

Abstract
Much data support a role for central nervous system antigen-specific antibodies in the pathogenesis of multiple sclerosis (MS). The effects of inducing a decrease in (auto)antibody levels on MS or experimental autoimmune encephalomyelitis (EAE) through specific blockade of FcRn, however, remain unexplored. We recently developed engineered antibodies that lower endogenous IgG levels by competing for binding to FcRn. These Abdegs (“antibodies that enhance IgG degradation”) can be used to directly assess the effect of decreased antibody levels in inflammatory diseases. In the current study, we show that Abdeg delivery ameliorates disease in an EAE model that is antibody dependent. Abdegs could therefore have promise as therapeutic agents for MS.
Keywords: autoantibody, EAE, FcRn, engineered antibodies, therapy
Introduction
Multiple sclerosis (MS) is a demyelinating, neurodegenerative disease involving autoreactive T and B cells.1,2 Evidence to support the involvement of antibodies in the pathogenesis of this disease includes the detection of Ig+ plasma cells in active MS lesions and the presence of central nervous system (CNS) antigen-specific antibodies in the serum and cerebrospinal fluid of MS patients.1,2 However, it is currently unknown whether a reduction in (auto)antibody levels can ameliorate MS or experimental autoimmune encephalomyelitis (EAE) in antibody-dependent models. Although B cell depleting antibodies such as the anti-CD20 antibody rituximab have been shown to have some efficacy in treating MS, this treatment does not delete long-lived, CD20-negative plasma cells and (auto)reactive antibody levels remain unaffected.3-6 The current study is directed toward analyzing the therapeutic effects of directly reducing endogenous IgG levels in a mouse model of EAE that is both T cell and antibody dependent.7,8
Several different classes of reagents that decrease antibody levels in vivo by inhibiting the MHC class I-related receptor, FcRn, have been described.9-14 FcRn regulates the levels and transport of antibodies by binding and recycling or by transcytosing IgG molecules, thereby preventing their lysosomal degradation.15 We have generated a novel class of antibody-based inhibitors of FcRn that are engineered to bind to this receptor through their Fc regions with increased affinity in the pH range of 6.0–7.4.9 In the presence of these inhibitors, competing IgGs are driven into the degradative, lysosomal pathway within cells.9,16 Consequently, Abdegs (“antibodies that enhance IgG degradation”) can be used to induce a rapid decrease in endogenous IgG levels in the body.9,17,18 High dose intravenous gammaglobulin (IVIG) can also compete for FcRn binding;10,19 however, the relatively low competitive activity of IVIG for FcRn necessitates the use of doses approaching the whole body load of IgG (i.e., ~1‒2 g/Kg body weight).10,19 By contrast, Abdegs inhibit FcRn at comparatively low doses (~50‒100 mg/Kg)9,18 due to their increased binding affinity for this receptor. As such, these engineered antibodies can be used as specific tools to reduce antibody levels in vivo.
Demyelinating antibodies specific for myelin oligodendrocyte glycoprotein (MOG) can contribute to pathogenesis in EAE models involving autoreactive CD4+ T cell responses.7,8,20,21 For example, immunization of B cell deficient mice with recombinant human MOG to induce a poorly encephalitogenic, MOG-specific CD4+ T cell response followed by the transfer of anti-MOG antibodies results in aggressive disease.7,20 In the study of the effect of (auto)antibody depletion reported here, we used a model recently developed in our laboratory in which encephalitogenic anti-MOG antibodies exacerbate EAE.8 This model involves the immunization of wild type (WT),8 rather than B cell deficient,7 mice with a weakly encephalitogenic peptide (residues 35–55 of human MOG, hMOG35–55), followed by the transfer of the demyelinating monoclonal antibody (mAb), 8–18C5.21,22 Delivery of 8–18C5 into hMOG35–55 immunized mice at day 15 post-immunization exacerbates EAE relative to the disease activity observed in phosphate-buffered saline (PBS) vehicle-treated mice (Fig. 1A). In addition, treatment with isotype control (mouse IgG1), anti-lysozyme antibody D1.323 does not exacerbate EAE (data not shown). Following injection, the accumulation and binding of 8–18C5 can be detected in spinal cord sections at two hours post-delivery, and the levels of this mAb in the CNS increase at six hours post-delivery (Fig. 1B). This EAE model is therefore instructive for the analysis of the effects of decreasing (auto)antibody levels in vivo.
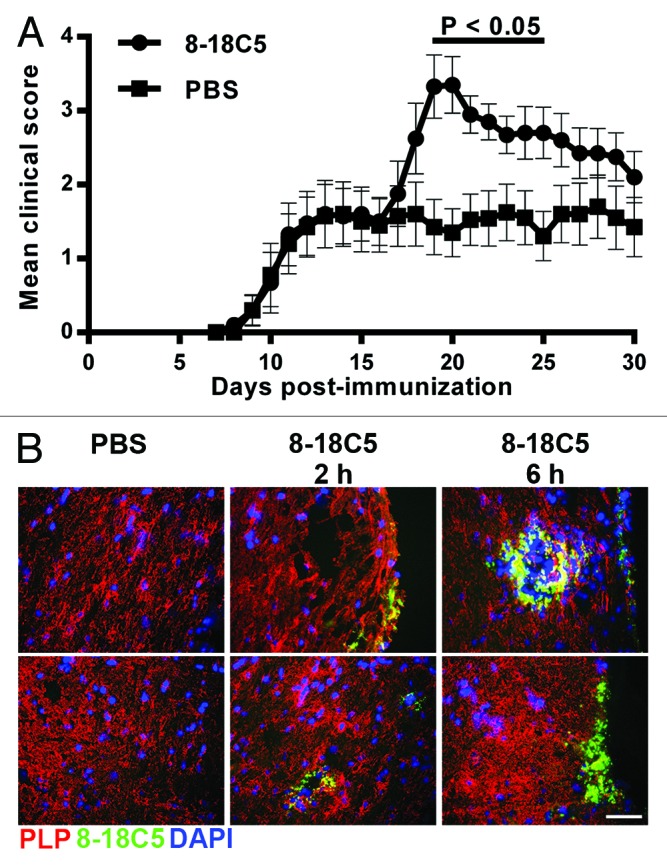
Figure 1. Exacerbation of EAE by transfer of 8–18C5 mAb into hMOG35–55-immunized C57BL/6 mice. C57BL/6 mice were immunized with hMOG35–55 (Materials and Methods) and treated with 200 ng pertussis toxin on days 0 and 2. On day 15, in (A) mice were sorted into equivalent groups (n = 10 mice/group; mean disease score of ~1.5) and were injected intravenously with 200 μg 8–18C5 or PBS vehicle. Mice were scored daily. Error bars indicate SEM. Significant differences (p < 0.05; Student’s t-test for pairwise comparison of groups) between 8–18C5 and PBS treated mice are indicated by a bar. (B) mice were sorted into equivalent groups (n = 6 mice/group for 8–18C5 and 2 mice/group for PBS) and were treated as in (A), except that 200 μg Alexa 647-labeled 8–18C5 or PBS vehicle was injected on day 15. Spinal cords were harvested at 2 or 6 h post-delivery of 8–18C5 or PBS, stained with anti-PLP antibody (pseudocolored red) and imaged. Cropped images of representative data are shown. Alexa 647-labeled 8–18C5 and DAPI stained nuclei are pseudocolored green and blue, respectively. For the PBS-injected group the upper and lower images correspond to spinal cord harvested at 2 h and 6 h post-delivery, respectively. The upper and lower panels in the 8–18C5 treated groups correspond to spinal cord harvested at either 2 h (middle column) or 6 h (right column) post-delivery. Bar = 50 μm.
To investigate the effects of Abdeg delivery on the clearance of 8–18C5 in mice with EAE, hMOG35–55 immunized mice were divided into equivalent groups with similar mean and median clinical scores and injected with radiolabeled (125I) 8–18C5 on day 17 post-immunization. The mean clinical score for each mouse group at the time of 8–18C5 injection was ~1.5. Two hours later, mice were injected with MST-HN Abdeg9 (human IgG1-derived) or, as controls, WT human IgG1 or PBS vehicle. A dose of 1.5 mg Abdeg (or WT IgG1) per mouse was chosen since the delivery of 1–2 mg MST-HN Abdeg per mouse induces a maximum decrease in endogenous IgG levels.18 The levels of labeled 8–18C5 remaining in the mice were determined by analysis of blood samples and whole body counting (Fig. 2A and B). Abdeg delivery induces a rapid decrease in 125I-labeled 8–18C5 levels in the blood and whole body relative to the slower clearance observed in control mice treated with WT IgG1 or PBS vehicle. Importantly, 125I-labeled 8–18C5 levels in the spinal cord and brain were also lower in mice following Abdeg delivery relative to those in WT IgG1-treated mice (Fig. 2C). Thus, Abdegs are effective in reducing accumulation of this encephalitogenic mAb in the CNS.
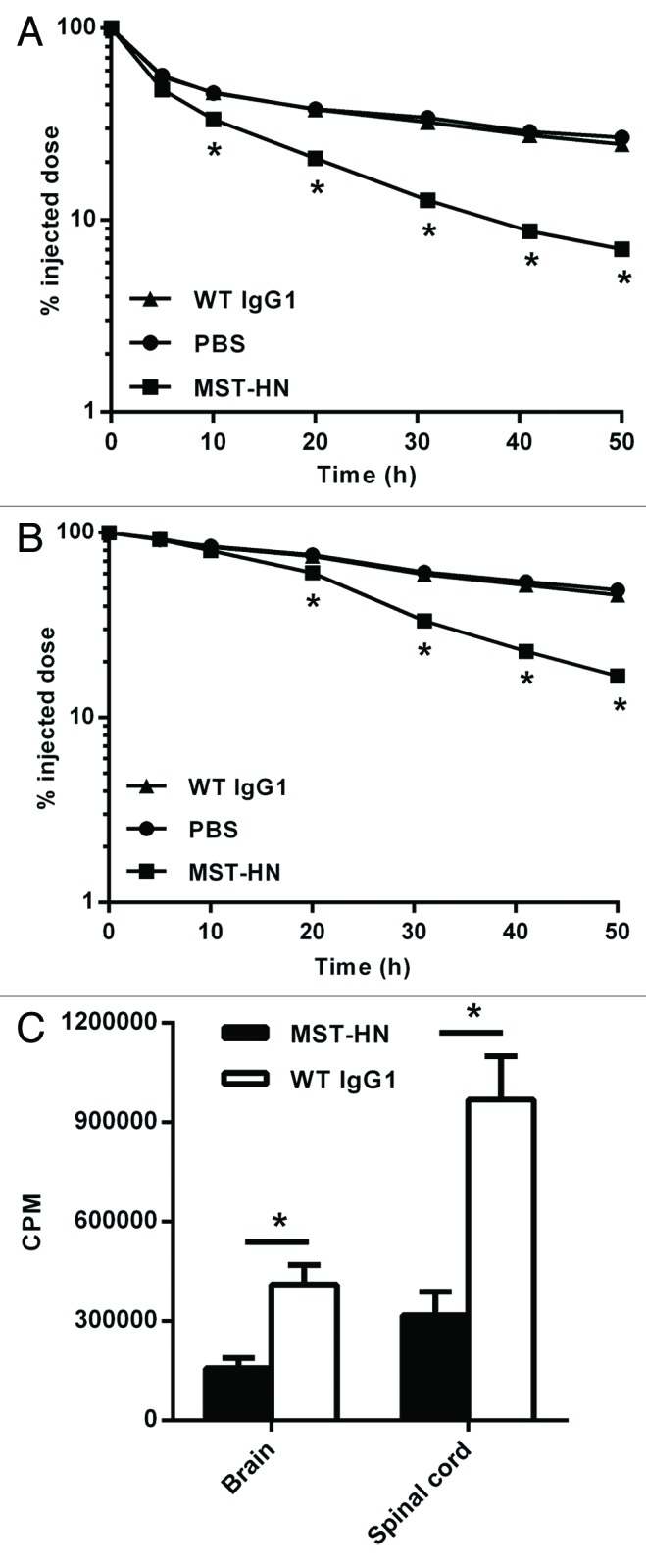
Figure 2. MST-HN Abdeg treatment induces a rapid decrease in the levels of 8–18C5 mAb in vivo. Mice were immunized with hMOG35–55 as in Figure 1, sorted into equivalent groups (n = 4–6 mice/group; mean disease score ~1.5) on day 17 and injected with a mixture of 125I-labeled 8–18C5 and unlabeled 8–18C5 (total of 200 μg mAb/mouse). Two hours later, mice were injected with 1.5 mg MST-HN, 1.5 mg WT IgG1 or PBS. Radioactivity levels were analyzed in blood (A) or by whole body counting (B) at the indicated times. (C) 48 h post-delivery of MST-HN or WT IgG1, mice were perfused with heparin/PBS and CNS tissue (brains and spinal cords) isolated. Radioactivity levels in these tissues were determined and mean values for each group are shown. Error bars indicate SEM and in panels (A) and (B) are obscured by the symbols. Significant differences (p < 0.05; Student’s t-test for pairwise comparison of groups) between MST-HN and WT IgG1/PBS treated mice are indicated by asterisks.
We next assessed the effect of Abdeg delivery on 8‒18C5-mediated exacerbation of EAE. In initial experiments, the anti-lysozyme human IgG1 antibody9 that differs from the Abdeg at the MST-HN mutation sites was used as a control. In contrast with our earlier studies in an arthritis model,18 however, delivery of this antibody resulted in a slight exacerbation of disease relative to control, vehicle-treated mice (data not shown). Trastuzumab, a human IgG1 anti-HER2 mAb that does not recognize mouse HER2, was therefore used as a WT IgG1 control. Mice were immunized with 100 μg hMOG35–55 and, at day 15, divided into groups with similar mean and median clinical scores (mean clinical score for each group ~2) and disease history prior to day 15. Two hours following the delivery of 8–18C5, when this mAb is detectable in the CNS (Fig. 1B), mice were treated with 1.5 mg MST-HN or as controls, WT IgG1 or PBS vehicle. The data presented in Figure 3 demonstrate that Abdeg delivery results in amelioration of EAE. The positive effect of the Abdeg on disease activity is rapid and is observed within one day of treatment, consistent with the dynamics of Abdeg-mediated clearance of 8–18C5 (Fig. 2). Although the delivery of WT IgG1 into mice results in reduced disease activity relative to vehicle-treated mice, possibly due to anti-inflammatory effects analogous to those described for high dose IVIG,24,25 the differences between WT IgG1- and PBS-treated mouse groups are not significant for the majority of days post-treatment (Fig. 3).
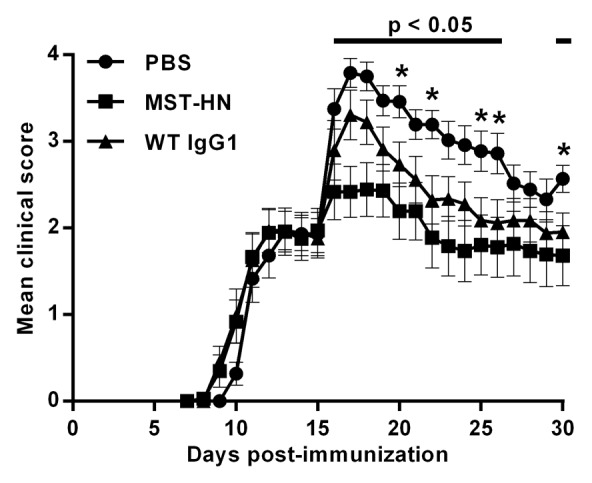
Figure 3. Delivery of the MST-HN Abdeg ameliorates EAE. Mice were immunized with hMOG35–55 as in Figure 1, sorted into equivalent groups (n = 8‒9 mice/group; mean disease score ~2) on day 15 and injected with 200 μg 8–18C5. Two hours later, mice in each group were treated with 1.5 mg MST-HN, 1.5 mg WT IgG1 or PBS vehicle. Mice were scored daily for disease activity. Data are combined from two independent experiments, totaling 17‒18 mice for each treatment group. Error bars indicate SEM. Significant differences (p < 0.05; Student’s t-test for pairwise comparison of groups) are indicated by bars (MST-HN vs. PBS treated mice) and asterisks (WT IgG1 vs. PBS treated mice).
Collectively, we demonstrate that a reduction of (auto)antibody levels ameliorates disease in a mouse model of EAE in which antibodies contribute to pathogenesis. Our studies indicate that FcRn blockade could have beneficial effects in MS, given the evidence to support a role for antibodies in pathogenesis.1,2 Currently available treatments that lower IgG levels are the use of IVIG or plasmapheresis26,27 which can both have adverse effects, and, in the case of IVIG, require high doses and are limited by supply.28 It will be of interest to analyze the effects of Abdegs on disease following the transfer of humanized anti-MOG antibodies into mice that transgenically express human FcRn.29 Abdeg-mediated inhibition of FcRn provides a new potential treatment for MS that, in combination with T cell-targeted treatments, could hold considerable promise for ameliorating this complex disease.
Materials and Methods
Mice
Female C57BL/6 mice were purchased from the Jackson Laboratory and used at 9‒10 weeks of age. Mice were housed in the animal facility at University of Texas (UT) Southwestern Medical Center and handled according to protocols approved by the IACUC.
Recombinant antibodies and MOG
Clinical grade WT human IgG1 trastuzumab (Herceptin®) was obtained from the pharmacy at UT Southwestern Medical Center. NS0 transfectants expressing WT humanized (IgG1) anti-hen egg lysozyme antibody30 and the MST-HN mutant derivative9 (Met252 to Tyr, Ser254 to Thr, Thr256 to Glu, His433 to Lys and Asn434 to Phe) were cultured and antibodies were purified using lysozyme-Sepharose as described previously.30 The MST-HN mutant was also scaled up in a bioreactor by BioXCell (West Lebanon, NH). Mouse IgG1 (anti-hen egg lysozyme, D1.323) was purified using lysozyme-Sepharose from hybridoma culture supernatants.
The VH and VL domain genes of the 8–18C5 mAb (PDB code, 1PKQ)31 were synthesized (Genscript USA Inc.) and used to generate full-length (mouse IgG1, kappa) expression constructs with codons encoding the leader peptide of the immunoglobulin heavy (MAVLVLFLCLVAFPSCVLS) and light (MKLPVRLLVLMFWIPASSS) chain genes of the anti-lysozyme D1.3 hybridoma23 appended by PCR to the 5′ ends of the VH and VL genes, respectively. PCR products encoding the complete 8–18C5 heavy and light chain genes were cloned into pOptiVEC-TOPO and pcDNA 3.3-TOPO (OptiCHO™ antibody express kit, Life Technologies Inc.), respectively.
Stable CHO DG44 transfectants were generated by first transfecting the 8–18C5 light chain expression construct and selecting the clone expressing the highest levels of light chain by ELISA. This clone was then transfected with the 8–18C5 heavy chain expression plasmid. Stably transfected clones were selected in Opti-CHO medium (Life Technologies Inc.) containing 500 μg/ml geneticin. The CHO DG44 clones expressing the highest levels of antibody were identified by ELISAs using 96-well plates coated with recombinant mouse MOG and rabbit anti-mouse immunoglobulin conjugated to HRPO (Life Technologies Inc.) for detection and cultured in increasing concentrations of methotrexate (MTX, 50 nM‒4 μM). For large scale production of 8–18C5 mAb, transfectants were expanded and antibody purified using protein G-Sepharose by BioXCell (West Lebanon, NH).
The extracellular domain (residues 1–121) of mouse MOG was expressed in recombinant form in baculovirus-infected High FiveTM insect cells (Life Technologies Inc.) using an analogous construct design to that described previously for the production of recombinant human MOG,32,33 except that codons encoding the honey bee melittin leader peptide sequence34 were appended to the 5′ end of the gene.
Antibody labeling
The 8–18C5 mAb was labeled with Alexa 647 carboxylic acid, succinimidyl ester (Life Technologies Inc.) using methods recommended by the manufacturer. Iodination (125I) of 8–18C5 was performed using Iodogen as previously described.35 The activity of the labeled 8–18C5 was verified by carrying out surface plasmon resonance (BIAcore) analyses.
EAE exacerbation, pharmacokinetics and treatment
C57BL/6 mice were immunized subcutaneously at four sites in the flanks with 100 μg hMOG35–55 peptide (MEVGWYRPPFSRVVHLYRNGK; CSBio Inc.) emulsified with complete Freund's adjuvant (Sigma Aldrich Inc.) containing an additional 4 mg/ml heat-inactivated Mycobacterium tuberculosis (strain H37RA, Difco). 200 ng of pertussis toxin (List Biological Laboratories) was injected i.p. on days 0 and 2 to disrupt the blood-brain barrier. Mice were monitored daily for disease and at day 15 were sorted into equivalent groups using a cost function (implemented in MATLAB) based on EAE scores prior to and including day 15. This cost function takes into account the similarity (in average scores and covariance) and the standard deviation of the disease scores. On day 15, mice were injected intravenously with 200 μg of Alexa 647-labeled or unlabeled 8–18C5 for immunofluorescence and treatment, respectively. For immunofluorescence, one group of mice was also injected with PBS as a control. Two or six hours later, mice in both groups were perfused with heparin/PBS and spinal cords isolated. For treatment, groups of mice were injected two hours following 8–18C5 delivery with MST-HN (1.5 mg/mouse), WT human IgG1 (1.5 mg/mouse) or PBS vehicle. Mice were monitored daily for clinical signs of EAE for 30 d post-immunization as described previously.36 Data are presented as mean clinical scores for each group, and dead animals were given a score of 6 from the day of death onwards.
To analyze the effects of MST-HN delivery on the levels of 8–18C5, mice were immunized as above and at day 17, sorted into equivalent groups and each mouse injected with a mixture of 15 μg 125I-labeled 8–18C5 and 185 μg unlabeled 8–18C5. Two hours later, groups of mice were injected with MST-HN (1.5 mg/mouse), WT human IgG1 (trastuzumab; 1.5 mg/mouse) or PBS vehicle. Levels of radioactivity were determined at the indicated times in 10 μl blood samples by gamma counting and by whole body counting using a Biodex Atom Lab 100 dose calibrator. To determine the levels of radioactivity in the CNS, mice were perfused with heparin/PBS 48 h following the delivery of MST-HN or WT human IgG1. Following perfusion, brains and spinal cords were isolated and the levels of 125I-labeled 8–18C5 in these tissues were determined by gamma counting.
Immunofluorescence analyses
Spinal cord tissue was embedded in Tissue-Tek® OCT compound (Sakura Finetek USA Inc.), sectioned (5 µm thick) using a Leica cryotome and stored at -80°C. Frozen sections were fixed in acetone (-20°C) and air-dried overnight. After washing with PBS, sections were blocked using 5% goat serum, followed by incubation with polyclonal rabbit anti-mouse proteolipid protein (PLP) antibody (Abcam). Bound anti-PLP antibody was detected using Alexa 555-labeled goat anti-rabbit IgG (Life Technologies Inc.). Following washing, coverslips were mounted using Vectashield mounting medium containing DAPI (Vector Laboratories).
Sections were imaged using a Zeiss Axiovert 200M inverted microscope equipped with a Zeiss 20X, 0.5 NA Plan-Neofluar objective and an ORCA CCD camera (Hamamatsu). Images were acquired with filtersets specific for Alexa 555 Fluor (TRITC-B-000-ZERO; Semrock), Alexa 647 Fluor (Cy5–4040C-ZERO; Semrock) and DAPI (Part No 31013v2; Chroma Technologies). The data were processed and displayed using the microscopy image analysis tool (MIATool) software package (www4.utsouthwestern.edu/wardlab/miatool.asp) in MATLAB (Mathworks). The acquired images were embedded in 16-bit grayscale format and overlaid for presentation. For comparative purposes, the intensities of the Alexa 647 Fluor channel were adjusted in an analogous manner across the data sets. Images were exported into Adobe Photoshop CS6 for final composition of the figures.
Statistical analyses
Statistical analyses of disease data were performed using Student’s t-test in the statistics toolbox of MATLAB (Mathworks). p values of less than 0.05 were taken to be significant.
Acknowledgments
We are grateful to Ramraj Velmurugan for assistance with fluorescence microscopy and to Dr Stephen Anthony for developing a computational algorithm for mouse grouping. This work was supported in part by grants from the National Multiple Sclerosis Society (RG 4308).
Glossary
Abbreviations:
- EAE
experimental autoimmune encephalomyelitis
- FcRn
neonatal Fc receptor
- mAb
monoclonal antibody
- MOG
myelin oligodendrocyte glycoprotein
- MS
multiple sclerosis
- WT
wild type
Disclosure of Potential Conflicts of Interest
E.S.W. is an inventor on a patent (owned by UT Southwestern Medical Center) related to the use of Abdegs as FcRn inhibitors. None of the other authors on this manuscript have any financial conflict of interest.
Footnotes
These authors contributed equally to this work.
Previously published online: www.landesbioscience.com/journals/mabs/article/25439
References
- 1.Hafler DA, Slavik JM, Anderson DE, O’Connor KC, De Jager P, Baecher-Allan C. Multiple sclerosis. Immunol Rev. 2005;204:208–31. doi: 10.1111/j.0105-2896.2005.00240.x. [DOI] [PubMed] [Google Scholar]
- 2.Popescu BF, Lucchinetti CF. Pathology of demyelinating diseases. Annu Rev Pathol. 2012;7:185–217. doi: 10.1146/annurev-pathol-011811-132443. [DOI] [PubMed] [Google Scholar]
- 3.Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, et al. HERMES Trial Group B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358:676–88. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- 4.Cross AH, Stark JL, Lauber J, Ramsbottom MJ, Lyons JA. Rituximab reduces B cells and T cells in cerebrospinal fluid of multiple sclerosis patients. J Neuroimmunol. 2006;180:63–70. doi: 10.1016/j.jneuroim.2006.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cross AH, Klein RS, Piccio L. Rituximab combination therapy in relapsing multiple sclerosis. Ther Adv Neurol Disord. 2012;5:311–9. doi: 10.1177/1756285612461165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang H, Benoist C, Mathis D. Rituximab specifically depletes short-lived autoreactive plasma cells in a mouse model of inflammatory arthritis. Proc Natl Acad Sci U S A. 2010;107:4658–63. doi: 10.1073/pnas.1001074107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lyons JA, Ramsbottom MJ, Cross AH. Critical role of antigen-specific antibody in experimental autoimmune encephalomyelitis induced by recombinant myelin oligodendrocyte glycoprotein. Eur J Immunol. 2002;32:1905–13. doi: 10.1002/1521-4141(200207)32:7<1905::AID-IMMU1905>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 8.Bansal P, Khan T, Bussmeyer U, Challa DK, Swiercz R, Velmurugan R, et al. The encephalitogenic, human myelin oligodendrocyte glycoprotein-induced antibody repertoire is directed towards multiple epitopes in C57BL/6-immunized mice. J Immunol. 2013 doi: 10.4049/jimmunol.1300019. [DOI] [PubMed] [Google Scholar]
- 9.Vaccaro C, Zhou J, Ober RJ, Ward ES. Engineering the Fc region of immunoglobulin G to modulate in vivo antibody levels. Nat Biotechnol. 2005;23:1283–8. doi: 10.1038/nbt1143. [DOI] [PubMed] [Google Scholar]
- 10.Hansen RJ, Balthasar JP. Effects of intravenous immunoglobulin on platelet count and antiplatelet antibody disposition in a rat model of immune thrombocytopenia. Blood. 2002;100:2087–93. [PubMed] [Google Scholar]
- 11.Getman KE, Balthasar JP. Pharmacokinetic effects of 4C9, an anti-FcRn antibody, in rats: implications for the use of FcRn inhibitors for the treatment of humoral autoimmune and alloimmune conditions. J Pharm Sci. 2005;94:718–29. doi: 10.1002/jps.20297. [DOI] [PubMed] [Google Scholar]
- 12.Liu L, Garcia AM, Santoro H, Zhang Y, McDonnell K, Dumont J, et al. Amelioration of experimental autoimmune myasthenia gravis in rats by neonatal FcR blockade. J Immunol. 2007;178:5390–8. doi: 10.4049/jimmunol.178.8.5390. [DOI] [PubMed] [Google Scholar]
- 13.Mezo AR, McDonnell KA, Hehir CA, Low SC, Palombella VJ, Stattel JM, et al. Reduction of IgG in nonhuman primates by a peptide antagonist of the neonatal Fc receptor FcRn. Proc Natl Acad Sci U S A. 2008;105:2337–42. doi: 10.1073/pnas.0708960105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuo TT, Baker K, Yoshida M, Qiao SW, Aveson VG, Lencer WI, et al. Neonatal Fc receptor: from immunity to therapeutics. J Clin Immunol. 2010;30:777–89. doi: 10.1007/s10875-010-9468-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ober RJ, Martinez C, Vaccaro C, Zhou J, Ward ES. Visualizing the site and dynamics of IgG salvage by the MHC class I-related receptor, FcRn. J Immunol. 2004;172:2021–9. doi: 10.4049/jimmunol.172.4.2021. [DOI] [PubMed] [Google Scholar]
- 16.Gan Z, Ram S, Vaccaro C, Ober RJ, Ward ES. Analyses of the recycling receptor, FcRn, in live cells reveal novel pathways for lysosomal delivery. Traffic. 2009;10:600–14. doi: 10.1111/j.1600-0854.2009.00887.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vaccaro C, Bawdon R, Wanjie S, Ober RJ, Ward ES. Divergent activities of an engineered antibody in murine and human systems have implications for therapeutic antibodies. Proc Natl Acad Sci U S A. 2006;103:18709–14. doi: 10.1073/pnas.0606304103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patel DA, Puig-Canto A, Challa DK, Perez Montoyo H, Ober RJ, Ward ES. Neonatal Fc receptor blockade by Fc engineering ameliorates arthritis in a murine model. J Immunol. 2011;187:1015–22. doi: 10.4049/jimmunol.1003780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bleeker WK, Teeling JL, Hack CE. Accelerated autoantibody clearance by intravenous immunoglobulin therapy: studies in experimental models to determine the magnitude and time course of the effect. Blood. 2001;98:3136–42. doi: 10.1182/blood.V98.10.3136. [DOI] [PubMed] [Google Scholar]
- 20.Marta CB, Oliver AR, Sweet RA, Pfeiffer SE, Ruddle NH. Pathogenic myelin oligodendrocyte glycoprotein antibodies recognize glycosylated epitopes and perturb oligodendrocyte physiology. Proc Natl Acad Sci U S A. 2005;102:13992–7. doi: 10.1073/pnas.0504979102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schluesener HJ, Sobel RA, Linington C, Weiner HL. A monoclonal antibody against a myelin oligodendrocyte glycoprotein induces relapses and demyelination in central nervous system autoimmune disease. J Immunol. 1987;139:4016–21. [PubMed] [Google Scholar]
- 22.Linnington C, Webb M, Woodhams PL. A novel myelin-associated glycoprotein defined by a mouse monoclonal antibody. J Neuroimmunol. 1984;6:387–96. doi: 10.1016/0165-5728(84)90064-X. [DOI] [PubMed] [Google Scholar]
- 23.Amit AG, Mariuzza RA, Phillips SE, Poljak RJ. Three-dimensional structure of an antigen-antibody complex at 2.8 Å resolution. Science. 1986;233:747–53. doi: 10.1126/science.2426778. [DOI] [PubMed] [Google Scholar]
- 24.Clynes R. IVIG therapy: interfering with interferon-gamma. Immunity. 2007;26:4–6. doi: 10.1016/j.immuni.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 25.Baerenwaldt A, Biburger M, Nimmerjahn F. Mechanisms of action of intravenous immunoglobulins. Expert Rev Clin Immunol. 2010;6:425–34. doi: 10.1586/eci.10.9. [DOI] [PubMed] [Google Scholar]
- 26.Kieseier BC, Wiendl H, Hemmer B, Hartung HP. Treatment and treatment trials in multiple sclerosis. Curr Opin Neurol. 2007;20:286–93. doi: 10.1097/WCO.0b013e3281568b80. [DOI] [PubMed] [Google Scholar]
- 27.Haas J, Maas-Enriquez M, Hartung HP. Intravenous immunoglobulins in the treatment of relapsing remitting multiple sclerosis--results of a retrospective multicenter observational study over five years. Mult Scler. 2005;11:562–7. doi: 10.1191/1352458505ms1224oa. [DOI] [PubMed] [Google Scholar]
- 28.Bayry J, Kazatchkine MD, Kaveri SV. Shortage of human intravenous immunoglobulin--reasons and possible solutions. Nat Clin Pract Neurol. 2007;3:120–1. doi: 10.1038/ncpneuro0429. [DOI] [PubMed] [Google Scholar]
- 29.Petkova SB, Akilesh S, Sproule TJ, Christianson GJ, Al Khabbaz H, Brown AC, et al. Enhanced half-life of genetically engineered human IgG1 antibodies in a humanized FcRn mouse model: potential application in humorally mediated autoimmune disease. Int Immunol. 2006;18:1759–69. doi: 10.1093/intimm/dxl110. [DOI] [PubMed] [Google Scholar]
- 30.Foote J, Winter G. Antibody framework residues affecting the conformation of the hypervariable loops. J Mol Biol. 1992;224:487–99. doi: 10.1016/0022-2836(92)91010-M. [DOI] [PubMed] [Google Scholar]
- 31.Breithaupt C, Schubart A, Zander H, Skerra A, Huber R, Linington C, et al. Structural insights into the antigenicity of myelin oligodendrocyte glycoprotein. Proc Natl Acad Sci U S A. 2003;100:9446–51. doi: 10.1073/pnas.1133443100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Devaux B, Enderlin F, Wallner B, Smilek DE. Induction of EAE in mice with recombinant human MOG, and treatment of EAE with a MOG peptide. J Neuroimmunol. 1997;75:169–73. doi: 10.1016/S0165-5728(97)00019-2. [DOI] [PubMed] [Google Scholar]
- 33.Lyons JA, San M, Happ MP, Cross AH. B cells are critical to induction of experimental allergic encephalomyelitis by protein but not by a short encephalitogenic peptide. Eur J Immunol. 1999;29:3432–9. doi: 10.1002/(SICI)1521-4141(199911)29:11<3432::AID-IMMU3432>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 34.Radu CG, Anderton SM, Firan M, Wraith DC, Ward ES. Detection of autoreactive T cells in H-2u mice using peptide-MHC multimers. Int Immunol. 2000;12:1553–60. doi: 10.1093/intimm/12.11.1553. [DOI] [PubMed] [Google Scholar]
- 35.Kim JK, Tsen MF, Ghetie V, Ward ES. Localization of the site of the murine IgG1 molecule that is involved in binding to the murine intestinal Fc receptor. Eur J Immunol. 1994;24:2429–34. doi: 10.1002/eji.1830241025. [DOI] [PubMed] [Google Scholar]
- 36.Minguela A, Pastor S, Mi W, Richardson JA, Ward ES. Feedback regulation of murine autoimmunity via dominant anti-inflammatory effects of interferon gamma. J Immunol. 2007;178:134–44. doi: 10.4049/jimmunol.178.1.134. [DOI] [PubMed] [Google Scholar]