

Abstract
We previously reported that a restrictive N-terminal truncation of cardiac troponin I (cTnI-ND) is up-regulated in the heart in adaptation to hemodynamic stresses. Over-expression of cTnI-ND in the hearts of transgenic mice revealed functional benefits such as increased relaxation and myocardial compliance. In the present study, we investigated the subsequent effect on myocardial remodeling. The alpha-smooth muscle actin (α-SMA) isoform is normally expressed in differentiating cardiomyocytes and is a marker for myocardial hypertrophy in adult hearts. Our results show that in cTnI-ND transgenic mice of between 2 and 3 months of age (young adults), a significant level of α-SMA is expressed in the heart as compared with wild-type animals. Although blood vessel density was increased in the cTnI-ND heart, the mass of smooth muscle tissue did not correlate with the increased level of α-SMA. Instead, immunocytochemical staining and Western blotting of protein extracts from isolated cardiomyocytes identified cardiomyocytes as the source of increased α-SMA in cTnI-ND hearts. We further found that while a portion of the up-regulated α-SMA protein was incorporated into the sarcomeric thin filaments, the majority of SMA protein was found outside of myofibrils. This distribution pattern suggests dual functions for the up-regulated α-SMA as both a contractile component to affect contractility and as possible effector of early remodeling in non-hypertrophic, non-failing cTnI-ND hearts.
Keywords: Troponin I, N-terminal truncation, α-SMA, Cardiac muscle remodeling, Cardiomyocyte, Transgenic mouse
Abbreviations: α-SMA, alpha-smooth muscle actin; cTnI, cardiac troponin I; cTnI-ND, N-terminal truncated cardiac troponin I; α-CA, alpha-cardiac actin; α-SKA, alpha-skeletal muscle actin; TnI, troponin I; TnT, Troponin T
Highlights
-
•
N-terminal truncated cardiac troponin I (cTnI-ND) upregulates α-smooth muscle actin.
-
•
This myocardial hypertrophy marker is expressed early in cardiomyocytes.
-
•
Increased relaxation by cTnI-ND has a potent effect on myocardial remodeling.
-
•
The majority of α-smooth muscle actin was found outside of myofibrils.
1. Introduction
Troponin I (TnI) is the inhibitory subunit of the troponin complex that plays a central function in the Ca2+ regulation of striated muscle contraction [1,2]. Three muscle type-specific isoforms of TnI are present in vertebrates. Cardiac TnI (cTnI) has an N-terminal extension of ∼30 amino acids, which is not present in the fast or slow skeletal muscle TnI isoforms. Embryonic hearts express slow skeletal muscle TnI with a perinatal switch to solely cardiac TnI [2]. The N-terminal extension of cTnI contains two Ser residues (Ser23 and Ser24) that are substrates of protein kinase A (PKA), and a regulatory site downstream of the β-adrenergic signaling pathway [3]. Therefore, the N-terminal extension of cTnI is an adult cardiac muscle-specific regulatory structure.
A posttranslational modification of cTnI via restrictive proteolysis was found as a molecular adaptation to cardiac stress such as that in rats following simulated microgravity [4], and in mice with β-adrenergic deficient failing hearts [5]. This restrictive proteolysis removes the cardiac-specific N-terminal extension (cTnI-ND). cTnI-ND is detectable only at low levels in normal hearts of multiple species [4] and the up-regulation adaptation to hemodynamic stresses indicates a functional significance.
cTnI-ND preserves the core structure of TnI and remains functional in the cardiac myofilaments. Double transgenic mice with endogenous cTnI gene deleted and solely cTnI-ND in the adult cardiac muscle survive well, demonstrating the non-destructive nature of cTnI-ND [6]. Transgenic mice over-expressing cTnI-ND in the hearts have apparently normal cardiac morphology and life span [7], yet exhibit increased myocardial relaxation and improved ventricular filling, consistent with a regulatory effect of removing the cardiac-specific N-terminal extension on reducing thin filament Ca2+ sensitivity and facilitated diastolic function of the cardiac muscle [7]. cTnI-ND hearts with enhanced relaxation exhibit increased systolic function and cardiac output through the Frank–Starling mechanism [7,8], providing a functional compensation in chronic heart failure [5].
The hemodynamic changes in cTnI-ND hearts may have effects on gene expression and structural remodeling in cardiomyocytes. In order to explore the beneficial function of cTnI-ND as a potential target in the treatment of heart failure, it is necessary to understand its potential effect on myocardial remodeling. In a search of remodeling markers in cTnI-ND mouse heart, the present study focused on alpha-smooth muscle actin (α-SMA), an established molecular marker of cardiac hypertrophy such as that provoked by aortic constriction [9]. Six isoforms of actin exist in mammalian cells, which can be divided in three classes [10]. The beta and gamma actins, which include β-cytosolic, γ-cytosolic, and γ-smooth muscle actin, are found in most cell types as major components of the cytoskeleton. Meanwhile, the three alpha actin isoforms, α-SMA, α-cardiac actin (α-CA), and α-skeletal muscle actin (α-SKA), are considered components of the contractile apparatus in muscle cells. During cardiac development, the three α-actins are expressed temporally, such that α-SMA expresses at the early stages and is replaced by α-SKA and α-CA in adult myocardium [11,12].
In experiments described here, we found an up-regulation of α-SMA in cTnI-ND hearts and determined its tissue type and cellular distributions. Our data demonstrate an increase in α-SMA in cardiomyocytes of young cTnI-ND mice, of which only a fraction is present in cardiac myofibrils. Our data suggest that the majority of up-regulated α-SMA may instead be part of early remodeling affectors that occur before overt adaptive changes such as hypertrophy and failure.
2. Results
2.1. Increased expression of α-SMA in cTnI-ND hearts
All mice used in the present study were between 2 and 3 months old. We have previously shown that transgenic expression of cTnI-ND to replace endogenous cTnI does not cause hypertrophy or failure [5,7,8].
Immunoblots of ventricular muscle homogenates detected a significant amount of α-SMA in cTnI-ND mouse hearts, which was not detectable in wild-type mouse hearts (Fig. 1). α-SMA was also not detectable in the ventricular homogenate of a control double transgenic mouse line that expresses a mutant cTnI on the endogenous cTnI gene null background (cTnI-K118C, [13]). These background controls indicate that (i) the endogenous cTnI gene null background did not contribute to the increased expression of α-SMA; and (ii) the contribution of vascular smooth muscle in the ventricular wall was insignificant in the amount of α-SMA detected in the cTnI-ND heart, which was also shown by the negative Western blots of calponin and SM22 (Fig. 1), two smooth muscle specific protein markers [2,14].
Fig. 1.
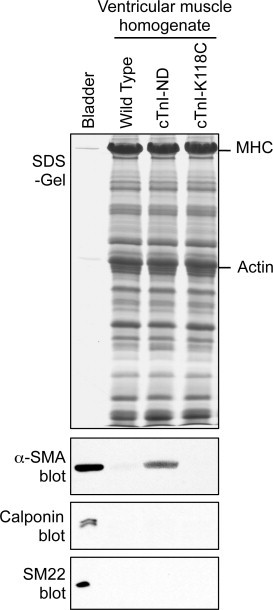
Significant expression of α-SMA in cTnI-ND mouse heart. Ventricular muscle homogenates of wild type, cTnI-ND, and cTnI-K118C (a double transgenic line expressing a cTnI mutant on the endogenous cTnI gene knockout background [13]) mice (∼80 μg total protein/lane), or mouse bladder (∼5 μg total protein/lane) were analyzed by SDS–PAGE (upper panel, Coomassie blue protein stain) and immunoblotting (lower three panels). The results detected a significant level of α-SMA in cTnI-ND hearts but not in the wild type and cTnI-118C control hearts. Smooth muscle-specific protein markers h1 and h2 calponins (RAH2 blot) and SM22 were examined as controls.
2.2. Increased blood vessel density in cTnI-ND hearts
α-SMA in adult cardiac muscle is a known marker of cardiac hypertrophy [9,15], we have previously demonstrated that cTnI-ND hearts exhibit no signs of hypertrophy [7]. To investigate the significance of increased α-SMA expression in the young and non-hypertrophic cTnI-ND mouse hearts, we carried out immunohistochemical analyses of transverse ventricular sections using an anti-α-SMA antibody (Fig. 2A). The results revealed changes in the vasculature in these young cTnI-ND mouse hearts. Quantification of the immunohistochemical images showed a significant (∼23%) increase in blood vessel density in cTnI-ND hearts as compared to wild type controls (Fig. 2B). This finding supported a possible role for cTnI-ND to induce myocardial vascular remodeling without cardiac muscle hypertrophy.
Fig. 2.
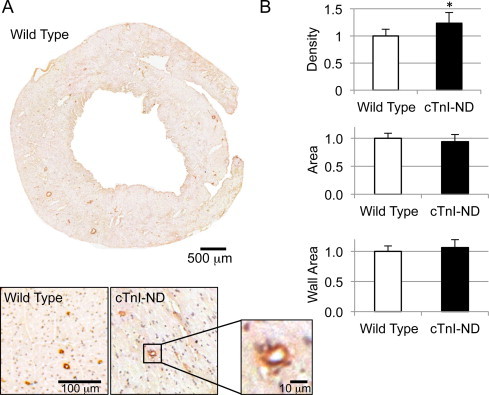
Analysis of vasculature in wild type and cTnI-ND mouse ventricle. (A) Immunohistochemical analysis of transverse heart sections from wild type and cTnI-ND hearts stained for α-SMA revealed vascular structures. A representative slice from a wild type heart is shown with magnified portions from wild type and cTnI-ND hearts to demonstrate the level of resolution used for quantification as described in Section 4. (B) Vessel density (upper plot) was increased in cTnI-ND hearts in comparison with that in wild type hearts. However, total vessel size (middle) and vessel wall area (bottom) was not significantly different between the two groups. Data are presented as mean ± SE. *P < 0.05. Wild type, 1149 vessels examined from 14 sections; cTnI-ND, 885 vessels examined from 11 sections. Wild type, N = 5; cTnI-ND, N = 4.
The data further showed that vessel size, i.e., cross-sectional area, in cTnI-ND hearts, was slightly smaller, while the relative cross-sectional areas of the vessel walls slightly increased, compared to wild type hearts, however neither difference was statistically significant (Fig 2B). Based upon the fact that α-SMA levels in vascular structures was similar in cTnI-ND hearts and wild-type hearts (Fig. 2), along with the absence of detectable smooth muscle protein markers (Fig. 1), we conclude that the significantly increased expression of α-SMA in cTnI-ND hearts was not from smooth muscle cells. The non-detectable levels of h1- and h2-calponins in cTnI-ND hearts (Fig. 1) are also consistent with the lack of significant contribution from myofibroblasts or fibroblasts. These cells have been reported to proliferate in remodeling cardiac muscle, such as that occurs following right ventricular pressure overload [15].
2.3. Expression of α-SMA in cTnI-ND cardiomyocytes
To further explore the cell source of up-regulated α-SMA in cTnI-ND hearts, freshly isolated cardiomyocytes from cTnI-ND and wild type hearts were examined by immunoblot analyses. The results, consistent with the immunohistochemical staining (Fig. 2), showed that wild-type cardiomyocytes were without detectable levels of α-SMA, whereas myocytes from all cTnI-ND hearts examined contained readily detectable levels of α-SMA (Fig. 3), albeit with significant variation among animals. The variation in expression level may reflect the variation in myocardial adaptation to the function of cTnI-ND and remains to be investigated.
Fig. 3.
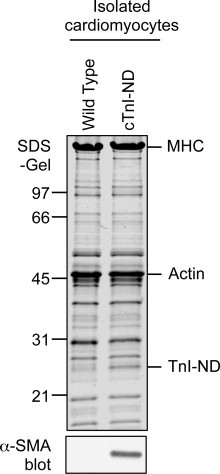
Significant expression of α-SMA in cTnI-ND mouse cardiomyocytes. Cardiomyocytes were isolated from 2 to 3 months old wild type and cTnI-ND mouse hearts and examined by SDS–PAGE (upper panel, Coomassie blue protein stain, ∼50 μg/lane) and immunoblot analyses using anti-α-SMA (lower panel). The results detected a significant level of α-SMA expressed in cTnI-ND but not wild type cardiomyocytes. Mobilities corresponding to molecular weight (MW) standards (in kDa) are indicated on the left.
Immunofluorescence analysis readily revealed striated distribution pattern for α-SMA immunofluorescence in cTnI-ND cardiomyocytes (Fig. 4A), whereas control staining in wild-type mouse cardiomyocytes produced low background signals similar to no-first-antibody controls. In comparison to α-SMA in wild-type control hearts, cTnI-ND cardiomyocytes were significantly higher in fluorescence intensity (3.3 ± 1.0-fold, Fig. 4B). The fluorescence pattern in the permeabilized cardiomyocytes suggests that the increased α-SMA protein detected by immunoblotting of the total protein extracts from the isolated cardiomyocytes involves myofilament association.
Fig. 4.
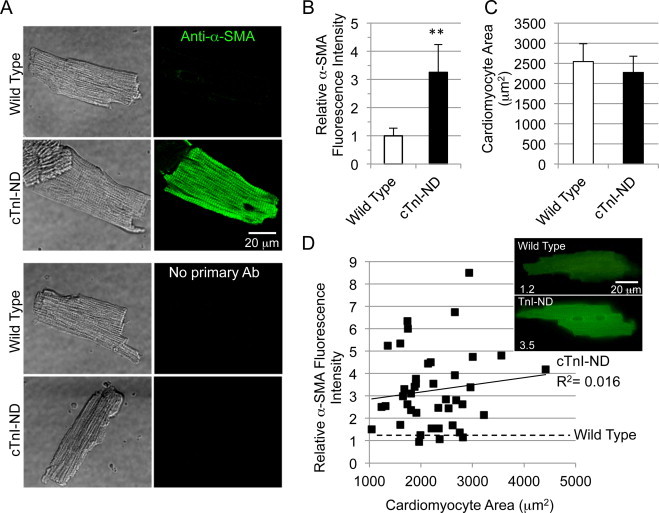
Immunofluorescence microscopy analysis of α-SMA expression in cTnI-ND cardiomyocytes. (A) Freshly isolated cardiomyocytes from wild type and cTnI-ND hearts were fixed on glass cover slips, stained with anti-α-SMA antibody and Alexa Fluor 488-conjugated second antibody. No-primary-antibody controls were included. Immunostained myocytes, also shown in phase contrast (left panels), were examined using identical imaging and off-line processing, which revealed significant levels of intracellular α-SMA in cTnI-ND but not wild type cardiomyocytes. (B) Total fluorescence intensity was determined for cells from wild type and cTnI-ND hearts by normalizing to the average level measured in wild type cells, to correct for the variable but low levels of autofluorescence within cardiomyocytes. Relative fluorescence intensities per cell area showed the significantly increased expression of α-SMA in cTnI-ND cardiomyocytes, while cell areas were similar to that of wild type cardiomyocytes (C), wild type, n = 35; cTnI-ND, n = 47. Data are presented as mean ± SE, **P < 0.01. (D) To determine whether anti-α-SMA immunofluorescence correlated with cell surface area, a measure of hypertrophic growth, cardiomyocyte area-normalized intensity was plotted against the area of each cell. Linear regression correlation confirmed that immunofluorescence intensity is independent of the cell size. Low levels of background fluorescence intensity measured for wild type cells was also independent of normalized cell area (least-squares fit of data, dashed line). Inset panels show representative cells with relative fluorescent intensity indicated on the bottom left. Wild type, N = 3; cTnI-ND, N = 3.
The area of cTnI-ND cardiomyocytes (2274 ± 399 μm2) was similar to that of wild type cells (2543 ± 439 μm2, Fig. 4C). Accordingly, there was no correlation between the levels of α-SMA and cell size (Fig. 4D), confirming that increased expression of α-SMA in cTnI-ND cardiomyocytes was not accompanied by a cellular level of hypertrophy.
2.4. Only a portion of the up-regulated α-SMA is localized to myofibrils
In adult cardiomyocytes, alpha-actins are a major constituent of the contractile apparatus, i.e., the thin filaments in the sarcomeres [16]. The significant amount of α-SMA expressed in cTnI-ND cardiomyocytes, therefore, potentially has myofilament incorporations and functions. To investigate this hypothesis, total homogenates and isolated myofibrils from ventricular muscle of wild type and cTnI-ND mouse hearts were examined for the relative level of α-SMA versus total actin.
SDS–PAGE and Western blot results showed that levels of total actin were similar for the two groups (Fig. 5A). Knowing that α-SMA was up-regulated in isolated cTnI-ND cardiomyocytes (Fig. 3), we wondered whether its level was increased proportionally in isolated myofibrils with enriched sarcomeric myofilaments. When the relative densitometry ratio of α-SMA (Western blots) to total actin (SDS-gel) was compared between total muscle homogenate and isolated myofibrils, a significantly lower proportion was found in the myofibrils (Fig. 5B). Thus, the low proportion of α-SMA observed in myofibrils isolated from cTnI-ND mouse hearts suggested that myofibrils were one source of α-SMA localization, but not the major source. An alternative explanation is that α-SMA in cTnI-ND cardiomyocytes, while a component of the myofibrillar complex, is structurally integrated differently from that of α-CA, so that the two forms of actin were not co-enriched.
Fig. 5.
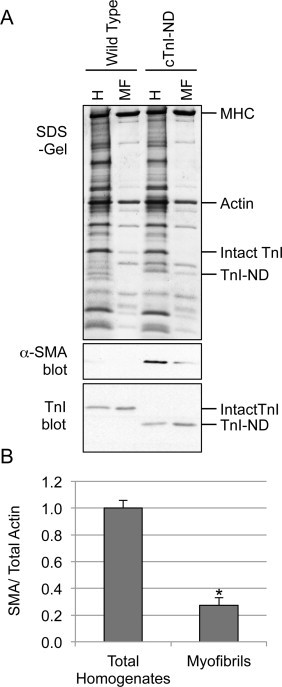
Limited myofibril-integration of α-SMA. (A) Ventricular muscle homogenates (H, 40 μg/lane) and isolated ventricular myofibrils (MF, protein levels adjusted to achieve similar actin bands on gel) were prepared from wild type and cTnI-ND mouse hearts and analyzed by SDS–PAGE (upper panel) and immunoblotting (lower two panels) using anti-α-SMA or anti-TnI antibody. (B) Anti-α-SMA immunoblot densitometry of total ventricular homogenates and isolated cardiac myofibrils from cTnI-ND hearts normalized to total actin band in the SDS–PAGE gel showed significantly lower proportion of α-SMA in the myofibrils than that in the total muscle homogenates. *P < 0.01. Wild type, N = 3; cTnI-ND, N = 3. Data are presented as mean ± SE.
To visualize the precise overlap of α-SMA with the myofibril, we compared indirect immunofluorescence for α-SMA and TnT, a predominant myofibril-specific protein. Confocal imaging of immunostained cTnI-ND cardiomyocytes (Fig. 6) showed non-complete overlap between the sarcomeric patterns for the two proteins, and a non-homogeneous appearance of α-SMA immunofluorescence among myofibrils across the cellular plane. Taken with the poor biochemical recovery of α-SMA with other myofibrillar components, the double immunofluorescence staining suggests that α-SMA resides in close proximity to the myofibrils, but its participation in the contractile function in these hearts remains to be investigated.
Fig. 6.
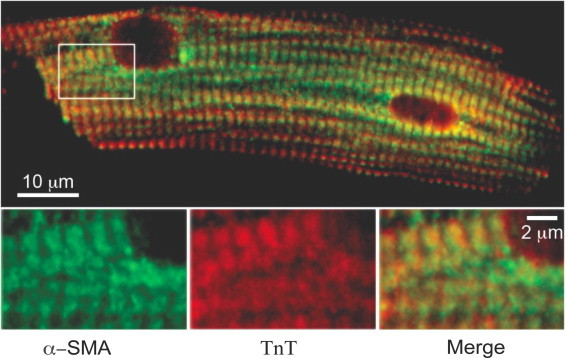
Intracellular localization of α-SMA shows non-complete overlap with the sarcomeric protein TnT. Cardiomyocytes from cTnI-ND hearts were immunostained for α-SMA (green) and TnT (red), followed by confocal imaging. Image of the cell shows apparent overlap of the two antigens across the cell plane. However, enlarged inset shows a non-complete overlap of the two antigens within individual sarcomeres, consistent with a non-identical association with the thin filaments. (For the interpretation of color mentioned in the caption of figure, the reader is requested to view the online version of this paper.)
3. Discussion
Myocardial remodeling occurs in response to various physiological and pathological conditions [17]. The mechanisms by which the contractile properties of the cardiac muscle result in myocardial remodeling are not fully understood. Our present study demonstrated and characterized an up-regulation of α-SMA in transgenic mouse hearts expressing cTnI-ND, a modification in the myofilament Ca2+-regulatory system shown to selectively facilitate diastolic function in the heart [5,7]. This unique model system allowed us to investigate the remodeling consequences of selectively increased myocardial relaxation with the following findings.
3.1. α-SMA expresses in cTnI-ND cardiomyocytes as an early remodeling response
It has been well established that α-SMA is expressed in cardiomyocytes during early stages of heart development but is soon thereafter replaced by α-SKA and α-CA [11,12]. Reactivation of its expression is considered a potential marker of ventricular hypertrophy [9,15,18]. In response to stresses or injury, the heart can adapt through a number of remodeling mechanisms that are manifested clinically as changes in size, shape, and function [17,19]. Remodeling mechanisms range from the reversible compensation resulting from athletic activity to the necrotic repair that follows infarction, and can modify cardiac function over time, from initially improved output to diminished function and progression to failure [17]. All myocardial remodeling is expected to require early changes in gene expression, however, the earliest such changes have been the most difficult to discover because animal models often have a rapid transition into cardiac hypertrophy or failure. In addition, myocardial remodeling often involves apoptotic or necrotic cell death and fibrosis [20].
As a unique model system, the cTnI-ND mouse hearts showed no signs of hypertrophy or destruction in function at the age investigated in the present study. Nonetheless, evidence of ventricular remodeling in the cTnI-ND hearts was indicated by the significant increased density but not size of the blood vessels (Fig. 2). There was no increase in the mass of vascular wall (Fig. 2) and no detection of smooth muscle-specific marker proteins SM22 and h1-calponin (Fig. 1), suggesting that the increased vasculature did not contribute substantively to the observed increase of α-SMA.
Having excluded smooth muscle as a significant source of the increased α-SMA in cTnI-ND hearts, myofibroblasts were similarly excluded based upon the absence of calponin in cTnI-ND ventricular muscle (Fig. 1), and on the absence of visible fibrosis in cTnI-ND hearts (unpublished observations). Our data then identified cardiomyocytes as the predominant cellular source of α-SMA in cTnI-ND hearts. The expression of α-SMA in adult cTnI-ND cardiomyocytes as an early sign of myocardial remodeling represents a fascinating result that has not previously been described in the literature. Since the primary functional impact of cTnI-ND is a facilitation of myocardial relaxation [7], this mouse line becomes a unique model in which the molecular mechanisms for diastolic function to induce myocardial remodeling may be investigated.
It is important to note that cTnI-ND hearts do not develop hypertrophy or failure during the life span of the animal, and exhibit beneficial effects on cardiac function with aging [8]. Early expression of α-SMA in cTnI-ND cardiomyocytes may therefore be a valid factor for investigations aimed at understanding pathological cardiomyocyte remodeling. In addition, the increased number of blood vessels in the cTnI-ND transgenic mouse hearts may represent not only an early index of cardiac remodeling but also a beneficial adaptation to improve energetic supply to the cardiomyocytes, a novel hypothesis worth further investigation.
3.2. α-SMA distributes to non-myofilament structures in cTnI-ND cardiomyocytes
Further investigation of the subcellular locations of α-SMA led to its detection within the myofibrils from cTnI-ND hearts, although its exact effects on cardiomyocyte contractile function will remain to be studied. Surprisingly, when we isolated myofibrils from these hearts, we found that they only contained a minor portion (27%) of the α-SMA expressed in the cTnI-ND heart (Fig. 5), indicative of a limited role in myofilament contractility. The remaining majority of α-SMA was either selectively lost from this preparation, or is simply not in the myofibril preparation along with other known components. The selective targeting of α-SMA in cardiomyocytes to more than a single compartment is interesting given its structural similarity to α-CA. Despite the significant non-myofilament association of α-SMA exhibited biochemically, the immunofluorescence of α-SMA appeared in a striated pattern (Fig. 6). However, the non-complete overlap observed with TnT in double labeling microscopy may indicate that while the sarcomeric portion of α-SMA was concentrated in the actin filaments as expected, non-sarcomeric α-SMA was enriched in a zoning pattern overlapping with the I-band of sarcomeres.
3.3. Implications for the function of α-SMA during early remodeling in non-hypertrophic and non-failing cTnI-ND hearts
Transient α-SMA expression in developing cardiomyocytes has been suggested to act as a scaffold for the organization of contractile proteins during the formation of new myofibrils [21] or during cardiac differentiation [22,23]. It has also been proposed that α-SMA plays a role in cellular tension and possibly regulates gene transcription during embryonic cardiac development [24].
Our findings reported here suggest that early stages of remodeling can precede development of cardiac complications. cTnI-ND mice exhibit normal life spans with no apparent change in cardiac morphology, and show functional benefits without evidence of hypertrophy or heart failure [5,7]. Moreover, these hearts show better-than-wild-type cardiac function at old age (16 months) [8]. Therefore, we propose that overexpression of cTnI-ND in mouse hearts may induce additional adaptive changes that contribute to a resetting of the equilibrium between force generation and relaxation, which in turn helps constrain myocardial remodeling to the level of physiological adaptation.
Besides muscle contraction, actin is essential for many cellular functions including cell division, adhesion, and migration. Although isoforms of actin are highly similar with minimal variations in amino acid sequence, recent findings suggest that each isoform has a specific cellular function [10]. α-SMA plays an important role in cardiac sarcomerogenesis, cardiac muscle differentiation, and influences cardiomyocyte rhythm [24]. The significant expression of α-SMA in cTnI-ND hearts may well have multiple functional impacts.
In summary, functions of N-terminal truncated cTnI in mouse hearts induced α-SMA expression in cardiomyocytes as an early remodeling response. Together with an increase in blood vessel density, this adaptation may be an early structural response to potent increases in diastolic function prior to any occurrence of myocardial hypertrophy. With its limited integration into the contractile apparatus, the expression of α-SMA may carry out multiple functional effects on cardiac function through effects on the cytoskeleton or other major non-myofilament compartment. Further studies are required to fully understand this adaptive regulation.
4. Experimental procedures
4.1. Animal models
Transgenic mice overexpressing cTnI-ND under control of the cardiac α-myosin heavy chain promoter were previously developed [7]. One of the cTnI-ND transgenic mouse lines that exhibited early postnatal expression of the transgene was used to cross with an endogenous cTnI gene deletion line to develop double transgenic mice expressing 100% cTnI-ND in the adult heart [6]. Mice used in the present studies were 2 to 3 months of age and without signs of cardiac hypertrophy or heart failure. Age-matched hearts from a double transgenic mouse line expressing a mutant cTnI on the same endogenous cTnI gene null background (cTnI-K118C, [13]) were used as control.
All animal procedures were approved by the Institutional Animal Care and Use Committee and were conducted in accordance with the Guiding Principles in the Care and Use of Animals, as approved by the Council of the American Physiological Society.
4.2. SDS–polyacrylamide gel electrophoresis (PAGE) and immunoblot analysis
Total protein was extracted from mouse ventricular muscle by homogenization of transverse heart blocks in 1% SDS using a PowerGen 1000 tissue homogenizer (Fisher Scientific). Adult cardiomyocytes were isolated using a previously established protocol [25–27] and lysed in SDS–PAGE sample buffer (50 mM Tris–HCl, pH 8.8, 2% SDS, 100 mM DTT, 0.1% bromophenol blue, 10% glycerol) [5].
Modified from a previously described method [28], mouse ventricular myofibrils were purified from fresh cardiac muscle after a 3 min perfusion of the heart with Krebs solution (12.6 mM NaCl, 25 mM NaHCO3, 1.2 mM NaH2PO4, 1.2 mM MgCl2, 2.5 mM CaCl2, pH 7.2) containing 50 mM KCl to relax the cardiac muscle. Total myofibril protein was extracted by lysis in SDS–PAGE sample buffer as above.
The samples were heated at 85 °C for 5 min followed by centrifugation to clarify the supernatant for gel loading. Protein extracts were resolved by SDS–PAGE using Laemmli buffer system [29]. 12% gels with an acrylamide:bisacrylamide ratio of 29:1 was used for analysis of calponin and SM22; 14% gels with an acrylamide:bisacrylamide ratio of 180:1 was used for analysis of α-SMA and cTnI. Electrophoretic transfer to nitrocellulose membrane was carried out using standard procedures [30] using 25 mM phosphate transfer buffer, pH 7.4, and transfer at 2 A for 60 min. The transferred protein bands were visualized using amido black prior to immunoblot analyses.
Antibodies and dilutions used in immunoblotting were as follows: an anti-α-SMA mouse monoclonal antibody (mAb) from Sigma–Aldrich (A5228) was used at 1:1000 dilution; a rabbit anti-calponin polyclonal antibody RAH2 was used at 1:4000; an anti-SM22 mAb 3F6 was used at 1:20; and an anti-TnI mAb TnI-1 was used at 1:2000. Phosphate-buffered saline (PBS) containing 0.2% Tween-20 (PBS-T) was used as the incubation and washing buffers. The blots were then detected using horseradish peroxidase (HRP)-conjugated goat anti-rabbit or anti-mouse IgG second antibody (Jackson ImmunoResearch Laboratories) at 1:30,000 and developed in ECL Western Blotting Substrate (Thermo Scientific) and the signals were revealed with autoradiography using BioMax MR film (Kodak). Densitometry analysis of Coomassie Blue stained SDS–PAGE gels, amido black stained nitrocellulose membranes, and autoradiography films was performed on images scanned at 500 pixels/in using NIH ImageJ software. Immunoblot band intensities were normalized to nitrocellulose protein stain.
4.3. Immunohistochemical analysis
Fresh hearts from wild type and cTnI-ND mice were flash frozen in O.C.T. compound (Fisher Scientific) and frozen sections were prepared as previously described [31]. The sections were incubated with 3% H2O2 at room temperature for 15 min, washed three times with PBS-T, and incubated with an anti-α-SMA rabbit polyclonal antibody (AbCam, ab5694) diluted 1:100 in PBS-T at room temperature for 2 h. Following three washes with PBS-T, sections were incubated with HRP-conjugated goat anti-rabbit antibody diluted 1:2000 in PBS-T at room temperature for 2 h, washed three times again in PBS-T, and developed at room temperature in DAB substrate solution (Sigma FAST 3,3′-Diaminobenzidine Tablet set). After washed in water, the sections were counter stained with Mayer's hematoxylin, following the manufacturer's protocol. All sections were mounted under #1 cover slip with Permount mounting medium (Fisher Scientific). The slides were maintained at 4 °C for long-term storage.
The immunohistological images were observed using a Zeiss Observer A1 AXIO microscope, and photographed through a 5× objective lens at a resolution of 1.5 pixels/μm. For quantification of vasculature, the complete transverse heart section was reconstructed from a series of overlapping microscope images using the photomerge function in Adobe Photoshop CS5 v12.1. NIH ImageJ software was used to pan around the entire cross-section and count all vesicular membranes and to measure the area of each vessel. Measurements of the vessel wall area was performed by assuming each vessel had a circular cross section, thus A = πR2. The outer diameter of the vessel was used to calculate the total area of the cross section and the inside radius of the vessel was used to calculate the lumen area. The vessel wall area was then determined by subtracting the inner area from the total cross sectional area.
4.4. Immunofluorescence microscopy
Cardiomyocytes isolated from 2 to 3 months old wild type and cTnI-ND mouse hearts as described above were fixed on #1 glass cover slips with 4% paraformaldehyde for 15 min, then permeabilized and washed three times in PBS-T before blocked with 1% goat serum in PBS-T at room temperature for 60 min. The cardiomyocytes were incubated with mouse anti-α-SMA mAb (Sigma–Aldrich A5228) diluted 1:100 at 4 °C overnight. Subsequent to a high salt wash (0.5 M NaCl in PBS-T, at room temperature for 10 min) and four 15 min washes with PBS-T, the cover slips were incubated with Alexa Fluor 488-conjugated goat anti-mouse IgG antibody (Life Technologies Corporation) diluted at 1:50 at room temperature for 2 h. After four 15-min washes, the cardiomyocytes were post-fixed in 4% paraformaldehyde for 15 min to preserve staining and mounted on a pre-cleaned microscope slide with Prolong Gold Antifade Reagent (Life Technologies Corporation). Images were captured using a laser (Point) scanning confocal microscope (Leica TCS SP5, excitation using the 458/476/488/496/514 nm multiline Argon laser). Fluorescence intensity analysis was performed on images taken with identical camera settings using NIH ImageJ software.
4.5. Data analysis
All measurements and analysis were performed blinded from the animal genotype. Quantitative data are plotted as mean ± SE (standard error of the mean, SD/√N, where N is number of animals). The statistical significance of differences was analyzed by two-tailed unpaired Student's t -test.
Acknowledgements
We would like to thank Geoff Cady and Hui Wang for technical support and genotyping of the transgenic mice. This study was supported by NIH/NHLBI grant HL098945 (to J.-P.J.) and an Incubator Grant from the Office of Vice President for Research, Wayne State University (to J.-P.J. and S.C.).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Perry S.V. Troponin I: inhibitor or facilitator. Mol. Cell. Biochem. 1999;190:9–32. [PubMed] [Google Scholar]
- 2.Jin J.P., Zhang Z., Bautista J.A. Isoform diversity, regulation, and functional adaptation of troponin and calponin. Crit. Rev. Eukar. Gene Expr. 2008;18:93–124. doi: 10.1615/critreveukargeneexpr.v18.i2.10. [DOI] [PubMed] [Google Scholar]
- 3.Solaro R.J., Sheehan K.A., Lei M., Ke Y. The curious role of sarcomeric proteins in control of diverse processes in cardiac myocytes. J. Gen. Physiol. 2010;136:13–19. doi: 10.1085/jgp.201010462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu Z.B., Zhang L.F., Jin J.P. A proteolytic NH2-terminal truncation of cardiac troponin I that is up-regulated in simulated microgravity. J Biol. Chem. 2001;276:15753–15760. doi: 10.1074/jbc.M011048200. [DOI] [PubMed] [Google Scholar]
- 5.Feng H.Z., Chen M., Weinstein L.S., Jin J.P. Removal of the N-terminal extension of cardiac troponin I as a functional compensation for impaired myocardial beta-adrenergic signaling. J. Biol. Chem. 2008;283:33384–33393. doi: 10.1074/jbc.M803302200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feng H.Z., Hossain M.M., Huang X.P., Jin J.P. Myofilament incorporation determines the stoichiometry of troponin I in transgenic expression and the rescue of a null mutation. Arch. Biochem. Biophys. 2009;487:36–41. doi: 10.1016/j.abb.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barbato J.C., Huang Q.Q., Hossain M.M., Bond M., Jin J.P. Proteolytic N-terminal truncation of cardiac troponin I enhances ventricular diastolic function. J. Biol. Chem. 2005;280:6602–6609. doi: 10.1074/jbc.M408525200. [DOI] [PubMed] [Google Scholar]
- 8.Biesiadecki B.J., Tachampa K., Yuan C., Jin J.P., de Tombe P.P., Solaro R.J. Removal of the cardiac troponin I N-terminal extension improves cardiac function in aged mice. J. Biol. Chem. 2010;285:19688–19698. doi: 10.1074/jbc.M109.086892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Black F.M., Packer S.E., Parker T.G., Michael L.H., Roberts R., Schwartz R.J., Schneider M.D. The vascular smooth muscle alpha-actin gene is reactivated during cardiac hypertrophy provoked by load. J. Clin. Invest. 1991;88:1581–1588. doi: 10.1172/JCI115470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perrin B.J., Ervasti J.M. The actin gene family: function follows isoform. Cytoskeleton (Hoboken) 2010;67:630–634. doi: 10.1002/cm.20475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwartz K., de la Bastie D., Bouveret P., Oliviero P., Alonso S., Buckingham M. Alpha-skeletal muscle actin mRNA's accumulate in hypertrophied adult rat hearts. Circ. Res. 1986;59:551–555. doi: 10.1161/01.res.59.5.551. [DOI] [PubMed] [Google Scholar]
- 12.Suurmeijer A.J., Clement S., Francesconi A., Bocchi L., Angelini A., Van Veldhuisen D.J., Spagnoli L.G., Gabbiani G., Orlandi A. Alpha-actin isoform distribution in normal and failing human heart: a morphological, morphometric, and biochemical study. J. Pathol. 2003;199:387–397. doi: 10.1002/path.1311. [DOI] [PubMed] [Google Scholar]
- 13.Wei B., Gao J., Huang X.P., Jin J.P. Mutual rescues between two dominant negative mutations in cardiac troponin I and cardiac troponin T. J. Biol. Chem. 2010;285:27806–27816. doi: 10.1074/jbc.M110.137844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li L., Miano J.M., Cserjesi P., Olson E.N. SM22 alpha, a marker of adult smooth muscle, is expressed in multiple myogenic lineages during embryogenesis. Circ. Res. 1996;78:188–195. doi: 10.1161/01.res.78.2.188. [DOI] [PubMed] [Google Scholar]
- 15.Leslie K.O., Taatjes D.J., Schwarz J., vonTurkovich M., Low R.B. Cardiac myofibroblasts express alpha smooth muscle actin during right ventricular pressure overload in the rabbit. Am. J. Pathol. 1991;139:207–216. [PMC free article] [PubMed] [Google Scholar]
- 16.Orlandi A., Hao H., Ferlosio A., Clement S., Hirota S., Spagnoli L.G., Gabbiani G., Chaponnier C. Alpha actin isoforms expression in human and rat adult cardiac conduction system. Differentiation. 2009;77:360–368. doi: 10.1016/j.diff.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 17.Cohn J.N., Ferrari R., Sharpe N. Cardiac remodeling–concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J. Am. Coll. Cardiol. 2000;35:569–582. doi: 10.1016/s0735-1097(99)00630-0. [DOI] [PubMed] [Google Scholar]
- 18.Friddle C.J., Koga T., Rubin E.M., Bristow J. Expression profiling reveals distinct sets of genes altered during induction and regression of cardiac hypertrophy. Proc. Natl. Acad. Sci. USA. 2000;97:6745–6750. doi: 10.1073/pnas.100127897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pfeffer M.A., Braunwald E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation. 1990;81:1161–1172. doi: 10.1161/01.cir.81.4.1161. [DOI] [PubMed] [Google Scholar]
- 20.Swynghedauw B. Molecular mechanisms of myocardial remodeling. Physiol. Rev. 1999;79:215–262. doi: 10.1152/physrev.1999.79.1.215. [DOI] [PubMed] [Google Scholar]
- 21.Donath M.Y., Zapf J., Eppenberger-Eberhardt M., Froesch E.R., Eppenberger H.M. Insulin-like growth factor I stimulates myofibril development and decreases smooth muscle alpha-actin of adult cardiomyocytes. Proc. Natl. Acad. Sci. USA. 1994;91:1686–1690. doi: 10.1073/pnas.91.5.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clement S., Pellieux C., Chaponnier C., Pedrazzini T., Gabbiani G. Angiotensin II stimulates alpha-skeletal actin expression in cadiomyocytes in vitro and in vivo in the absence of hypertension. Differentiation. 2001;69:66–74. doi: 10.1046/j.1432-0436.2001.690107.x. [DOI] [PubMed] [Google Scholar]
- 23.Ehler E., Fowler V.M., Perriard J.C. Myofibrillogenesis in the developing chicken heart: role of actin isoforms and of the pointed end actin capping protein tropomodulin during thin filament assembly. Dev. Dyn. 2004;229:745–755. doi: 10.1002/dvdy.10482. [DOI] [PubMed] [Google Scholar]
- 24.Clement S., Stouffs M., Bettiol E., Kampf S., Krause K.H., Chaponnier C., Jaconi M. Expression and function of alpha-smooth muscle actin during embryonic-stem-cell-derived cardiomyocyte differentiation. J. Cell Sci. 2007;120:229–238. doi: 10.1242/jcs.03340. [DOI] [PubMed] [Google Scholar]
- 25.Yu Z.B., Gao F., Feng H.Z., Jin J.P. Differential regulation of myofilament protein isoforms underlying the contractility changes in skeletal muscle unloading. Am. J. Physiol. Cell Physiol. 2007;292:C1192–C1203. doi: 10.1152/ajpcell.00462.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O’Connell T.D., Rodrigo M.C., Simpson P.C. Isolation and culture of adult mouse cardiac myocytes. Methods Mol. Biol. 2007;357:271–296. doi: 10.1385/1-59745-214-9:271. [DOI] [PubMed] [Google Scholar]
- 27.Yu Z.B., Wei H., Jin J.P. Chronic coexistence of two troponin T isoforms in adult transgenic mouse cardiomyocytes decreased contractile kinetics and caused dilatative remodeling. Am. J. Physiol. Cell Physiol. 2012;303:C24–C32. doi: 10.1152/ajpcell.00026.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang Q.Q., Brozovich F.V., Jin J.P. Fast skeletal muscle troponin T increases the cooperativity of transgenic mouse cardiac muscle contraction. J. Physiol. 1999;520(Pt 1):231–242. doi: 10.1111/j.1469-7793.1999.00231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 30.Towbin H., Staehelin T., Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jacob S., Sleiman N.H., Kern S., Jones L.R., Sala-Mercado J.A., McFarland T.P., Sabbah H.H., Cala S.E. Altered calsequestrin glycan processing is common to diverse models of canine heart failure. Mol. Cell. Biochem. 2013;377:11–21. doi: 10.1007/s11010-013-1560-7. [DOI] [PubMed] [Google Scholar]