000) The accuracy of statistical methods for estimation of haplotype frequencies: an example from the CD4 locus. Am J Hum Genet 67:518–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Deng M, Chen T, Waterman MS, Sun F (2002) A dynamic programming algorithm for haplotype block partitioning. Proc Natl Acad Sci USA 99:7335–7339 [DOI] [PMC free article] [PubMed] [Google Scholar]